
Abstract
Background
Magnesium ions (Mg2+) increase and prolong opioid analgesia in chronic and acute pain. The nature of this synergistic analgesic interaction has not yet been explained. Our aim was to investigate whether Mg2+ alter tramadol pharmacokinetics. Our secondary goal was to assess the safety of the combination.
Methods
Tramadol was administered to healthy Caucasian subjects with and without Mg2+ as (1) single 100-mg and (2) multiple 50-mg oral doses. Mg2+ was administered orally at doses of 150 mg and 75 mg per tramadol dosing in a single- and multiple-dose study, respectively. Both studies were randomized, open label, laboratory-blinded, two-period, two-treatment, crossover trials. The plasma concentrations of tramadol and its active metabolite, O-desmethyltramadol, were measured.
Results
A total of 25 and 26 subjects completed the single- and multiple-dose study, respectively. Both primary and secondary pharmacokinetic parameters were similar. The 90% confidence intervals for Cmax and AUC0-t geometric mean ratios for tramadol were 91.95–102.40% and 93.22–102.76%. The 90% confidence intervals for Cmax,ss and AUC0-τ geometric mean ratios for tramadol were 93.85–103.31% and 99.04–105.27%. The 90% confidence intervals for primary pharmacokinetic parameters were within the acceptance range. ANOVA did not show any statistically significant contribution of the formulation factor (p > 0.05) in either study. Adverse events and clinical safety were similar in the presence and absence of Mg2+.
Conclusions
The absence of Mg2+ interaction with tramadol pharmacokinetics and safety suggests that this combination may be used in the clinical practice for the pharmacotherapy of pain.
Graphic abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pain, especially chronic, decreases the quality of life and results in economic burdens for both individuals and the society in general [1]. Thus, it is important to continue research efforts for an effective pharmacotherapy of pain [2]. Opioids are still among the most powerful painkillers. Unfortunately, the use of these compounds is burdened with many side effects, such as respiratory depression, opioid hyperalgesia, development of tolerance, constipation, and addiction risk, which limits their use. In fact, despite a number of opioid prescription restrictions imposed several decades ago, we have witnessed opioid addiction that has reached epidemic proportions in the United States in the twentieth and twenty-first centuries. To reduce opioid doses (so-called sparing dose), and thus, reduce the risk of side effects, attempts have been made to combine opioids with adjuvant analgesics, such as anticonvulsants (gabapentin), antidepressants (amitriptyline), and N-methyl-d-aspartate (NMDA) receptor antagonists (ketamine). It is worth noting that magnesium ions [Mg2+] belong to the class of physiological antagonists of NMDA receptors [3, 4]. Results of own studies and the literature data demonstrated that Mg2+ increased opioid analgesia in experimental animal models of neuropathic [5,6,7], inflammatory [5], as well as acute [8,9,10] pain. Furthermore, the results of several clinical trials confirmed that parenterally administered Mg2+ reduces opioid consumption and improves postoperative pain scores without increasing opioid side effects [11,12,13,14]. Thus, the co-administration of opioids and Mg2+ may represent a valuable therapeutic option [15], but more data on the efficacy and safety of this combination are needed. Moreover, oral administration of opioids combined with magnesium salts would extend the use of this combination therapy in outpatient care.
The nature of synergistic analgesic interactions between opioids and Mg2+ remains unclear. However, it should be emphasize that Ionotropic NMDA receptors have been implicated in the underlying mechanisms of pain. The activation of these receptors removes the Mg2+ block in the channel. This results in calcium entering the cell and neuronal sensitization [16]. It was suggested that NMDA and opioid receptors are co-localized on the cell membrane of the same neuron and their C-termini are associated [17]. Moreover, the morphine-induced activation of the mu-opioid receptor leads to the phosphorylation of the Ser890 residue of the NMDA receptor’s C-terminus. This causes a dissociation of both receptors, an activation of the NMDA receptor, and an induction of further mechanisms leading to mu receptor phosphorylation and receptor desensitization [17, 18]. Thus, it can be assumed that Mg2+ reduces NMDA receptor-induced processes, decreases opioid receptor phosphorylation, and consequently enhances opioid analgesia [15].
Another hypothesis on the mechanism of Mg2+-potentiating opioid analgesia is the influence on opioid pharmacokinetics. The absorption process seems most likely to be affected, but increased Mg2+ content in the body may also influence distribution or metabolism, e.g., by changing conformation of proteins. Not all processes involved in tramadol distribution or metabolism are known and described. Thus, before defining the mechanism of hypothetical pharmacokinetic interaction, the first question is whether such an interaction exists. Our work focuses on one of the group of opioid painkiller—tramadol. The clinical pharmacology of this compound was reviewed by Grond and Sablotzky [19] and by Leppert [20]. After oral administration, the drug is rapidly and almost completely absorbed. A different enantiomeric ratio for tramadol and its main metabolites has been observed after intravenous and oral administration [21] and some gender differences have also been reported [22]. The mean plasma elimination half-life in healthy subjects after a single-dose administration ranges from 4.7 to 7.9 h [23,24,25]. After multiple dosing (50 mg every 6 h), the plasma steady state is reached within 3 days [26]. The influence of different pharmaceutical forms on the pharmacokinetics was investigated in elderly patients [27] and in patients who underwent total gastric resection [28]. Although the pharmacokinetics of tramadol and O-desmethyltramadol in humans is well described, we have failed to find any report describing how the pharmacokinetics may be altered by the co-administration of Mg2+.
Therefore, the aim of this study was to investigate whether the increased and prolonged tramadol analgesia in the presence of Mg2+ has any pharmacokinetic background. For this purpose, we have measured the concentrations of tramadol and its active metabolite, O-desmethyltramadol, after single and multiple oral administrations to healthy volunteers. The secondary goal of our study was to assess the safety of this combination.
Methods
Ethics and study design
The clinical study protocols were approved by an Independent Ethics Committee of the Warsaw Medical Chamber, Warsaw, Poland. Each subject signed a subject information and informed consent form prior to the screening procedures performed in accordance with the most recent version of the Declaration of Helsinki, current Good Clinical Practice guidelines, Polish laws governing the conduct of clinical investigations, and standard operating procedures of the clinical site. The studies were approved by the Polish Ministry of Health, registered by the Polish Regulatory Authorities – Central Register of Clinical Trials. The EudraCT numbers were 2014-004716-11 and 2014-004717-82 for the single- and multiple-dose studies, respectively.
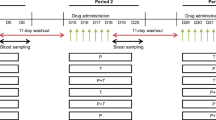
Similar procedures were applied in both single- and multiple-dose studies, thus, the “Methods” section is combined. Both comparative pharmacokinetic studies were designed according to the European Medicines Agency (EMA) guideline on bioequivalence [29] as a randomized, open label, two-period, two-treatment, crossover trial. These single-site studies were conducted by BioVirtus Research Site Sp. z o.o. in Phase 1 Unit in Kajetany, Poland. Block randomization with block size of four was applied. The studies were laboratory-blinded with a wash-out period from 7 to 14 days. The same standard procedures were applied in both study periods, schemes for both studies are presented in the Supplementary Information section (Supplementary Figs. S1 and S2).
Subjects
The following assumptions were adopted for the single-dose study: (1) within subject variation for Cmax of no more than 20% [26], (2) ratio of pharmacokinetic parameters of no more than 1.05, and (3) a priori power of 0.80. The calculated sample size of 24 to complete the study was increased to 26 subjects to be randomized to account for the possible dropout. A total of 38 subjects were screened to meet this goal.
The following assumptions were made for the multiple-dose study: (1) within subject variation for Cmax,ss of no more than 15% [26], (2) ratio of pharmacokinetic parameters of no more than 1.10, and (3) a priori power of 0.80. The calculated sample size of 22 to complete the study was increased to 30 subjects to be randomized to account for the possible dropout. A total of 56 subjects were screened to meet this goal.
The subjects enrolled met the following inclusion criteria: males and non-pregnant, non-lactating females 18–55 years old, body mass index (BMI) in the range of 18.5–30.0 kg/m2, non-smoker for at least 3 months before screening, and good general health condition assessed by medical history showing no co-morbidities, physical examination, normal electrocardiogram (ECG), and standard clinical laboratory tests within normal values.
Subjects were excluded if they met any of the exclusion criteria: history of clinically significant medical condition including arrhythmias and cardiac disease, hematologic condition and coagulation disorders, immunologic conditions, diseases of the musculoskeletal system, significant lung disease (bronchospastic respiratory disease), diabetes mellitus, kidney or liver insufficiency, endocrine disorders, neurologic or psychiatric disease, and active infection.
Subjects were also excluded in the following cases: drug abuse within last 5 years or alcohol abuse within 1 year, hypersensitivity or confirmed allergy to tramadol and/or to any excipient of the study products, history of gastrointestinal dysfunction, clinically significant abnormal or uncontrolled values of hematology, clinical chemistry or urinalysis that would affect the interpretation of the study data or the subject’s participation in the study, use of non-prescription drugs (including vitamins, herbal and dietary supplements) within 7 days or a prescription drug (excluding contraceptive pills and hormone replacement therapy) within 14 days or 5 half-lives (whichever longer) prior to the first dose of the study medication, or if the subject’s diet was deemed non-compliant by the investigator because of the inappropriate content of protein, fat and carbohydrates, or donation of at least 400 mL of blood within 90 days before the commencement of the study.
Study products
The same products were used in both studies. The test product—50 mg rac-tramadol hydrochloride tablets with magnesium lactate corresponding to 75 mg of Mg2+—was manufactured by PozLab Sp. z o.o. (batch No. F03005, expiry date 09.2016). The reference product—without Mg2+—commercially available Tramadol Vitabalans® 50-mg tablets was manufactured by Vitabalans Oy (batch No. 1197502, expiry date 12.2017).
Study product administration
In both studies, the study product was administered orally with 250 mL of water at room temperature in the sitting position to be swallowed completely. Each subject’s oral cavity was checked following drug administration to confirm swallowing of the tablet. No drinking (except the water used for the product administration) was allowed from 1 h before to 1 h after the drug administration.
In the single-dose study, two 50-mg tablets of the test product or the reference product were administered in a fasted state on day 0 of each period. No food was allowed until 4 h after drug administration. In the multiple-dose study, one 50-mg tablet of the test product or the reference product was administered every 6 h on days 1 (two doses), 2, 3, 4, 5 and 6 (two doses) of each period. The total dose of tramadol in each period was 1000 mg. Subjects were fasted on day 1 for at least 4 h before the first dose and for 10 h before and 4 h after the last dose administration on day 6.
Blood sampling
In both studies, the subjects had an intravenous cannula since day 0. Blood samples were centrifuged at 1500×g at 4 °C for 10 min, immediately after collection. The plasma was frozen in polypropylene vials at no less than − 65 °C for storage until the analysis.
In the single-dose study, blood samples were collected at 18 time points: pre-dose, 0.17; 0.5; 0.75; 1; 1.5; 2; 2.5; 3; 3.5; 4; 5; 6; 9; 12; 16; 24, and 36 h post-dose. In the multiple-dose study, blood samples were collected at 16 time points: 0 (pre-dose), and at 96, 102, 108, 114, 114.25, 114.5, 114.75, 115, 115.5, 116, 116.5, 117, 118, 119 and 120 h after the first dose administration. In total, a maximum of 187 mL and 175 mL of blood was collected from a single subject throughout the duration of the single- and multiple-dose studies, respectively.
Bioanalysis of tramadol and O-desmethyltramadol
Tramadol and O-desmethyltramadol plasma concentrations were determined using the achiral liquid chromatography–mass spectrometry. Citrate was used as an anticoagulant for clinical samples, calibration standards, and quality control samples. Tramadol-d6 and O-desmethyltramadol-d6 were used as internal standards. Liquid–liquid extraction with tert-butyl methyl ether and sodium hydroxide was used for sample preparation (adapted from Tao et al. [30] and Godoy et al., [31], see also Supplementary Information). Novel conditions of chromatographic separation were applied using Kinetex biphenyl column (150 × 3.0 mm, 2.6 μm; Phenomenex, Torrance, CA, USA) at 45 ºC. The mobile phase was a mixture of 0.1% aqueous acetic acid and methanol (4:6 v/v). Ions of tramadol, O-desmethyltramadol, tramadol-d6, and O-desmethyltramadol-d6 were monitored at m/z ratio of 264.0, 250.0, 270.0, and 256.0, respectively. The study was conducted in compliance with the principles of Good Laboratory Practice.
Pharmacokinetic and statistical analysis
The primary endpoints of the single-dose study were the maximum plasma concentration (Cmax) and the measured area under the plasma concentration vs. time curve (AUC0-t) for tramadol. The area under the plasma concentration vs. time curve extrapolated to infinity (AUC0-∞), the time to reach maximum plasma concentration of the drug (tmax), and the elimination half-life (t1/2) for tramadol were selected as secondary parameters, which also included Cmax, AUC0-t, AUC0-∞, t1/2, and tmax for O-desmethyltramadol.
The primary endpoints of the multiple-dose study were the maximum plasma concentration in steady state (Cmax,ss) and the area under the plasma concentration vs. time curve in a dosing interval (AUC0-τ) for tramadol. Steady-state parameters, namely the time to reach maximum plasma concentration of the drug (tmax,ss), minimum plasma concentration (Cmin,ss), average concentration (Cav,ss), and peak trough fluctuation (PTF) for tramadol were selected as secondary parameters, which also included Cmax,ss, AUC0-τ, tmax,ss, Cmin,ss, Cav,ss, and PTF for O-desmethyltramadol.
The pharmacokinetic parameters were determined using Phoenix® WinNonlin™ version 6.4 (Pharsight Corp.). Cmax and tmax were obtained directly from experimental data. The elimination rate constant (k) was estimated from 3 to 6 points by least square regression analysis. The t1/2 was calculated as ln(2)/k. The AUC0-t was calculated by the trapezoidal rule up to the last measurable plasma concentration (Ct). The AUC0-∞ was calculated as the sum of AUC0-t and the extrapolated area (AUCrest = Ct/ k).
The pharmacokinetics at steady state was studied for the dosing interval at 114–120 h after the first drug administration in a given period. The values of Cmax,ss, Cmin,ss, and tmax,ss were obtained directly from experimental data. The AUC0-τ values were calculated by the trapezoidal rule. Cav,ss was calculated as AUC0-τ/τ. PTF was calculated as (Cmax,ss−Cmin,ss)/Cav,ss and presented as a percent value. The linear regression approach was used to verify whether the steady state was attained.
The SAS System for Windows version 9.4 was used for statistical analysis. Normal distribution of the primary pharmacokinetic parameters was tested using the Shapiro–Wilk, Kolmogorov–Smirnov, Cramer–von Mises and Anderson–Darling procedures at α = 0.05 significance level. The primary and secondary parameters except tmax were logarithmically transformed and subjected to the analysis of variance (ANOVA). The 90% confidence intervals of the test over the reference product’s geometric mean ratio of the primary pharmacokinetic parameters (AUC0-t and Cmax for tramadol in the single-dose study and Cmax,ss and AUC0-τ for tramadol in the multiple-dose study) were constructed. Mg2+ was assumed to have no influence when 90% confidence intervals were within the range of 80.00–125.00% [29]. Such analysis is equivalent to two one-sided Student’s t tests proposed by Schuirmann [32] with the null hypothesis of bioinequivalence (level of significance α = 0.05).
Safety assessment
In both the studies, clinical safety had been assessed since the first drug administration and until the end of the study including subjects that did not complete the study. Monitoring of adverse events was complemented by the evaluation of standard clinical laboratory parameters, physical examinations, vital signs, and 12-lead ECG. The adverse events were classified by the system organ class and the preferred term using the Medical Dictionary for Regulatory Activities version 15.1. The adverse event severity was assessed subjectively and classified by investigators according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.02. Information on the adverse events was obtained from medical observations, laboratory test analyses, and spontaneous subject reporting after product administration. For every adverse event, the following data were recorded: type, severity, onset and resolution date, concomitant medications, relationship to the investigational products or procedures, and actions taken.
Results
Study population
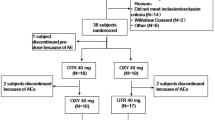
In the single-dose study, 26 subjects (Table 1) met the inclusion criteria, and 25 subjects completed the study (Supplementary Fig. S3). One subject was withdrawn due to the positive result of toxicology on day 1 of Period 2.
In the multiple-dose study, 30 subjects were enrolled (Table 1) and 26 of them completed the study (Supplementary Fig. S4). Three subjects discontinued the study during Period 1 due to adverse events. One subject was withdrawn at the beginning of Period 2 due to the positive toxicology test for opiates.
Bioanalytical method validation
All validation tests recommended by the EMA guideline [33] met the acceptance criteria (Supplementary Table S1). Calibration curves constructed by plotting the analyte-to-internal standard peak area ratios against the nominal concentrations were linear within the range of 5.0–750.0 ng/mL for tramadol and 2.5–150.0 ng/mL for O-desmethyltramadol. To confirm the method selectivity, blank human plasma from six different sources (including hemolyzed and hyperlipidemic plasma) were analyzed. There were no peaks influencing the quantification near the retention times of tramadol, O-desmethyltramadol, and the internal standards. The chromatograms of blank plasma samples containing N-desmethyltramadol, ibuprofen, and paracetamol did not show any significant interferences at the retention times of tramadol, O-desmethyltramadol, and the internal standards. Intra-run (within 1 day) and inter-run (within 3 days) accuracy and precision for both tramadol and O-desmethyltramadol met the acceptance criteria (Supplementary Tables S2 and S3). The stability of both analytes was confirmed using 90% confidence intervals (Supplementary Table S4, [34]). Relative standard deviation of normalized matrix factor was below 3% for 6 studied plasma sources including hemolyzed and hyperlipidemic plasma.
Quality control samples analyzed during the study met the acceptance criteria in all sequences. The method’s reliability during the study was further confirmed by the incurred samples reanalysis (ISR). The acceptance criteria, i.e., reanalysis within ± 20% of the mean concentration [33], in the single-dose study were met for 84 out of 92 samples (91%) in the case of tramadol, and for 85 out of 92 samples (92%) in the case of O-desmethyltramadol. The acceptance criteria in the multiple-dose study were met for 84 out of 86 samples (98%) in the case of tramadol, and for 81 out of 86 samples (94%) in the case of O-desmethyltramadol. No trends were observed on the complementary plots: %difference versus mean concentration and cumulative ISR plot (Supplementary Fig. S5 and S6) [35].
Pharmacokinetic and statistical analysis
A hypothesis on the log-normal distribution of the primary parameters (Cmax, AUC0-t, Cmax,ss and AUC0-τ) for tramadol for the test product and the reference product (p > 0.15) could not be rejected. The mean values of the primary and secondary pharmacokinetic parameters were similar for both formulations (Tables 2 and 3). The mean and individual plasma concentrations vs. time profiles in the presence and absence of Mg2+ were similar for both tramadol and O-desmethyltramadol (Figs. 1, 2 and S7–S14). A graphical presentation for the single-dose study was truncated at 24 h because only a few samples with the concentrations above the lower limit of quantification were recorded for both analytes at 36 h. In the single-dose study, the AUC0-t to AUC0-∞ ratio was over 0.9 in all profiles for tramadol. As for O-desmethyltramadol, this ratio was higher than 0.8 in 47 out of 50 profiles (94%). In the multiple-dose study, the linear regression of the concentration vs. time was calculated at three time points (96, 102 and 108 h after the first dose administration in a given period). The slopes of the regression lines could not be differentiated from zero. Hence, the steady state was attained.

Mean tramadol (solid line) and O-desmethyltramadol (dotted line) plasma concentration vs. time curves in 25 healthy subjects following a single oral dose of tramadol (2 × 50 mg) with (filled points) and without magnesium ions (unfilled points) presented in the linear/linear scale (A) and log/linear scale (B)

Mean tramadol (solid line) and O-desmethyltramadol (dotted line) plasma concentration vs. time curves in 26 healthy subjects following multiple oral doses of tramadol (50 mg) with (filled points) and without magnesium ions (unfilled points) presented in the linear/linear scale (a) and log/linear scale (b)
For the single-dose study, the 90% confidence intervals for both primary pharmacokinetic parameters (Cmax and AUC0-t for tramadol) were within the acceptance range of 80.00–125.00% (Table 2, estimated power > 0.999). The ANOVA indicated that the primary sources of variation for both tramadol and O-desmethyltramadol are attributed to the sequence and inter-subject effects, which were significant for all parameters (p ≤ 0.05, Table 4). The formulation effect, which corresponds to the Mg2+ influence, was not significant for the listed parameters (p > 0.05, Table 4).
For the multiple-dose study, the 90% confidence intervals for both primary pharmacokinetic parameters (Cmax,ss and AUC0-τ for tramadol) were within the acceptance range of 80.00–125.00% (Table 3, estimated power > 0.999). The ANOVA indicated that the primary sources of variation for both tramadol and O-desmethyltramadol are attributed to the sequence and inter-subject effects that were significant for all parameters (p ≤ 0.05, Table 4). The formulation effect for the listed parameters was not significant (p > 0.05, Table 4).
Safety
There were neither serious adverse events nor deaths recorded during both studies. All adverse events were mild to moderate in severity. There were no clinically significant changes in the mean clinical laboratory test results, electrocardiogram findings, or vital signs. The physical examination results remained unchanged for all the participants.
In the single-dose study, 20 adverse events were reported in 12 subjects (46%). Out of 26 subjects who received the study products in Period 1, 9 subjects (35%) experienced 12 adverse events; 5 adverse events were classified as possibly related and 7 as not related to the study products. After the product administration in Period 2, 6 subjects (23%) experienced a total of 8 adverse events; 4 adverse events were classified as possibly related and 4 as not related to the study products. The types of adverse events were similar across both the treatments (Table 5, Supplementary Table S5).
In the multiple-dose study, 136 adverse events were reported in 26 subjects (87%). Out of 30 subjects who received the study products in Period 1, 26 subjects experienced 91 adverse events (14 adverse events were classified as probably related, 36 as possibly related, 15 as unlikely related, and 26 as not related to the study products). After the product administration in Period 2, 15 subjects out of 26 continuing the study experienced a total of 33 adverse events (1 adverse event was classified as probably related, 11 as possibly related, 6 as unlikely related, and 15 as not related to the study products). After being discharged from the clinic in Period 2 and before the follow-up visit, 10 subjects out of 26 continuing the study experienced a total of 12 adverse events (2 adverse events were classified as probably related, 6 as possibly related, 1 as unlikely related and, 3 as not related to the study products). The types of adverse events were similar across both treatments (Table 5, Supplementary Table S6).
Discussion
The results of the study indicate that Mg2+ ions do not alter tramadol pharmacokinetics. Both primary and secondary pharmacokinetic parameters were similar, with overlapping average plasma profiles. The 90% confidence intervals for the geometric mean ratios of the primary pharmacokinetic parameters met the acceptance criteria, and the formulation factor in ANOVA was not significant. The adverse events were also similar in the subjects receiving tramadol with and without Mg2+.
Magnesium deficiency is a common condition in Poland and it may mask the influence of Mg2+. Therefore, a single-dose approach would not be sufficient to draw valid conclusions. However, it would be unethical and unnecessary to conduct a multiple-dose study if the single-dose study revealed a significant influence of Mg2+ on tramadol pharmacokinetics. Since Mg2+ effect was not observed in the single-dose study, a confirmatory study after a multiple-dose administration was conducted. The design of both studies enabled us to draw valid conclusions. The bioanalytical method had an appropriate range to measure tramadol and O-desmethyltramadol concentrations. Sampling schedules enabled proper characterization of the pharmacokinetic profiles. Tramadol and O-desmethyltramadol pharmacokinetics reported in this paper are in line with literature data for achiral methods [23, 24]. Pharmacokinetics in overweight subjects were similar to normal weight subjects, in line with observations of Porażka et al. [36]. The studies were conducted according to the EMA bioequivalence guideline [29] because its methodology is appropriate for comparing drug formulations with and without Mg2+.
The population consisting of healthy subjects may be considered a limitation of the study. Healthy subjects were selected to avoid any influence of co-morbidities and concomitant medications on tramadol pharmacokinetics. Hence, just as in bioequivalence studies, some differences in pharmacokinetics between patients and healthy subjects can be anticipated. In our study, we have observed very narrow confidence intervals and failed to find any indication that the interaction of Mg2+ with tramadol would be different in both the groups.
The CYP2D6 gene—responsible for the metabolism of tramadol to O-desmethyltramadol—is highly polymorphic. We decided to avoid genotyping and exclusion of poor and ultra-rapid CYP2D6 metabolizers to make the study population more representative for the general population. In the single-dose study, Cmax for tramadol ranged from 132.51 to 458.16 ng/mL for the test product and from 155.54 to 435.22 ng/mL for the reference product. These values suggest differences in metabolism of tramadol in particular subjects. However, they did not influence the study results as 90% confidence intervals for geometric means test to reference ratio were within the limits for primary pharmacokinetic parameters. Still, this issue may be interesting for further research.
In our studies, Mg2+ was not defined as an active substance, thus, its determination was not required. We hope that this paper will inspire future research on Mg2+ levels monitoring in opioid-treated patients. In addition, one may argue that Mg2+ is present in numerous medicinal products containing opioids—including the reference product in these studies—as magnesium stearate. Both very low solubility and low content in tablets disqualify this excipient as a source of Mg2+ and preclude its interaction with opioids.
As expected, the frequency of adverse events was considerably higher in the multiple-dose study (87%) than in the single-dose study (46%). Typical adverse events for tramadol such as nausea and vomiting were observed and classified as probably related to the product. The results confirmed that the prophylaxis for these symptoms was not required in any subjects. There were no novel or atypical adverse event related to the study product observed (Table 5, Supplementary Tables S5 and S6).
The results of our studies do not support the hypothesis that the increased and prolonged analgesia of tramadol in the presence of Mg2+ is based on the pharmacokinetics of the parent drug and its main metabolite. Moreover, the analyses of the adverse events indicate that Mg2+ does not alter the safety of tramadol therapy. The absence of Mg2+ influence on the pharmacokinetics and adverse events of tramadol confirms the safety of this combination in clinical practice.
In a multicenter, single blinded, parallel-group study to assess efficacy, safety and tolerability of the combination of tramadol with magnesium lactate in the management of chronic pain in subjects with osteoarthritis of the hip or/and knee, it was demonstrated that the combined oral administration of magnesium salt with tramadol at a lower dose induced a comparable analgesic effect as a reference product—tramadol at a higher dose (data to be published in the future). Unfortunately, the increase of opioid analgesia after oral administration of Mg2+ has not been confirmed in cancer patients receiving morphine [37]. Due to these discrepancies, further clinical studies focused on other types of pain are needed; however, the results of our pharmacokinetic research suggest that the co-administration of opioids (particularly tramadol) with Mg2+ may be considered as a promising novel pharmacotherapy of pain. This research is particularly important in the light of the over-prescription of opioids in recent years, and thus, the spreading wave of opioid addiction worldwide. [4, 38,39,40,41,42,43]. The addition of Mg2+ may help to adequately control opioid treatment by reducing consumption, and thus, reducing adverse events [11,12,13].
Conclusions
In both single- and multiple-dose studies, the absence of Mg2+ influence on tramadol pharmacokinetics and safety was observed after oral administration to healthy subjects. Pharmacokinetic parameters and profiles for tramadol and O-desmethyltramadol were similar. The lack of pharmacokinetic interaction between Mg2+ and tramadol suggests that the combination of tramadol and Mg2+ is safe in clinical practice, supporting the co-administration of opioids with Mg2+ as a promising novel strategy for the pharmacotherapy of pain.
References
Fornasari D. Pharmacotherapy for neuropathic pain. A review. Pain Ther. 2017;6:25–33.
Knezevic NN, Mandalia S, Raasch J, Knezevic I, Candido KD. Treatment of chronic low back pain - new approaches on the horizon. J Pain Res. 2017;10:1111–23.
Karanges EA, Blanch B, Buckley NA, Pearson SA. Twenty-five years of prescription opioid use in Australia: a whole-of-population analysis using pharmaceutical claims. Br J Clin Pharmacol. 2016;82(1):255–67.
Floyd CN, Warren JB. Opioids out of control. Br J Clin Pharmacol. 2018;84(5):813–5.
Begon S, Pickering G, Eschalier A, Dubray C. Magnesium increases morphine analgesic effect in different experimental models of pain. Anesthesiol. 2002;96:627–32.
Bujalska M, Malinowska E, Makulska-Nowak H, Gumułka SW. Magnesium ions and opioid agonist activity in streptozotocin-induced hyperalgesia. Pharmacol. 2008;82:180–6.
Bujalska M, Makulska-Nowak H, Gumułka SW. Magnesium ions and opioid agonists in vincristine-induced neuropathy. Pharmacol Rep. 2009;61:1096–104.
Bujalska-Zadrożny M, Duda K. Additive effect of combined application of magnesium and MK-801 on analgesic action of morphine. Pharmacol. 2014;93:113–9.
Bujalska-Zadrożny M, Kulik K, Ordak M, Sasinowska-Motyl M, Gąsińska E, de Corde A, et al. Micronized organic magnesium salts enhance opioid analgesia in rats. PLoS ONE. 2016;11:e0161776.
McCarthy RJ, Kroin JS, Tuman KJ, Penn RD, Ivankovich AD. Antinociceptive potentiation and attenuation of tolerance by intrathecal co-infusion of magnesium sulfate and morphine in rats. Anesth Analg. 1998;86:830–6.
Bolcal C, Iyem H, Sargin M, Mataraci I, Yildirim V, Doganci S, et al. Comparison of magnesium sulfate with opioid and NSAIDs on postoperative pain management after coronary artery bypass surgery. J Cardiothorac Vasc Anesth. 2005;19:714–8.
Ferasatkish R, Dabbagh A, Alavi M, Mollasadeghi G, Hydarpur E, Moghadam AA, et al. Effect of magnesium sulfate on extubation time and acute pain in coronary artery bypass surgery. Acta Anaesthesiol Scand. 2008;52:1348–52.
Mentes O, Harlak A, Yigit T, Balkan A, Balkan M, Cosar A, et al. Effect of intraoperative magnesium sulphate infusion on pain relief after laparoscopic cholecystectomy. Acta Anaesthesiol Scand. 2008;52:1353–9.
Jabbour HJ, Naccache NM, Jawish RJ, Abou Zeid HA, Jabbour KB, Rabbaa-Khabbazet LG, et al. Ketamine and magnesium association reduces morphine consumption after scoliosis surgery: prospective randomised double-blind study. Acta Anaesthesiol Scand. 2014;58:572–9.
Bujalska-Zadrożny M, Tatarkiewicz J, Kulik K, Filip M, Naruszewicz M. Magnesium enhances opioid-induced analgesia—what we have learnt in the past decades? Eur J Pharm Sci. 2017;99:113–27.
McCartney CJL, Sinha A, Katz J. A qualitative systematic review of the role of N-methyl-D-aspartate receptor antagonists in preventive analgesia. Anesth Analg. 2004;98:1385–400.
Rodríguez-Muñoz M, Sánchez-Blázquez P, Vicente- Sánchez A, Berrocoso E, Garzón J. The Mu-Opioid Receptor and the NMDA Receptor Associate in PAG Neurons: Implications in Pain Control. Neuropsychopharmacol. 2012;37:338–49.
Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, et al. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem. 1997;272:5157–66.
Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43:879–923.
Leppert W. Tramadol as an analgesic for mild to moderate cancer pain. Pharmacol Rep. 2009;61:978–92.
García-Quetglas EG, Azanza JR, Cardenas E, Sádaba B, Campanero MA. Stereoselective pharmacokinetic analysis of tramadol and its main phase I metabolites in healthy subjects after intravenous and oral administration of racemic tramadol. Biopharm Drug Dispos. 2007;28:19–33.
Hui-Chen L, Yang Y, Na W, Ming D, Jian-Fang L, Hong-Yuan X. Pharmacokinetics of the enantiomers of trans-tramadol and its active metabolite, trans-O-demethyltramadol, in healthy male and female Chinese volunteers. Chirality. 2004;16:112–8.
Dyderski S, Szkutnik D, Zgrabczyńska M, Drobnik L. Bioavailability of tramadol hydrochloride from tramadol capsules 50 mg. Acta Pol Pharm. 2001;58:345–9.
Silva Mde F, Schramm SG, Kano EK, Mori Koono EE, Porta V, dos Reis Serra CH. Bioequivalence evaluation of single doses of two tramadol formulations: a randomized, open-label, two-period crossover study in healthy Brazilian volunteers. Clin Ther. 2010;32:758–65.
Zhou X, Liu J. Fluorescence detection of tramadol in healthy Chinese volunteers by high-performance liquid chromatography and bioequivalence assessment. Drug Des Devel Ther. 2015;9:1225–31.
Karhu D, Fradette C, Potgieter MA, Ferreira MM, Terblanche J. Comparative pharmacokinetics of a once-daily tramadol extended-release tablet and an immediate-release reference product following single-dose and multiple-dose administration. J Clin Pharmacol. 2010;50:544–53.
Likar R, Wittels M, Molnar M, Kager I, Ziervogel G, Sittl R. Pharmacokinetic and pharmacodynamic properties of tramadol IR and SR in elderly patients: a prospective, age-group-controlled study. Clin Ther. 2006;28:2022–39.
Szałek E, Karbownik A, Murawa D, Połom K, Urbaniak B, Grabowski T, et al. The pharmacokinetics of the effervescent vs conventional tramadol/paracetamol fixed-dose combination tablet in patients after total gastric resection. Pharmacol Rep. 2014;66:159–64.
European Medicines Agency. Guideline on the investigation of bioequivalence (CPMP/EWP/QWP/1401/98/Rev. 1/Corr**). Published January 2010.
Tao Q, Stone DJ Jr, Borenstein MR, Jean-Bart V, Codd EE, Coogan TP, et al. Gas chromatographic method using nitrogen-phosphorus detection for the measurement of tramadol and its O-desmethyl metabolite in plasma and brain tissue of mice and rats. J Chromatogr B. 2001;763:165–71.
Godoy AL, De Moraes NV, Martinez EZ, Carvalho TM, Marques MP, Lanchote VL. Simultaneous analysis of tramadol, O-desmethyltramadol, and N-desmethyltramadol enantiomers in rat plasma by high-performance liquid chromatography-tandem mass spectrometry: application to pharmacokinetics. Chirality. 2011;23:287–93.
Schuirmann DJ. A comparison of the 2 one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15:657–80.
European Medicines Agency. Guideline on bioanalytical method validation (EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2**). Published July 2011.
Rudzki PJ, Leś A. Application of confidence intervals to bioanalytical method validation—drug stability in biological matrix testing. Acta Pol Pharm. 2008;65:743–7.
Rudzki PJ, Biecek P, Kaza M. Comprehensive graphical presentation of data from incurred sample reanalysis. Bioanalysis. 2017;9:947–56.
Porażka J, Szałek E, Połom W, Czajkowski M, Grabowski T, Matuszewski M, et al. Influence of obesity and type 2 diabetes mellitus on the pharmacokinetics of tramadol after single oral dose administration. Eur J Drug Metab Pharmacokinet. 2019;44:579–84.
Baaklini LG, Arruda GV, Sakata RK. Assessment of the analgesic effect of magnesium and morphine in combination in patients with cancer pain: a comparative randomized double-blind study. Am J Hosp Palliat Care. 2017;34:353–7.
Brząkała J, Leppert W. The role of rapid onset fentanyl products in the management of breakthrough pain in cancer patients. Pharmacol Rep. 2019;71:438–42.
Zatorski H, Salaga M, Zielińska M, Wasilewski A, Misicka A, Sacharczuk M, et al. High activity of endogenous opioid system protects against gastric damage development in mouse models of gastric mucosal injury. Pharmacol Rep. 2019;71:218–24.
LaVigne J, Keresztes A, Chiem D, Streicher JM. The endomorphin-1/2 and dynorphin-B peptides display biased agonism at the mu opioid receptor. Pharmacol Rep. 2020;72:465–71.
Szymaszkiewicz A, Włodarczyk J, Mazur M, Olczak J, Fichna J, Zielińska M. Cyclic derivatives of morphiceptin possess anti-transit effect in the gastrointestinal tract and alleviate abdominal pain in mice. Pharmacol Rep. 2020;72:314–21.
Safakhah HA, Damghanian F, Bandegi AR, Miladi-Gorji H. Effect of crocin on morphine tolerance and serum BDNF levels in a rat model of neuropathic pain. Pharmacol Rep. 2020;72:305–13.
Maharaj AR, Wu H, Zimmerman KO, Speicher DG, Sullivan JE, Watt K, et al. Dosing of continuous fentanyl infusions in obese children: a population pharmacokinetic analysis. J Clin Pharmacol. 2020;60:636–47.
Acknowledgements
This work was supported by the National Centre for Research and Development (Poland) [Grant number INNOTECH-K2/IN2/70/183154/NCBR/13]. The supporting institution was not involved in the study. The authors would like to thank the volunteers and the medical, nursing and laboratory staff for their participation in this study. The authors gratefully acknowledge Dr. W. Zieliński for his scientific advice, Dr. D. Rabczenko for the statistical evaluation of the safety parameters, and Dr. G. Garbacz for his critical review of the manuscript. The authors gratefully acknowledge Rose Soskind, PharmD, MS from Rutgers University and KMK Translation Company (Warsaw, Poland) for reviewing grammar and spelling.
Author information
Authors and Affiliations
Contributions
MBZ is an author of research hypothesis. All the authors except KBK participated in the trial design. KJD was the principal investigator in the clinical part of the trial. MF, EG, KBK, and PJR conducted the bioanalytical and pharmacokinetic part of the trial. AL conducted statistical analysis. All the authors interpreted the data. PR, KJD, EG, KBK, and MBZ participated in writing. All the authors have read the manuscript critically and approved the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
MSM, ŁN, and MBZ are full-time employees of the Medical University of Warsaw. PJR is a contract lecturer at the Medical University of Warsaw. All the other authors have stated explicitly that there are no conflicts of interest in connection with this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rudzki, P.J., Jarus-Dziedzic, K., Filist, M. et al. Evaluation of tramadol human pharmacokinetics and safety after co-administration of magnesium ions in randomized, single- and multiple-dose studies. Pharmacol. Rep 73, 604–614 (2021). https://doi.org/10.1007/s43440-021-00239-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-021-00239-x