
Abstract
Cytokinin oxidase/dehydrogenase (CKX) is the key enzyme that has been observed to catalyze irreversible inactivation of cytokinins and thus modulate cytokinin levels in plants. CKX gene family is known to have few members which are, expanded in the genome mainly due to duplication events. A total of nine MiCKXs were identified in Morus indica cv K2 with almost similar gene structures and conserved motifs and domains. The cis-elements along with expression analysis of these MiCKXs revealed their contrasting and specific role in plant development across different developmental stages. The localization of these enzymes in ER and Golgi bodies signifies their functional specification and property of getting modified post-translationally to carry out their activities. The overexpression of MiCKX4, an ortholog of AtCKX4, displayed longer primary root and higher number of lateral roots. Under ABA stress also the transgenic lines showed higher number of lateral roots and tolerance against drought stress as compared to wild-type plants. In this study, the CKX gene family members were analyzed bioinformatically for their roles under abiotic stresses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cytokinins are important plant hormones that contribute to the regulation of numerous biological processes such as cell division and differentiation, vascular and flower development, nutrient allocation, leaf expansion, root development, leaf senescence, and seed germination (Matsumoto-Kitano et al. 2008; Merewitz et al. 2010). Additionally, they help plants to militate against the severe consequences of abiotic and biotic stresses (Smigocki 1995; Liu et al. 2002; Babosha et al. 2009; Ikeda et al. 2021; Zhu et al. 2022; Argueso and Kieber 2024). Cytokinins (CKs) form a complex regulatory system along with other plant hormones such as auxin, gibberellins, abscisic acid, brassinosteroids, strigolactones and jasmonic acid (Hai et al. 2020). CKs are derivatives of purine bases with an isoprenoid or aromatic side chain at the N6 position. The natural isoprenoid CKs are N6-(Δ2-isopentenyl) adenine (iP), dihydrozeatin (DZ) and zeatin (cis-Zeatin and trans-Zeatin) whereas natural aromatic CKs include ortho-topolin, meta-topolin, 6-benzyladenine (6-BA) and 6-BA derivatives (Frébort et al. 2011; Argueso and Kieber 2024). The control of cytokinin levels in a particular tissue is important for its impact on growth and development. Homeostasis of CKs in cells is maintained via major three processes that are related to the rate of biosynthesis, inactivation/activation, and degradation. Isopentenyltransferases (IPT) are the major biosynthetic enzymes for the production of CKs via mevalonate (MVA) and methylerythritol (MEP) pathways. The inactivation and activation of CKs is a reversible process and primarily occurs through their conjugation with β-glucose, facilitated by enzymes such as zeatin O-glucosyltransferase (ZOG) and β-glucosidase (GLU), respectively. Meanwhile, the irreversible degradation of CKs and their derivatives is catalyzed by cytokinin oxidase/dehydrogenase (CKXs) encoded by a small multigene family in plants (Avalbaev et al. 2012; Jameson and Song 2016).
CKX is the only enzyme associated with CKs' breakdown to regulate their levels in plant development. CKX is a flavin adenine dinucleotide (FAD)–containing oxidoreductase that functions by oxidative cleavage of CKs carrying unsaturated N6-side chain, producing adenine (or its corresponding derivative for N9-substituted cytokinins) and a side chain-derived aldehyde (Sharma et al. 2022). Consequently, CKX regulates the level of free ribose of iP, zeatin, and also their ribosylated derivatives. Conversely, it is unable to act on CKs with saturated side chains like dihydrozeatin or O- glycosylated conjugates with glycosyl or xylosyl residues, and CKs with aromatic rings on their side chain. However, recent studies from Arabidopsis thaliana and Zea mays reported cleavage of aromatic cytokinins via CKXs although at a very low turnover rate when compared with that of iP (Galuszka et al. 2007; Frébortová et al. 2010). Earlier CKX activity was considered to be based on molecular oxygen, although many other electron acceptors were established to perform efficiently via other oxidizers and dehydrogenation. Thus, CKXs were classified as both cytokinin oxidase and cytokinin dehydrogenase (Galuszka et al. 2001). CKX enzymes comprise conserved FAD- and CK- binding domains located mainly on the N- and C-terminal of protein, respectively. The presence of a covalently bound FAD molecule is found in all known plant CKXs. The cofactor bonds through the 8-methyl group of the isoalloxazine ring to a histidine residue located in the conserved GHS motif in the N-terminal of the enzymes. CKX enzymes contain several consensus N-glycosylation sites that allow their post-translational modifications. This imparts CKXs regulation based on their enzymatic activity at different optimal pH i.e., 6.0 to 9.0, stability of protein, molecular weight, and localization. The presence of CKX inside or outside a cell thus depends highly on its specificity towards its substrate (Frébort et al. 2011).
Mulberry (Morus spp.) is a perennial, fast-growing, heterozygous, dioecious, and woody tree cultivated worldwide. Morus leaves are the major source of food with higher protein content for silkworm Bombyx mori which produces lustrous silk fibers. The use of mulberry for the production of silk makes it a significantly valuable tree for large-scale cultivation. Mulberry species like Morus alba, Morus multicaulis, and Morus notabilis are widely distributed in China. In India, M. alba is distributed in northern parts of the Himalayas and Punjab, while M. indica varies in range from temperate to sub-tropical and tropical regions. Morus laevigata is found naturally throughout India and Morus serrata is confined mainly to high-altitude regions and in northern India. The plantation of mulberry gets hampered by various unfavourable conditions including abiotic stresses such as heat, drought, and salt stress, as well as biotic stresses such as pests which destroy the plant completely. The draft genome sequence of M. indica cv K2 and RNA-seq data helped in the study of the expression of various genes in different developmental stages of plants (Jain et al. 2022a). In this particular study, we identified and studied CKX gene family members based on their structural, evolutionary, and functional analysis. CKX genes are reported to enhance tolerance against abiotic stresses including salt, drought, and heat in many plants. Therefore, we aimed to study the CKX gene family to provide a better understanding of mulberry growth and yield.
Results
Identification and sequence analysis of CKX in M. indica cv K2
The HMM approach was used for identifying the CKX proteins in M. indica cv K2 resulting in nine CKXs which were further confirmed using the SMART database and NCBI CDD. All nine MiCKXs were found to have two major conserved domains of CKXs, i.e., N-terminal FAD and C-terminal cytokinin binding domains (Fig. S1). The names of identified MiCKX were given according to the orthologs present in A. thaliana. The length of MiCKXs varied between 1506 to 1689 bp corresponding to their protein length ranging from 513 to 562 aa. The isoelectric points (pI) displayed variations from the lowest at 5.51 to the highest at 8.53. Other physiochemical properties like instability index, aliphatic index, and Grand average of hydropathicity (GRAVY) of MiCKX proteins were predicted. The subcellular localization predicted in different organelles and cytosol indicates their diverse role in cellular homeostasis. Additionally, the N-glycosylation sites of all MiCKXs were found to be present in the range of 2 to 4 (Table 1 and Table S1).
Gene structure, conserved and functional motifs
The gene structure deciphered not only the position of exons and introns but also helped in understanding the evolutionary factors that influence the function of CKX family members. The number of introns and exons were found to be 4 and 5, respectively in all MiCKXs except MiCKX7.A and MiCKX7.B where the introns were 3 and exons were 4 (Fig. 1a). The online software, MEME, predicted a total of 10 conserved motifs in all MiCKX proteins, designated as Motifs 1 to 10 (Fig. 1b).

Gene structure and conserved motif analysis. a Illustration of the gene structure of nine MiCKX genes, showing the distribution of exons and introns. Exons are shown using solid boxes while introns are shown using solid lines. b Motif analysis
Multiple sequence alignment and phylogeny analysis
The sequence alignment of all MiCKX proteins along with ZmCKXs and AtCKXs proteins showed that FAD binding domain was well conserved among all known CKX sequences. Moreover, conserved motifs like GHS were also well conserved, where the FAD cofactor is linked through the 8-methyl group of isoalloxazine ring to the histidine residue (Frébort et al. 2011; Sharma et al. 2022). Further, motifs like PHPWLNL and PGQxIF were also found in all CKXs analyzed in this study. These conserved amino acid sequences suggest their potential functions in the recognition of substrate and electron transport (Fig. 2).

Alignment of amino acid sequences of MiCKXs. The position of the motif for covalent binding of FAD cofactor (GHS, where H is the binding residue in all plant CKXs) was indicated. Amino acid residues predicted to bind to the cytokinin substrate were marked with an asterisk. The cytokinin N9 hydrogen-bonding residue that affected the substrate specificity was marked with a red arrow
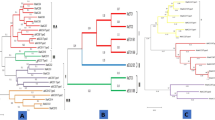
The aligned sequences were used for phylogenetic analysis and they were categorized into three main clades namely Clade I, II, and III. Clade I contained two MiCKXs i.e., MiCKX7.A and MiCKX7.B along with MnCKX7 and AtCKX7, 4 ZmCKXs, 2 OsCKXs, and 2 MdCKXs. The Clade II was divided into two subclades where Clade IIa contained CKX only from dicot species i.e., 3 AtCKXs, 4 MdCKXs, and 2 each from MiCKXs and MnCKXs; Clade IIb contained only monocot CKXs with 5 each of OsCKXs and ZmCKXs. Further, Clade III was also divided into two subclades which include Clade IIIa with 8 CKXs and Clade IIIb with 16 CKXs from all species (Fig. 3).

Phylogenetic analysis of CKX proteins. Amino acid sequences from M. indica cv K2, M. notabilis, A. thaliana, Oryza sativa, Zea mays and Malus domestica were aligned using MUSCLE and the phylogenetic tree was constructed using neighbor joining (NJ) method from MEGA-X. The numbers on the nodes denote bootstrap values from 1000 replicates. The tree here has been divided into four different clades.
 indicates MiCKXs and
indicates MiCKXs and
 indicates MnCKXs
indicates MnCKXs
Gene duplication and collinearity analysis
The mechanism of gene duplication involves three major types of duplications i.e., proximal, tandem and segmental. In our study, three out of nine MiCKXs namely, MiCKX7.A/MiCKX7.B, MiCKX6.A/MiCKX6.B and MiCKX1.A/MiCKX1.B possessed highest similarity amongst each other. MiCKX1.A/MiCKX1.B are predicted to be a case of proximal duplication as the distance between the two is less than 20 genes and they are present on the same scaffold. MiCKX7.A/MiCKX7.B and MiCKX6.A/MiCKX6.B are predicted to be a part of tandem duplications as they are present on the same scaffold in more than 200 kb range (Fig. 4a). Collinearity analysis was performed between Morus genotypes i.e., M. indica cv K2, M. alba and M. notabilis for CKX genes to identify orthologs and characterize the evolutionary patterns of MiCKX genes within Morus genus. According to the synteny analysis, there are 7 orthologous MiCKX genes present in M. alba and 5 in M. notabilis (Fig. 4b).

Duplication analysis of MiCKXs. a Mapping of duplicated MiCKXs on the Super Scaffolds (SS) and Scaffolds (S) using Circos. Grey ribbons represent collinear relationships among the blocks in the whole genome and red ribbons represent segmentally duplicated genes. b Synteny analysis of CKX genes M. indica cv K2 with M. notabilis and M. alba
Cis-acting elements of MiCKXs promoter region
To reveal the potential role of MiCKX genes in various regulatory pathways of the plant life cycle, the promoter regions were studied. For this study, 2 kb upstream region of genes was taken, and based on the PLACE database, different elements were depicted that implicated the role of MiCKX in various stages of plant development. The promoter contained phytohormone-responsive responsive i.e., cytokinin and auxin-responsive elements, gibberellin, abscisic acid, and jasmonic acid-related elements. Cytokinin-mediated induction elements like ARR1 and CPBCSPOR were found which signifies their role in mediating cytokinin-related pathways. Various abiotic and biotic stress-related elements like drought, wounding, and defense implicated the role of CKX genes in the mitigation of different stresses. The identification of heat and low-temperature responsive elements along with light-related elements, further emphasizes the diverse regulatory functions of MiCKX genes. Additionally, the presence of distinct tissue-specific elements, such as those found in fruit, seed, and endosperm, denotes the fundamental significance of CKXs in tissue development processes in plants. Some of the distinct cis-elements were also observed. MiCKX3, MiCKX1.A and MiCKX1.B genes contained QARBNEXTA which is found in the promoter of extA extension genes and is induced under pathogenic attack (Hao et al. 2019). Moreover, there was one negative regulatory element NRRBNEXTA (Nakaminami et al. 2009) found only in MiCKX3. The element related to vascular development, XYLAT, was also found in MiCKX7.A, MiCKX1.A and MiCKX1.B along with BS1EGCCR in MiCKX7.A and MiCKX7.B indicating their role in vascular differentiation. The P1BS element related to phosphate starvation was found only in MiCKX6.A and IRO2OS element, induced exclusively in iron deficiency, was found in MiCKX5, MiCKX7.A, MiCKX6.A and MiCKX6.B implicating their role in nutrient deficiency conditions. One of the major cis-elements related to ER stress, UPRMOTIFIIAT, was also found in MiCKX5 and MiCKX3, indicating their function in the unfolded protein response (UPR) which occurs in response to the accumulation of unfolded or misfolded proteins within the endoplasmic reticulum (ER) due to various changes in the cellular environment. The biotic stress-related elements like GCCCORE and WBBOXPCWRKY were also found, reinforcing the essential role of MiCKX genes in the plant defense mechanism against pathogen attack. The structural variation in the promoter regions of duplicated gene pairs, as in the case of MiCKX7.A/MiCKX7.B and MiCKX6.A/MiCKX6.B, highlights the differential regulatory pathways governing the roles of these genes in plant development (Fig. 5).

2 kb Promoter region of all nine MiCKXs in M. indica cv K2. Different colors represent different cis-elements
Gene ontology (GO) analysis gives detailed information regarding the three categories of gene properties, namely biological process, molecular function, and cellular components via GO terms. GO for all MiCKXs regarding the biological processes showed cytokinin and cellular hormone metabolic processes and regulation of hormone levels. The molecular functions enriched with GO terms suggest the role of CKXs in cytokinin dehydrogenase and oxidoreductase activity along with flavin adenine dinucleotide activity and ion binding activities. GO terms related to cellular components indicated their presence in the cytoplasm, intracellular part, and vacuoles. All these GO terms signify the role of CKXs in various aspects of plant growth and development (Fig. S2).
Differential expression analysis of MiCKXs in different genotypes and tissues of Morus
The expression patterns of CKX genes across different Morus genotypes and different tissues were analyzed using FPKM values procured from RNA-Seq data. In M. indica cv K2, amongst the nine CKX genes, four MiCKX genes, including MiCKX1.A/MiCKX1.B, MiCKX3, and MiCKX4, exhibited negligible expression levels. Conversely, MiCKX7.B/MiCKX7.A demonstrated the highest expression, followed by MiCKX6.A/MiCKX6.B and MiCKX5. These observations offer valuable insights into the differential expression patterns of CKX genes in M. indica cv K2, elucidating their variability in transcriptional regulation. Analogous expression profiles of CKX genes were observed in M. laevigata, with notable upregulation in MiCKX7.A/MiCKX7.B, followed by MiCKX3 and MiCKX6.A. Noteworthy, MiCKX3 displayed the highest expression level in M. laevigata compared to other genotypes, suggesting its specific involvement in the molecular landscape of this genotype. MiCKX7.B, followed by MiCKX6.B/MiCKX6.A, demonstrated the highest expression levels, among all other MiCKX genes in M. notabilis. Interestingly, the expression levels of MiCKX7.A were found to be lower in M. notabilis, contrasting with its higher expression in other genotypes, suggesting its distinct role in M. notabilis. In M. serrata and M. indica cv V1, all MiCKX genes displayed negligible expression levels, except MiCKX7.B/MiCKX7.A, which exhibited higher expression. Collectively, the differential expression of MiCKX genes highlights the functional diversification of these genes across Morus and indicates their possible role in local adaptation in response to various environmental conditions where these genotypes are flourishing (Fig. 6a).

Heatmaps showing differential expression of MiCKXs. a Expression analysis in different genotypes of Morus. b Expression analysis in different developmental stages of M. indica cv K2
The differential expression of genes across various tissues suggests their potential involvement in the development of different organs and implies their role in tailoring the needs and functions of different tissues within an organism. In this study, distinct patterns of MiCKX genes have been observed in M. indica cv K2, across various developmental stages which reflects their role in orchestrating specific processes at different stages of development during the plant growth. Amongst the nine MiCKX genes examined, MiCKX genes examined, MiCKX7.B/MiCKX7.A revealed a unique pattern of exclusive upregulation displaying the highest expression levels in the root and stem tissues, as compared to other MiCKX genes. This result emphasizes the possible importance of MiCKX7.B/MiCKX7.A in developing roots and shoots within the Morus genome. Additionally, in the leaf tissue, MiCKX7.A/MiCKX7.B showed elevated expression levels followed by MiCKX6.B/MiCKX6.A and MiCKX5, in contrast to the negligible expression levels observed for other MiCKX genes within the same tissue. Concerning dormant buds, MiCKX6.A/MiCKX6.B demonstrated the highest upregulation, followed by MiCKX5 and MiCKX7.A/MiCKX7.B. Examining the female inflorescence tissues of M. indica cv K2, the expression levels of MiCKX5 was the highest, followed by MiCKX7.A/MiCKX7.B. In the male inflorescence tissues, noteworthy elevation in the transcript levels was observed for all MiCKX genes except MiCKX3, with MiCKX4 exhibiting the highest expression, followed by MiCKX1.A and MiCKX5. Moreover, it is important here to mention that MiCKX1.A showed higher expression specific to male inflorescence in contrast to other tissues, thereby, indicating its potential role in the development of male reproductive organs in Morus. In the fruit tissue, MiCKX7.A/MiCKX7.B displayed higher transcript levels as compared to MiCKX3, MiCKX4, and MiCKX5, whereas MiCKX1.A showed a negligible expression level (Fig. 6b). Altogether, the differential expression pattern of MiCKX genes provides insights into their role in respective tissues, warranting further investigation.
Expression of CKX gene family at different hormonal levels and under drought stress
The expression analysis at different hormonal and under drought stress for MiCKXs was performed using the RT-qPCR method that allowed monitoring of the expression of the target gene under different sets of conditions. The transcripts of three genes are MiCKX7.A/MiCKX7.B, MiCKX6.A/MiCKX6.B, and MiCKX1.A/MiCKX1.B are highly conserved among themselves and hence, these were analyzed jointly for both the copies. To study the effect of exogenous cytokinin, MiCKX transcript levels were checked after providing 6-Benzylaminopurine (BAP) at the concentration of 50 µM to leaves of M. indica cv K2 against the control conditions. The MiCKX3, followed by MiCKX5 showed the highest upregulation under cytokinin treatment by 150 and 70 folds, respectively. Other MiCKXs were also found to be upregulated indicating the induction of CKXs upon higher levels of cytokinin in leaf tissue (Fig. 7a). In continuation with understanding the role of CKXs under hormonal stress, IAA treatment was given to the leaves at 100 µM concentration and it was observed that MiCKX3 and MiCKX4 showed maximum upregulation followed by MiCKX1.A/MiCKX1.B and MiCKX7.A/MiCKX7.B after the IAA treatment. The expression of MiCKX6.A and MiCKX5 showed less upregulation as compared to other MiCKXs (Fig. 7b). The presence of ARF binding sites in the promoter region of MiCKX3 and MiCKX4 may contribute to this high expression. The results here suggest the crucial crosslink between the two hormonal signaling pathways.

Relative expression analysis of MiCKXs under different conditions. a BAP (50 µM). b IAA (100 µM). c Drought stress via air drying of leaves. Three biological and three triplicates were used. Error bars indicate values ± SD. Asterisks on top of the error bars represent the significance levels (Student’s t-test; p-value ≤ 0.05); ns represents non-significant
To comprehend more about the purpose of CKX under abiotic stress and more specifically during drought stress, Morus leaves were air-dried and then analyzed for CKX transcript levels. MiCKX4 was recorded to be highly upregulated, up to approximately 150-fold under drought stress evincing its indispensable role in conferring resistance against drought stress in Morus. MiCKX3 was the second highly upregulated gene under drought stress followed by MiCKX7.A/B and MiCKX.1/A (Fig. 7c). The presence of elements which respond to drought stress probably relates to their higher expression levels.
Subcellular localization of MiCKX proteins
The subcellular localization was analyzed for four MiCKXs via transient expression studies, using particle bombardment procedure in onion epidermal peels. The genes CDS were fused with YFP at the C-terminal. Expression of these chimeric genes was analyzed by and upon observation under confocal microscopy. The proteins were found to be present in cytoplasmic structures resembling endoplasmic reticulum (ER) and Golgi bodies. To validate these results, the co-expression of MiCKXs with ER and Golgi markers were checked. ER marker (ER-ck/CD3-953) and Golgi marker (G-ck/CD3-961) were used with CFP tags using appropriate gene constructs, respectively. The fluorescence was observed for both markers and gene samples, in both ER and Golgi bodies, confirming the localization of MiCKX6.A/B, MiCKX3, MiCKX4, and MiCKX7.A/B in these organelles (Fig. 8a and b).

Subcellular localization of four MiCKXs with organelle markers. a Endoplasmic reticulum (CD3-953) marker. b Golgi bodies (CD3-961) marker. MiCKXs are tagged with YFP after cloning in pSITE3CA:YFP vector
Overexpression of MiCKX4 enhances drought tolerance
To investigate the role of MiCKX genes in plants, transgenic Arabidopsis plants overexpressing MiCKX4 were generated and three confirmed transgenic lines were used for analysis (Fig. S3). Under ½ MS media, the overexpression (OE) lines showed robust growth, characterized by longer primary roots and higher number of lateral roots as compared to wild-type (Fig. 9a and b). The expansion of cotyledon was also found greater in OE lines in comparison with wild-type Arabidopsis plants (Fig. 9c). The primary root phenotypes were noticed on the 6th day post-seed germination and analysis of lateral roots was carried out after the 10th day (Fig. 9d-f). To assess the drought stress tolerance of OE lines, the 6-day-old seedlings which were grown on ½ MS media, were transferred to ABA concentration of 10 µM and grown vertically for 2 days. The phenotypic analysis after two days of ABA stress revealed a notable increase in the number of lateral root production in OE lines as compared to wild-type plants under ABA-induced stress conditions. Additionally, the length of lateral roots was also significantly longer than the wild-type which indicates their role in conferring tolerance against drought stress mediated by ABA (Fig. 10a-d).

Phenotypic analysis of MiCKX4 overexpression lines in A. thaliana. a Longer primary root length and larger cotyledon expansion in transgenics as compared to WT in 6-day-old seedlings. b and c Statistical analysis of primary root lenghth (b), cotyledon expansion (c). d Longer primary root length and higher number of lateral roots of transgenics as compared to WT in 10-day-old seedlings. e and f Statistical analysis of primary root length (e), number of lateral roots (f) d. Error bars indicate values ± SD. Asterisks on top of the error bars represent the significance levels (Student’s t-test; p-value ≤ 0.05); ns represents non-significant

Phenotypic analysis of MiCKX4 overexpression lines in A. thaliana in ABA (10 µM). a Higher number of lateral roots in transgenics as compared to WT in 6-day old seedlings before ABA treatment. b Statistical analysis of lateral root numbers of a. c Higher number of lateral roots in transgenics as compared to WT after 3 days of ABA treatment in transgenic plants. d Statistical analysis of lateral root numbers of c. e NBT assay for ABA-treated seedlings. Error bars indicate values ± SD. Asterisks on top of the error bars represent the significance levels (Student’s t-test; p-value ≤ 0.05); ns represents non-significant
Reactive oxygen species (ROS) are the by-products of different metabolic pathways which are produced by the incomplete reduction of oxygen, thereby, producing highly reactive molecules. ROS act as important signaling molecules in response to stress and can be detected by NBT staining. NBT is an artificial electron acceptor and interacts with superoxide generated during various stresses. In our study, seedlings were subjected to ABA for 3 days followed by nitro blue tetrazolium (NBT) staining to assess the level of injury to the cell under stress conditions. The OE lines were found to have significantly lower amounts of ROS relative to wild-type under ABA stress (Fig. 10e). These results suggest that OE lines are producing less ROS and experiencing attenuated stress which contributes in enhancing drought tolerance in OE lines as compared to wild-type seedlings.
To illuminate more about the response of MiCKX4 overexpression under drought stress, we checked the expression of stress-related genes that are known to get upregulated under multiple stress conditions in plants. The levels of AtRD29A, AtRD29B, AtRD26, and AtERD6 were found to be higher in transgenics as compared to WT after ABA stress. Further, the levels of ROS-scavenging genes AtCAT1 and AtCAT2 were also higher in transgenics under stressed conditions (Fig. 11). These results suggest that MiCKX4 imparts drought stress tolerance by the upregulation of stress-related genes and mediates ROS scavenging through the upregulation of antioxidant-generating enzymes.

Relative expression analysis of drought stress marker genes. All the expression analyses were checked using RT-qPCR, where actin was used for normalizing values between WT and OE lines. Three biological and three triplicates were used for both control and treated samples. Error bars indicate values ± SD. Asterisks on top of the error bars represent the significance levels (Student’s t-test; p-value ≤ 0.05); ns represents non-significant
Discussion
Cytokinins are well known to display a significant impact at every stage of the plant life cycle by coordinating complex processes of growth involving differentiation, and morphogenesis by regulating development from roots to shoots, leaves to flowers, and fruits (Argueso and Kieber 2024). Moreover, extensive research has established the crucial role of cytokinins in plants under diverse environmental conditions including abiotic and biotic stresses (Argueso and Kieber 2024). CKXs are the major regulator of cytokinin in plants as they maintain the level of CKs via their irreversible degradation. Several members of the CKX gene family have been identified in various crops and their role in maintaining hormone homeostasis in the plants has been emphasized. In the present study, a total of nine CKX gene family members were identified which possess both FAD and cytokinins binding domain which are the signature domains of CKXs. The previous reports in CKX identification revealed that it is a gene family with a small number of members with conserved motifs and gene structure, which is also found in our study.
The exon–intron structure aids insights into the rate of gene expression and explores the diversification of functions associated with genes. The genes with the lower number of introns have been shown to rapidly change their expression levels in response to stress in Arabidopsis, yeasts, and mice (Jeffares et al. 2008). In the MiCKX gene family, the number of introns ranges from 3 to 4, which may contribute to their higher expression levels during various stress conditions. Moreover, the number of exons and introns was the same for all genes except, MiCKX7.A/B, suggesting the conservation of gene structure. The motif analysis helps in understanding the molecular and biological mechanisms of genes as these motifs serve as binding sites between molecules which affect the structure of molecules and provide proper enzymatic activities (Kemp 1980). In this study, MiCKX gene family members were found to have conserved motifs and domains that signify their functional conservation and evolutionary relationships amongst the gene family members.
The phylogenetic tree was constructed using amino acid sequences of five plant species (Arabidopsis, rice, maize, apple, and two genotypes of Morus) to explore their divergence during evolution. A previous study on CKX genes from A. thaliana reported seven AtCKXs, divided into three major clades (Schmülling et al. 2003). In this study, nine members of the MiCKX gene family were also divided into three major clades Clade I, II, and III with two sub-clades for Clade II and Clade III. The groups contained members with similar sequences and structures which conveys their similar role in plant development across the species. AtCKX7 is the only CKX present from Arabidopsis in Clade I which is considered one of the ancient CKX (Wang et al. 2020) in terms of its evolutionary analysis and the orthologs of AtCKX7 in other angiosperms are also considered ancient CKXs. Accordingly, we can predict MiCKX7.A and MiCKX7.B are the ancient CKX as they carry the same position in Clade I. All AtCKXs present in other clades were considered non-ancient. The Clade II contains two subclades that divide CKXs among monocots and dicots, indicating their genetic divergence during evolution and speciation. These non-ancient CKXs are known to provide tolerance to various abiotic stresses like drought, high temperature, salinity, etc. (Wang et al. 2020).
Gene duplication is an important mechanism that allows the expansion and diversification of gene families (Jaillon et al. 2009). The contribution of duplicated genes in producing genes with novel structures and functions, along with novel evolutionary traits has also been known for a role in crop improvement (Panchy et al. 2016). The genome sequence of M. indica cv K2 was revealed to consist of more than half of the genes duplicated through whole genome and tandem gene duplication events (Jain et al. 2022a). In this study, using circos analysis, however, we found that out of nine MiCKX genes, three are present as duplicated gene pairs due to proximal, tandem, and segmental duplication events. Further, the collinearity analysis between three genotypes of Morus revealed a closer relationship of MiCKX genes with M. alba followed by M. notabilis. The duplication events in CKXs suggest that functional redundancy may be more common among CKX genes in M. indica cv K2, which has also been found in other plant species like maize, rice, finger millet, etc., (Gu et al. 2010; Jain et al. 2022b; Blume et al. 2022).
Analysis of cis-regulatory elements renders understanding of the gene expression in deeper ways. In recent studies, promoters were considered to be the most important factor that may play a crucial role in crop improvement by regulating gene expression and influencing agronomic traits (Kummari et al. 2020; Villao-Uzho et al. 2023). The study of promoter sequences helps in understanding the role of a gene in various pathways by regulating their expression not only in a tissue-specific manner but also during abiotic and biotic stresses (Villao-Uzho et al. 2023). In this study, we utilized nucleotide sequences present 2000 bp upstream of the initiation codon of the gene and found various potential elements signifying the role of CKXs in various stages of plant development and stress mitigation pathways. Two major CK-induced elements i.e., ARR1AT and CPBCSPOR were found to be present in the promoters of MiCKX genes (Sakai et al. 2000; Ross et al. 2004; Fusada et al. 2005). The presence of more than 20 ARR1 sites in all MiCKXs makes it evident that they are induced highly by cytokinin and thus maintain cytokinin equilibrium in the plant. CPBCSPOR was found only in four MiCKXs, varying from one to three, further implicating their role in cytokinin-dependent events. Further, various other hormone-related elements like auxin, gibberellin, jasmonic acid, and abscisic acid were found in the promoter regions of CKX, indicating their roles in hormonal crosstalk. ARF binding sites present in CKXs promoter have been earlier reported to control crown root formation in rice. Moreover, the presence of various stress-related elements signifies the crucial role of CKXs in stress-mitigating pathways. Also, they contained elements related to iron and phosphate deficiency i.e., IRO2OS and P1BS (Rubio et al. 2001; Ogo et al. 2006), respectively, suggesting their roles in improving plant tolerance against nutrient deficiency, which is already reported in multiple crops like barley, rapeseed, and maize (Argueso and Kieber 2024). The tissue-specific elements further suggest the expression of CKXs is crucially regulated for the proper development of plant organs. It was also seen that duplicated genes except MiCKX1.A/MiCKX1.B do not share common elements suggesting functional divergences and the acquisition of novel biological functions of MiCKX genes.
Extensive studies related to the expression patterns of CKXs have been reported for maize and rice, where different CKXs have specific roles in maintaining cytokinin levels in different tissues (Schmülling et al. 2003; Rong et al. 2022). Expression analysis of CKXs in different developmental stages revealed their putative role in vegetative and reproductive development in Morus. Many MiCKXs exhibited distinguished tissue-specific expression patterns. MiCKX7.A and MiCKX7.B exhibited the highest expression in roots as compared to other MiCKXs which implies their role in root development. AtCKX1 and AtCKX2 were also found to play a major role in root development which was observed after their constitutive expression in Nicotiana tabacum (Khandal et al. 2020). Some MiCKXs were found to have higher expression in female and male inflorescence depicting the role of CKXs in flower development. Most of the CKXs had lower expression in dormant buds except MiCKX4 and MiCKX1.A which revealed that cytokinin accumulation is a prerequisite for the growth of fruit. Further, the promoter activity exerts significant control over the regulation of gene expression, managing the complex process of turning genes on or off within a cell. The understanding of various regulatory pathways associated with a gene can be correlated with the presence of cis-elements within its promoter region. In our study, the expression levels of MiCKX4 were observed to be very low in vegetative tissues including roots, stems, and leaves. This observation may be attributed to the presence of the unique cis-element NRRBNEXTA, a negative regulatory element known to modulate the development of roots, stems, and petioles, and alteration of this region leads to the expression of genes in tissues as mentioned above. Additionally, the presence of BS1EGCCR element, which is required for vascular expression and stem development, exclusively in MiCKX7.A/MiCKX7.B, demonstrates its elevated expression levels in the stem, distinguishing it from other MiCKX genes. Furthermore, hormone-related elements present in MiCKX genes associate their differential expression in vegetative and reproductive tissues which imply their role in the development and differentiation of various plant tissues.
The modulation of CKX enzyme activity seems to correlate with the diverse subcellular localization patterns demonstrated by these enzymes. Hence, the presence of intracellular CKX suggests its involvement in maintaining the movement of cytokinin within specific tissues or organs. Further, extracellular CKXs are anticipated to participate in degrading cytokinins that regulate the cell cycle (Avalbaev et al. 2012). In our study, four MiCKXs were studied for analysis of their subcellular localization, and their presence was detected in ER and Golgi bodies. The presence of MiCKXs in ER emphasizes their role in unfolded protein responses under stressed conditions, as detected earlier for AtCKX1 (Niemann et al. 2015, 2018). Further, their localization in Golgi bodies indicates their post-translational modifications as they are known to have N-glycosylation sites which contribute to regulation of their enzymatic activities, translocation, and protein stability (Frébort et al. 2011). Most of the CKXs in maize and other plants are glycoproteins with one to eight glycosylation sites (Schmülling et al. 2003). The pH difference was observed for glycosylated and non-glycosylated CKXs which suggests that the protein stability, enzymatic activity, localization, and translocation are highly dependent on the glycosylation properties of CKXs (Schmülling et al. 2003). However, in the present study, all nine MiCKXs were found to have glycosylation sites varying from a range of 2 to 4.
Genetic manipulation of CKXs has shown substantial improvement in agronomic traits by providing tolerance against various abiotic stresses that include salinity, drought as well as in nutrient deficiency. RNAi-mediated knockdown using OsCKX2 delayed leaf senescence in transgenic lines along with the increase in panicle branches, grain number per panicle, and plant height, thereby, increasing seed yield (Yan et al. 2022). Further, CRISPR/Cas9 mediated gene knockout of OsCKX11 also increased in tiller number, grains per panicle, and grains per plant while decreasing the fertility rate (Zhang et al. 2021). Further, the knockdown of TaCKX1 and HvCKX1 increased the number of spikes and overall grain yield in Triticum aestivum and Hordeum vulgare, respectively (Holubová et al. 2018; Jabłoński et al. 2020). In dicots like A. thaliana, Brassica napus and Medicago truncatula, mutants of CKXs produced a greater number of flowers and has larger flower sizes with a higher number of ovules per gynoecium, higher siliques and seed number per siliques (Bartrina et al. 2011; Schwarz et al. 2020; Wang et al. 2021). In terms of gain-of-function, the constitutive expression of CKXs in Arabidopsis, Nicotiana, Medicago sativa, etc., were observed to possess longer primary roots with increased number and length of lateral roots with dwarfed shoot phenotype and a smaller number of flowers (Sharma et al. 2022). The enhanced root growth indicates the importance of CKXs under drought and salinity stress as higher root biomass contributes to great tolerance against these stresses. Also, there are recent reports that indicate the vital role of CKXs in nutrient and minerals consumption in plants where the overexpression of the CKX gene resulted in higher content of various micro- and macro- elements which can be again a response due to increased size of the root systems (Sharma et al. 2022; Argueso and Kieber 2024). In the present report also, we see that overexpressed MiCKX4 under the constitutive promoter in Arabidopsis resulted in robust root growth in transgenic lines as compared to wild-type Col-0. Both the primary roots and lateral roots exhibited increased length and number, respectively. Moreover, under simulated drought stress, transgenic lines displayed vigorous growth in lateral roots as compared to wild-type. The ROS production in transgenic lines was also very low as compared to WT, thus supporting the fact that overexpression of MiCKX4 leads to lower stress in plants during drought conditions. Moreover, genes like RD29A and Rd29B (Msanne et al. 2011), which are known to express very low under optimal conditions and get upregulated under stress conditions, were also found to express at higher levels in the transgenic lines as compared to wild-type. Our findings on MiCKXs, thus, provide more supporting evidence that CKX genes play an essential role in mitigating abiotic stresses and their function is conserved across diverse plant species.
Conclusions
In the present study, we identified nine MiCKXs and found three duplicated genes which shows the expansion of the CKX gene family in M. indica cv K2. To get more understanding of the evolutionary significance, gene structure, phylogenetic relationship, and synteny as well as cis-elements were investigated for all MiCKX genes. The expression analysis of MiCKXs displayed variations in their levels during different developmental stages of plants thus suggesting their crucial role in plant organ development at both vegetative and reproductive stages. Additionally, the overexpression of the MiCKX4 gene resulted in increased root mass, thereby conferring enhanced tolerance against drought stress.
Materials and methods
Identification and analysis of MiCKX genes
The information on the reference genome and annotated proteins of M. indica cv K2 was obtained from http://tgsbl.jnu.ac.in/MindGP/genome.php (Jain et al. 2022a) The Hidden Markov model (HMM) search (E-value equal to 10) and the Pfam database were used to screen the potential candidates for MiCKX proteins using FAD-binding domain and cytokinin binding domain with Pfam id PF01565 and PF09265, respectively. The retrieved sequences from HMM profiling were then taken forward to check the presence of conserved domains in these sequences. For this, two different software were used namely, NCBI-CDD and SMART, which analyze the presence of domains in the given protein sequences. The sequences with conserved FAD and cytokinin binding domains were then proceeded for further study. The protein sequences of the confirmed MiCKXs were then utilized to explore their physiochemical properties including molecular weight, isoelectric point (pI), instability index, aliphatic index, and GRAVY using the online available tool ProtParam (https://web.expasy.org/protparam/). Additionally, the N-glycosylation sites were determined using https://services.healthtech.dtu.dk/services/NetNGlyc-1.0/ for all MiCKX proteins.
Gene structure analysis and conserved motif of MiCKX gene family
The CDS and genomic sequences of all MiCKXs were obtained and Gene Structure Display Server 2.0 (https://gsds.gao-lab.org/) was used to analyze the organization of exons and introns in MiCKX genes. The protein sequences of MiCKXs were utilized to understand the conserved motifs among CKXs using the MEME Suite 5.5.5 tool (https://meme-suite.org/meme/index.html) where the number of motifs was kept at 10 with optimum motif width between 8 and 50 residues. The derived result of motifs was further used in TBtools-II software to represent their conserved positions in MiCKX proteins (Chen et al. 2020).
Phylogenetic analysis of the CKX Gene family
The multiple sequence alignment was performed using full-length protein sequences of various plant species consisting of monocots like Zea mays, Oryza sativa, and dicot species like Arabidopsis thaliana, Malus domestica and Morus notabilis against Morus indica cv K2. The protein sequences of 13 ZmCKXs, 11 OsCKXs, 7 AtCKXs, and 12 MdCKXs were retrieved using the Phytozome database (https://phytozome-next.jgi.doe.gov/) (Schmülling et al. 2003; Gu et al. 2010; Tan et al. 2018; Rong et al. 2022). The CKX protein sequences of Morus notabilis (MnCKXs) were searched by applying the HMM approach in the Morus Genome Database- MorusDB (https://morus.biodb.org/). Six MnCKXs with conserved FAD and cytokinin binding domains were found and used for analysis. ClustalW from MEGA-X was employed with default parameters for alignment and illustrated using BioEdit. The conserved FAD and cytokinin binding domains were highlighted in the alignment. The aligned file was used for constructing a phylogenetic tree using the neighbor-joining (NJ) tool of MEGA-X with bootstrap support of 1000 replicates. The Newick file output of the generated tree was uploaded to the online tool EvolView (https://www.evolgenius.info/evolview/#/treeview) for representation.
Gene duplication and Synteny analysis of MiCKX
The duplication of MiCKX genes was studied using MCScanX tool available in Tbtools-II software (Wang et al. 2012). The file containing total linked genes present in M. indica cv K2 was generated and used to make CIRCOS using Advanced Circos application for selected super-scaffolds containing MiCKXs. The collinearity analysis was performed between three different Morus genotypes including M. notabilis, M. alba, and M. indica cv K2. The Multiple Synteny Plot modules of Tbtools-II were used to visualize the CKX gene among the above-mentioned species.
Promoter and Gene Ontology analysis of CKX gene family
To identify the cis-elements in the promoter regions, the 2 kb region upstream from the start of the gene was retrieved for all MiCKX genes and used in the New PLACE database (https://www.dna.affrc.go.jp/PLACE/?action=newplace). Further, the Gene Ontology analysis was conducted using the PlantRegMap tool, and terms enriched for biological processes, molecular functions, and cellular components were studied. The expression levels of CKXs were checked using the transcriptome data present in http://tgsbl.jnu.ac.in/MindGP/genome.php for different genotypes and tissues of Morus. The FPKM values of CKXs were then used for representation using TB-ToolsII.
Plant growth conditions and stress treatment
The freshly harvested seeds of M. indica cv K2 were imbibed overnight and then sterilized using 0.1% mercuric chloride for 8 min and subsequently washed with autoclaved RO water five times. The sterilized seeds were allowed to dry on Whatman paper for 2–3 h. The seeds were grown in MS media in a growth chamber maintained at 26 °C in the dark for 6 days. The germinated seeds were then shifted to 16/8 h light/dark period for further growth. The plants were allowed to grow till the four leaves stage and then transferred to pots. Two-month-old mature leaves were then used for various hormonal treatments and drought stress. For hormonal stress, IAA (100 µM) and BAP (50 µM) were used as auxin and cytokinin for 4 h, respectively. The leaves were air-dried for 4 h in the laminar hood to provide simulated drought stress. The tissues were then flash-frozen in liquid nitrogen and stored at -80 °C until further use.
Subcellular localization of MiCKXs
The CDS of four MiCKXs were cloned in pENTR D-TOPO and the plasmid was sequenced. The clones were then transferred to the destination vector pSITE-3CA-YFP for detecting their subcellular localization. The individual plasmids DNA samples having the target gene or the organelle markers were coated with gold particles and bombarded at 900 psi pressure on the onion epidermal peels. The peels were observed under a confocal microscope (Leica, Germany) after 12 h. The organelle markers used for endoplasmic reticulum and Golgi bodies were ER-ck/CD3-953 and G-ck/CD3-961, respectively (Nelson et al. 2007).
Generation of MiCKX4 overexpression lines in Arabidopsis thaliana
The full-length coding region of MiCKX4 was cloned in pENTR D-TOPO and confirmed via sequencing after which it was transferred to pMDC32 vector via Gateway cloning under CamV 35S promoter. The recombinant plasmid was transformed into Agrobacterium strain GV3101 and colonies were screened after 3 days. The wild-type Arabidopsis (Col-0) was transformed using the floral dipping method (Clough and Bent 1998). T0 transgenic seeds were screened on ½ MS containing 15 mg/L hygromycin for confirmation of plants with transgene. T3 homozygous seedlings were then confirmed for the presence of transgene by performing PCR of genomic DNA using MiCKX4 specific primers and also via qPCR to check the transcript abundance of MiCKX4 in transgenic lines. The confirmed lines were further utilized for analysis.
Phenotypic study of MiCKX4 overexpression lines under control and stress conditions
The wild-type seeds of Col-0 along with T3 seeds of MiCKX4 overexpressing lines were sterilized using 0.4% sodium hypochlorite for 10 min and then washed with autoclaved RO five times. The sterilized seeds were then placed on ½ MS and kept on stratification at 4 °C in the dark for 2 days. On the third day, the seeds were transferred to the culture room at 22 °C with 16/8-h light/dark cycle. After 6 days of incubation, the primary root lengths were measured. The lateral root numbers were also calculated after 13 days of seed germination. For studying the stress tolerance, the 7-day-old seedlings of both Col-0 and MiCKX4 overexpression (OE) lines were transferred to ABA concentrations of 10 µM. The phenotype was photographed after 3 days of stress.
Histochemical analysis for ROS detection
The amount of reactive oxygen species (ROS) generated after ABA treatment in both wild-type Col-0 and MiCKX4 OE lines was checked by staining the seedlings with nitro blue tetrazolium (NBT). The ABA-treated seedlings were incubated in NBT stain (NBT powder and 20 mM phosphate buffer) in the dark overnight. Next day the seedlings were transferred to a bleaching solution containing ethanol, acetic acid and glycerol in a ratio of 3:1:1 for chlorophyll removal. The seedlings were then visualized under the Leica S8 APO stereo microscope.
RNA extraction for RT-qPCR analysis
The total RNA of mulberry leaves was extracted using RNeasy plant mini kit (Qiagen, Germany) according to the instructions provided by the manufacturer, including on-column DNaseI treatment for removing any genomic DNA contamination. Arabidopsis seedlings were isolated using Trizol reagent (Sigma), as per the manufacturer’s protocol. An aliquot having 2 µg RNA was then used for each sample to synthesize cDNA using the High-capacity cDNA Reverse Transcription kit (Applied Biosystems). The RT-qPCR analysis was performed using the ABI Prism 7000 Sequence Detection System and Software on ABI QuantStudio™ 3 Real-Time PCR machine. The obtained Ct values were used to calculate relative expression levels via the 2-ΔΔCt method, using Actin as internal control gene. The primers used are listed in Table S2.
Statistical analysis
The GraphPad Prism 8 was used to perform a t-test for evaluating the statistical significance. P < 0.05, 0.01, and 0.001 were regarded as significant, very significant, and extremely significant, respectively, from the control conditions.
Availability of data and materials
Not applicable.
Abbreviations
- ABA:
-
Abscisic acid
- ARR:
-
Arabidopsis Response Regulator
- ARF:
-
Auxin Response Factor
- BAP:
-
6-Benzylaminopurine
- CAT1:
-
Catalase 1
- CAT2:
-
Catalase 2
- CDS:
-
Coding sequences
- CFP:
-
Cyan Fluorescent Protein
- CK:
-
Cytokinins
- CKX:
-
Cytokinin oxidases/dehydrogenases
- ER:
-
Endoplasmic reticulum
- ERD6:
-
Early response to dehydration 6
- FAD:
-
Flavin adenine dinucleotide
- FPKM:
-
Fragments Per Kilobase
- GRAVY:
-
Grand average of hydropathy
- GO:
-
Gene ontology
- HMM:
-
Hidden Markov model
- IAA:
-
Indole-3-acetic acid
- MEME:
-
Multiple Em for Motif Elicitation
- NCBI-CDD:
-
National Center for Biotechnology Information Conserved Domain Database
- OE:
-
Overexpression
- PLACE:
-
Plant cis-acting regulatory DNA elements
- RD29A:
-
Responsive to desiccation 29A
- RD29B:
-
Responsive to desiccation 29B
- ROS:
-
Reactive oxygen species
- RT-qPCR:
-
Reverse transcription-quantitative polymerase chain reaction
- SMART:
-
Simple Modular Architecture Research Tool
- YFP:
-
Yellow Fluorescent Protein
References
Argueso CT, Kieber JJ (2024) Cytokinin: From autoclaved DNA to two-component signaling. Plant Cell. https://doi.org/10.1093/plcell/koad327
Avalbaev AM, Somov KA, Yuldashev RA et al (2012) Cytokinin oxidase is key enzyme of cytokinin degradation. Biochemistry (Mosc) 77:1354–1361. https://doi.org/10.1134/s0006297912120024
Babosha AV, Ryabchenko AS, Avetisyan TV (2009) Effect of exogenous cytokinins on dynamics of development and differentiation of infectious structures of the pathogen of wheat powdery mildew. Cell Tissue Biol 3:387–396. https://doi.org/10.1134/S1990519X09040117
Bartrina I, Otto E, Strnad M et al (2011) Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana. Plant Cell 23:69–80. https://doi.org/10.1105/tpc.110.079079
Blume R, Yemets A, Korkhovyi V et al (2022) Genome-wide identification and analysis of the cytokinin oxidase/dehydrogenase (ckx) gene family in finger millet (Eleusine coracana). Front Genet 13:963789. https://doi.org/10.3389/fgene.2022.963789
Chen C, Chen H, Zhang Y et al (2020) TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol Plant 13:1194–1202. https://doi.org/10.1016/j.molp.2020.06.009
Clough SJ, Bent AF (1998) Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16:735–743. https://doi.org/10.1046/j.1365-313X.1998.00343.x
Frébort I, Kowalska M, Hluska T et al (2011) Evolution of cytokinin biosynthesis and degradation. J Exp Bot 62:2431–2452. https://doi.org/10.1093/jxb/err004
Frébortová J, Novák O, Frébort I et al (2010) Degradation of cytokinins by maize cytokinin dehydrogenase is mediated by free radicals generated by enzymatic oxidation of natural benzoxazinones. Plant J 61:467–481. https://doi.org/10.1111/j.1365-313X.2009.04071.x
Fusada N, Masuda T, Kuroda H et al (2005) Identification of a novel cis-element exhibiting cytokinin-dependent protein binding in vitro in the 5′-region of NADPH-protochlorophyllide oxidoreductase gene in cucumber. Plant Mol Biol 59:631–645. https://doi.org/10.1007/s11103-005-0579-x
Galuszka P, Frébort I, Šebela M et al (2001) Cytokinin oxidase or dehydrogenase? Mechanism of cytokinin degradation in cereals. Eur J Biochem 268:450–461. https://doi.org/10.1046/j.1432-1033.2001.01910.x
Galuszka P, Popelková H, Werner T et al (2007) Biochemical characterization of cytokinin oxidases/dehydrogenases from Arabidopsis thaliana expressed in Nicotiana tabacum L. J Plant Growth Regul 26:255–267. https://doi.org/10.1007/s00344-007-9008-5
Gu R, Fu J, Guo S et al (2010) Comparative Expression and Phylogenetic Analysis of Maize Cytokinin Dehydrogenase/Oxidase (CKX) Gene Family. J Plant Growth Regul 29:428–440. https://doi.org/10.1007/s00344-010-9155-y
Hai NN, Chuong NN, Tu NHC et al (2020) Role and Regulation of Cytokinins in Plant Response to Drought Stress. Plants (Basel) 9:422. https://doi.org/10.3390/plants9040422
Hao Q, Zhang L, Yang Y et al (2019) Genome-wide analysis of the wox gene family and function exploration of GmWOX18 in soybean. Plants (Basel) 8:215. https://doi.org/10.3390/plants8070215
Holubová K, Hensel G, Vojta P et al (2018) Modification of barley plant productivity through regulation of cytokinin content by reverse-genetics approaches. Front Plant Sci 9:1676. https://doi.org/10.3389/fpls.2018.01676
Ikeda Y, Zalabák D, Kubalová I et al (2021) Interpreting Cytokinin Action as Anterograde Signaling and Beyond. Front Plant Sci 12:641257. https://doi.org/10.3389/fpls.2021.641257
Jabłoński B, Ogonowska H, Szala K et al (2020) Silencing of TaCKX1 mediates expression of other TaCKX genes to increase grain yield in wheat. Int J Mol Sci 21:4809. https://doi.org/10.1101/2020.01.07.897421
Jaillon O, Aury JM, Wincker P (2009) “Changing by doubling”, the impact of Whole Genome Duplications in the evolution of eukaryotes. C R Biol 332:241–253. https://doi.org/10.1016/j.crvi.2008.07.007
Jain M, Bansal J, Rajkumar MS et al (2022a) Draft genome sequence of Indian mulberry (Morus indica) provides a resource for functional and translational genomics. Genomics 114:110346. https://doi.org/10.1016/j.ygeno.2022.110346
Jain P, Singh A, Iquebal MA et al (2022b) Genome-Wide Analysis and Evolutionary Perspective of the Cytokinin Dehydrogenase Gene Family in Wheat (Triticum aestivum L.). Front Genet 13:931659. https://doi.org/10.3389/fgene.2022.931659
Jameson PE, Song J (2016) Cytokinin: A key driver of seed yield. J Exp Bot 67:593–606. https://doi.org/10.1093/jxb/erv461
Jeffares DC, Penkett CJ, Rg Bä Hler J (2008) Rapidly regulated genes are intron poor. Trends Genet 24:375-378. https://doi.org/10.1016/j.tig.2008.05.006
Kemp BE (1980) Wilhelm Roux’s Arch. Cambridge University Press 18 Nieuwkoop
Khandal H, Gupta SK, Dwivedi V et al (2020) Root-specific expression of chickpea cytokinin oxidase/dehydrogenase 6 leads to enhanced root growth, drought tolerance and yield without compromising nodulation. Plant Biotechnol J 18:2225–2240. https://doi.org/10.1111/pbi.13378
Kummari D, Palakolanu SR, Kishor PBK et al (2020) An update and perspectives on the use of promoters in plant genetic engineering. J Biosci 45:119. https://doi.org/10.1007/s12038-020-00087-6
Liu X, Huang B, Banowetz G (2002) Cytokinin effects on creeping bentgrass responses to heat stress: I. Shoot and Root Growth. Crop Sci 42:457–465. https://doi.org/10.2135/cropsci2002.4570
Matsumoto-Kitano M, Kusumoto T, Tarkowski P et al (2008) Cytokinins are central regulators of cambial activity. Proc Natl Acad Sci U S A 105:20027-20031. https://doi.org/10.1073/pnas.0805619105
Merewitz EB, Gianfagna T, Huang B (2010) Effects of SAG12-ipt and HSP18.2-ipt Expression on Cytokinin Production, Root Growth, and Leaf Senescence in Creeping Bentgrass Exposed to Drought Stress. J Amer Soc Hort Sci 135:230-239. https://doi.org/10.21273/JASHS.135.3.230
Msanne J, Lin J, Stone JM et al (2011) Characterization of abiotic stress-responsive Arabidopsis thaliana RD29A and RD29B genes and evaluation of transgenes. Planta 234:97–107. https://doi.org/10.1007/s00425-011-1387-y
Nakaminami K, Hill K, Perry SE et al (2009) Arabidopsis cold shock domain proteins: Relationships to floral and silique development. J Exp Bot 60:1047–1062. https://doi.org/10.1093/jxb/ern351
Nelson BK, Cai X, Nebenführ A (2007) A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 51:1126–1136. https://doi.org/10.1111/j.1365-313x.2007.03212.x
Niemann MCE, Bartrina I, Ashikov A et al (2015) Arabidopsis ROCK1 transports UDP-GlcNAc/UDP-GalNAc and regulates ER protein quality control and cytokinin activity. Proc Natl Acad Sci U S A 112:291–296. https://doi.org/10.1073/pnas.1419050112
Niemann MCE, Weber H, Hluska T et al (2018) The cytokinin oxidase/dehydrogenase CKX1 is a membrane-bound protein requiring homooligomerization in the endoplasmic reticulum for its cellular activity. Plant Physiol 176:2024–2039. https://doi.org/10.1104/pp.17.00925
Ogo Y, Itai RN, Nakanishi H et al (2006) Isolation and characterization of IRO2, a novel iron-regulated bHLH transcription factor in graminaceous plants. J Exp Bot 57:2867–2878. https://doi.org/10.1093/jxb/erl054
Panchy N, Lehti-Shiu M, Shiu SH (2016) Evolution of gene duplication in plants. Plant Physiol 171:2294–2316. https://doi.org/10.1104/pp.16.00523
Rong C, Liu Y, Chang Z et al (2022) Cytokinin oxidase/dehydrogenase family genes exhibit functional divergence and overlap in rice growth and development, especially in control of tillering. J Exp Bot 73:3552–3568. https://doi.org/10.1093/jxb/erac088
Ross EJH, Stone JM, Elowsky CG et al (2004) Activation of the Oryza sativa non-symbiotic haemoglobin-2 promoter by the cytokinin-regulated transcription factor, ARR1. J Exp Bot 55:1721–1731. https://doi.org/10.1093/jxb/erh211
Rubio V, Linhares F, Solano R et al (2001) A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Genes Dev 15:2122–2133. https://doi.org/10.1101/gad.204401
Sakai H, Aoyama T, Oka A (2000) Arabidopsis ARR1 and ARR2 response regulators operate as transcriptional activators. Plant J 24:703–711. https://doi.org/10.1111/j.1365-313x.2000.00909.x
Schmülling T, Werner T, Riefler M et al (2003) Structure and function of cytokinin oxidase/dehydrogenase genes of maize, rice, Arabidopsis and other species. J Plant Res 116:241-252. https://doi.org/10.1007/s10265-003-0096-4
Schwarz I, Scheirlinck MT, Otto E et al (2020) Cytokinin regulates the activity of the inflorescence meristem and components of seed yield in oilseed rape. J Exp Bot 71:7146–7159. https://doi.org/10.1093/jxb/eraa419
Sharma A, Prakash S, Chattopadhyay D (2022) Killing two birds with a single stone—genetic manipulation of cytokinin oxidase/dehydrogenase (CKX) genes for enhancing crop productivity and amelioration of drought stress response. Front Genet 13:941595. https://doi.org/10.3389/fgene.2022.941595
Smigocki AC (1995) Phenotype Modification and Enhanced Tolerance to Insect Pests by Regulated Expression of a Cytokinin Biosynthesis Gene. HortScience 30:967-969. https://doi.org/10.21273/HORTSCI.30.5.967
Tan M, Li G, Qi S et al (2018) Identification and expression analysis of the IPT and CKX gene families during axillary bud outgrowth in apple (Malus domestica Borkh.). Gene 651:106–117. https://doi.org/10.1016/j.gene.2018.01.101
Villao-Uzho L, Chávez-Navarrete T, Pacheco-Coello R (2023) Plant Promoters: Their Identification, Characterization, and Role in Gene Regulation. Genes 14:1226. https://doi.org/10.3390/genes14061226
Wang Y, Tang H, Debarry JD et al (2012) MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40:e49. https://doi.org/10.1093/nar/gkr1293
Wang X, Ding J, Lin S et al (2020) Evolution and roles of cytokinin genes in angiosperms 2: Do ancient CKXs play housekeeping roles while non-ancient CKXs play regulatory roles? Hortic Res 7:29. https://doi.org/10.1038/s41438-020-0246-z
Wang C, Wang H, Zhu H et al (2021) Genome-wide identification and characterization of cytokinin oxidase/dehydrogenase family genes in Medicago truncatula. J Plant Physiol 256:13308. https://doi.org/10.1016/j.jplph.2020.153308
Yan H, Wang Y, Chen B et al (2022) OsCKX2 regulates phosphate deficiency tolerance by modulating cytokinin in rice. Plant Sci 319:111257. https://doi.org/10.1016/j.plantsci.2022.111257
Zhang W, Peng K, Cui F et a (2021) Cytokinin oxidase/dehydrogenase OsCKX11 coordinates source and sink relationship in rice by simultaneous regulation of leaf senescence and grain number. Plant Biotechnol J 19:335–350. https://doi.org/10.1111/pbi.13467
Zhu M, Wang Y, Lu S et al (2022) Genome-wide identification and analysis of cytokinin dehydrogenase/oxidase (CKX) family genes in Brassica oleracea L. reveals their involvement in response to Plasmodiophora brassicae infections. Hortic Plant J 8:68–80. https://doi.org/10.1016/j.hpj.2021.05.003
Acknowledgements
We acknowledge the Central Instrumentation Facility (CIF), University of Delhi South Campus, New Delhi, India for providing the confocal and sequencing facilities used in this manuscript.
Funding
CS and AS are thankful to the Council of Scientific and Industrial Research, India for fellowships. This work has been supported by grants from the Department of Biotechnology and the JC Bose fellowship award, Science and Engineering Research Board (SERB), Government of India.
Author information
Authors and Affiliations
Contributions
CS Conceptualization, Methodology, Formal analysis, Investigation, Data curation, Writing- Original Draft. AS: Review and editing. PK and AKS: Conceived the idea and provided logistic support. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All the authors have given their consent for publication of this manuscript by Stress Biology.
Competing interests
The authors declare that they have no competing interests.
Additional information
Handling Editor: Mukesh Jain.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
44154_2024_173_MOESM1_ESM.pptx
Additional file 1: Table S1. Information on MiCKX proteins with their physiochemical parameters with pI, instability index, aliphatic index, and GRAVY along with their subcellular localization and N-glycosylation.
44154_2024_173_MOESM2_ESM.pptx
Additional file 2: Fig. S1. Domain analysis of MiCKXs depicting FAD and cytokinin binding domains. Fig. S2. Gene ontology analysis. a Biological processes. b Molecular processes. c Cellular components. Fig. S3. Transgenic confirmation of MiCKX4 overexpression lines in A. thaliana Col-0 Wild type. a Genomic DNA confirmation using MiCKX4 gene-specific primers. b Relative transcript levels of MiCKX4 in transgenics as compared to wild-type.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singhal, C., Singh, A., Sharma, A.K. et al. Identification of CKX gene family in Morus indica cv K2 and functional characterization of MiCKX4 during abiotic stress. Stress Biology 4, 35 (2024). https://doi.org/10.1007/s44154-024-00173-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44154-024-00173-x