
Abstract
Cisplatin, an anticancer drug, has limited its clinical application due to its severe nephrotoxicity, such as acute kidney injury (AKI). Damaged or dysfunctional mitochondria caused by cisplatin are toxic to the cell by producing reactive oxygen species and releasing cell death factors. Mitophagy is the mechanism of selective degradation of these damaged mitochondria via autophagy, that is critical to cellular homeostasis and viability. In this study, the protective functions of inorganic nitrate against cisplatin-induced nephrotoxicity are assessed. Our results in vitro show that nitrate significantly reduced the apoptosis of HK2 or NRK52E cells induced by cisplatin treatment. Furthermore, dietary nitrate notably alleviates the tubular and glomerular damages as well as the loss of renal function in cisplatin-induced AKI mice models. These protective effects are closely related to downregulation of cell apoptosis and reduction of reactive oxygen species (ROS) generation. Mechanistically, inorganic nitrate treatment promotes the activation of mitophagy mediated by the PINK1-PRKN/PARK2 pathway, which plays an important role in the maintenance of mitochondrial quality, helping renal tubular cells to survive and recover from cisplatin stress. These novel findings suggest that inorganic nitrate supplementation deserve further exploration as a potential treatment in patients with cisplatin-induced renal injury.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Acute kidney injury (AKI), characterized by a rapid decrease in renal function, is a global public health problem that poses a significant economic burden on health finances (Gonsalez et al. 2019; Lewington et al. 2013). Cisplatin, as a first-line chemotherapeutic drugs, has commonly been used for treating numerous solid tumours, such as cervical, ovarian, head-neck, and non-small-cell lung cancers (Rottenberg et al. 2021; Goldberg et al. 2022; Wen et al. 2023). However, the clinical application of cisplatin is largely restricted due to its severe undesirable toxic effects, particularly nephrotoxicity, such as AKI (Miller et al. 2010; Tang et al. 2023; Klumpers et al. 2022). Recent research reports that cisplatin caused renal tubular cell injury through several factors (Volarevic et al. 2019), such as the mitochondria, which is an important involved organelle (Oh et al. 2017). Specifically, cisplatin can induce mitochondria DNA damage in renal proximal tubular cells and evoke excessive production of reactive oxygen species (ROS). These effects ultimately damage the mitochondrial redox balance, decrease membrane potential and lead to kidney injury (Ichinomiya et al. 2018; Jia et al. 2019).
Autophagy is a process of engulfing its own cytoplasmic proteins or organelles, wrapping them into vesicles and merging with the lysosome to degrade the contents of them, so as to realize the metabolic needs of the cell itself and the renewal of certain organelles. Many studies have proved that downregulation of autophagy in the kidney is associated with serious AKI because autophagy is crucial for normal homeostasis of the renal proximal tubules (Kaushal and Shah 2016). In the AKI model, loss of autophagy in the proximal tubule due to abnormalities in autophagy-related genes worsen tubular injury and renal function by exacerbating mitochondrial damage (Tang et al. 2018; Lin et al. 2019). Conversely, certain methods that promote autophagy could protect renal tubular cells from multiple stressors (Zhang et al. 2019; Liu et al. 2018). Mitophagy, as a form of selective autophagy, can selectively isolate and degrade damaged or incomplete mitochondria, is vital for maintaining the integrity of mitochondrial function and cell homeostasis (Pickles et al. 2018). Some researches reveal that stimulation of mitophagy ameliorate the mitochondrial function in cisplatin-induced AKI mice (Zhu et al. 2020). Therefore, safe and effective approaches that activate mitophagy, and reduce oxidative stress may provide a new strategy for the prevention and treatment of cisplatin-induced AKI.
Inorganic nitrate, derived from our daily diet (such as green leafy vegetables), can be converted into nitrite by symbiotic bacteria in the mouth, and then nitrite is swallowed, absorbed in the intestine and metabolized to nitrogen oxide (NO) and other bioactive nitrogen oxides in tissues (Lundberg et al. 2008; Duncan et al. 1995). Many studies have shown the antioxidant properties of nitrate which protect cells against ROS-related injury efficiently through the nitrate-nitrite-NO pathway in adipocytes of diabetes mellitus type 2 models (Ohtake et al. 2015). In the rat model of renal and cardiovascular disease induced by high-salt diet, dietary inorganic nitrate has been proved to prevent proteinuria and histological signs of kidney injury and attenuate the cardiac hypertrophy and fibrosis via decreasing the levels of ROS (Carlström et al. 2011). Moreover, Carlstrom et al. (2011) report that dietary nitrate alleviates renal ischemia–reperfusion (IR) injury, suggesting that patients with high risk of renal IR damage may benefit from a high nitrate diet (Yang et al. 2017). Consistently, our previous studies have proved that dietary inorganic nitrate exerts protective role on irradiation-induced systemic injury model by reducing ROS levels (Chang et al. 2019). However, the effect of inorganic nitrate on kidney injury in cisplatin-induced AKI models and its influence on mitophagy have not been studied.
In this study, we constructed in vitro and in vivo cisplatin-induced renal injury model to explore the protective effects of inorganic nitrate on renal injury and whether these effects were relied on the activation of mitophagy.
2 Materials and methods
2.1 Cell culture
The human renal cell line HK2 and mouse renal cell line NRK25E cells were purchased from the National Infrastructure of Cell Line Resource (Beijing, China) and Procell Life Science&Technology Co.,Ltd (Wuhan, China). HK2 cells was incubated in MEM medium and NRK25E cells was incubated in DMEM medium, supplemented with 10% fetal bovine serum (complete medium) and 1% penicillin/streptomycin under a humidified atmosphere of 5% CO2 at 37 °C. All cells were maintained in culture for no more than 25 continuous passages and were verified by short tandem repeat DNA fingerprinting and tested for mycoplasma before using.
2.2 Cell viability measurement and cell apoptosis analysis
Cell viability was measured using a CCK8 assay kit (Dojindo, Kumamoto, Japan). In short, 10 µL of CCK8 solution was added to each well containing 90 µL of medium. After incubating for 1 h, the absorbance was detected at 450 nm using an EnSpire label microplate reader (PerkinElmer, Waltham, MA). Cell Apoptosis Detection Kit were purchased from Solarbio (Beijing, China). The cells were digested using trypsin without EDTA and then resuspended in PBS. After centrifugation, the cells were then stained with 5 μL fluorescein isothiocyanate (FITC)-Annexin V, incubated at room temperature in a dark place, and then stained with 10 μL propidium iodide (20 μg /mL). The apoptotic cells were analyzed by flow cytometry (SBeckman Coulter, USA).
2.3 Drugs and chemicals
Cisplatin (P4394), Sodium nitrate (S5506) were purchased from Sigma-Aldrich. Hydroxychloroquine (HCQ, HY-W031727), Rapamycin (Rapa, HY-10219) were purchased from MedChemExpress. These compounds are dissolved and stored according to the corresponding instructions.
2.4 Animals and experimental models
A total of 60 male C57BL/6 mice (6–8 weeks) were obtained from the Capital Medical University, Beijing, China. This study was conducted following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Animal Use and Care Committee of Capital Medical University (ethical code: AEEI-2018–216). The animals were acclimatised to the surrounding environment for 1 week prior to the study, maintained at a 12-h light/dark cycle, and provided regular water and food. To assess the protective role of inorganic nitrate on cisplatin-induced AKI, the mice were divided into 4 groups (normal control, Ctrl: n = 15; nitrate-treated, NaNO3: n = 15; cisplatin-treated, Cisp: n = 15; cisplatin + nitrate, Cisp + NaNO3: n = 15). The control mice were given drinking water supplemented with sodium chloride (Nacl, 2 mmol) and the nitrate-treated group mice were subjected to drinking water supplemented with NaNO3 (2 mmol). The cisplatin-treated mice were single intraperitoneal injection of cisplatin (20 mg/kg) and given drinking water supplemented with sodium chloride (2 mmol). The cisplatin + nitrate group mice were subjected to 2 mmol NaNO3 drinking water for 1 week before cisplatin injection. The normal control and nitrate-treated group mice received saline injection with equal volume of cisplatin. At the end of the experiment, the animals were sacrificed and the blood and kidneys were collected for related testing.
2.5 Sample harvest
The mice were anesthetized with isoflurane and blood were isolated from the ophthalmic vein. Whole blood with 2 mmol EDTA was centrifuged immediately with 6000 × g at 4 °C for 15 min to obtain the serum and stored at -80 °C. The kidneys were weighed and the tissues were mixed with 0.9% NaCl solution and homogenized by a tissue homogenate breaker, and then the tissue homogenate was centrifuged at 6500 × g for 15 min at 4 °C. The supernatant of kidney tissue was collected to test the levels of superoxide dismutase (SOD), catalase (CAT), glutathione (GSH) and malondialdehyde (MDA) using commercial kits. The mice kidneys were harvested and fixed in 4% neutral paraformaldehyde, and then embedding with paraffin or optimal cutting temperature compound. The remaining kidney tissues were stored at -80 °C for other detection.
2.6 Serum biochemistry analysis
Plasma samples from mice were centrifuged with 1000 × g for 15 min, and then serum creatinine (Crea) and blood urea nitrogen (BUN) were measured by Zhong Yan Biolab Company (Shenzhen, China).
2.7 Histopathological examination
The mice kidneys were harvested and cut at 4-μm thickness. Serial sections were subjected to hematoxylin–eosin (H&E) and periodic acid-schiff (PAS) staining. Tubular damage was assessed in PAS-stained kidneys by an expert pathologist and scored according to the severity of tubular injury indicated by tubular epithelial cell vacuolisation, cell necrosis, cast formation, brush border loss, and sloughing of cells: 0, normal; 1, 1%–10%; 2, 11%–25%; 3, 26%–45%; 4, 46%–75%; and 5, 76%–100%. Quantification was determined by ImageJ software (National Institutes of Health, Bethesda, MD).
2.8 p-H2AX and TUNEL staining
Phosphorylated-H2A.X variant histone (p-H2AX) was detected as previously described (Chang et al. 2019). Kidney tissue section were incubated with primary anti-p-H2AX antibody for 1 h at room temperature, then stained with secondary antibody (Table S1), and counterstained with DAPI (1,002,339,028; Sigma-Aldrich). Cell apoptosis was measured by a TUNEL apoptosis detection kits according to the manufacturer’s instructions (C1098; Beyotime Institute of Biotechnology). Images were obtained from a confocal system and a digital camera (SP8; Leica), and the positive cells were calculated by the ImageJ software.
2.9 Immunofluorescence
After treating with cisplatin and/or nitrate, HK2 cells cultured in 6-well plate were further incubated with 200 nM MitoTracker Deep Red FM (M22426, Invitrogen) in 37° C incubator for 30 min. After staining, wash the cells in fresh, pre-warmed growth medium, then carefully remove the medium covering the cells, washed with ice-cold PBS, fixed with 4% paraformaldehyde (SigmaAldrich, P6148) and permeabilized with 0.5% Triton X-100 (MP Blomedicals, 194,854). Cells were then blocked with 2% FCS and followed by incubation with anti-LC3B antibody (CST, 83506S; diluted 1:100) overnight at 4 °C. Next day cells were washed with TBST and incubated with goat anti-mouse IgG Alexa Fluor 488-labeled secondary antibody (2,120,125, Invitrogen) for 1 h at room temperature. Finally, the images were captured using confocal microscopy (Leica DM6000 CS, Germany).
2.10 Immunohistochemistry staining
Immunohistochemistry assay was performed as described previously (Wang et al. 2022). Briefly, the 4-μm sections were placed in 3% hydrogen peroxide, blocked with 5% BSA, and incubated with anti- kidney injury marker-1 (KIM-1) antibody (Table S1) at 4 °C overnight. Then, the sections were incubated with HRP-conjugated secondary antibody for 1 h at room temperature and further incubated with 0.06% 3,3-diaminobenzidine tetrahydrochloride (P0203; Beyotime Institute of Biotechnology, Shanghai, China). Quantification was determined by the ImageJ software (10 random fields per section, × 400).
2.11 Autophagic flux detection assay
Autophagic flux activity was detected by the mCherry-GFP-LC3B reporter. HK2 cells were transfected with adenovirus expressing mCherry-GFP-LC3B (C3011, Beyotime Biotechnology) according to the manufacturer’s protocol. At 24 h post-transfection, the cells were treated with cisplatin and/or nitrate. The cells were fixed with 4% paraformaldehyde for 1 h, and washed three times with PBS. Autophagic flux was analysed by detecting the fluorescent intensity of autophagosomes and autolysosomes using confocal system (Leica DM6000 CS, Germany). DAPI staining was used to visualize the nucleus. The ratio of the fluorescence of mCherry+ puncta to the fluorescence of GFP+ puncta is used as the autophagic index. More than 10 cells were counted for each condition.
2.12 Transmission Electron Microscopy (TEM)
To observe the morphology of autophagosomes at different stages, HK2 cells were fixed with 2% paraformaldehyde and 2.5% glutaraldehyde and post fixed with 1% OsO4 in 0.1 mol/L cacodylate buffer for 2 h. The samples were then cut into 0.12-μm sections after embedded, and stained with 0.2% lead citrate and 1% uranium acetate. To morphologically demonstrate the structure of mitochondria in renal tubular epithelial cells of cisplatin-induced AKI mice models, kidney tissues from mice were cut into 1 mm × 1 mm × 1 mm sized patch and fixed in paraformaldehyde and glutaraldehyde, post fixed in osmium tetroxide, dehydrated in ethanol, subjected to resin penetration and embedded in Epon. Ultrathin sections were stained with uranyl acetate (E. Merck, Darmstadt, Germany) and lead citrate (Sigma-Aldrich, St. Louis, MO, USA). Transmission electron microscope (HT7700, HITACHI, Japan) was used for observation. All analysis was performed blind to the observer.
2.13 Western blot analysis
Western blot was carried out as described previously (Ma et al. 2017). In short, the cell and tissue were harvested and homogenized with RIPA buffer. Samples were incubated with primary antibodies overnight at 4 °C and then incubated with horseradish peroxidase-linked secondary antibodies (Table S1). Immunoreactive bands were detected by NIH Image 1.62 (National Institutes of Health, Bethesda MD). Quantification of protein expression was performed using Image J software.
2.14 siRNA transfection
Cells were seeded into a 6-well plate and then transfected with 80–100 nmol/L siRNA using Lipofectamine 3000 according to the manufacturer's instructions, and cells were collected for protein extraction at 48 h after transfection. siRNA were synthesized and purchased from Sangon Biotech Co., Ltd (Shanghai, China). The sequences of siRNA oligonucleotides were as follows: control siRNA, 5'-uucuccgaacgugucacgu; PINK1 siRNA, 5'-ccaggctgggccgcaggaccg; PRKN siRNA, 5'-ggatcagcagagcattgttca.
2.15 ROS measurement
ROS detection was accomplished following the procedures of Chang et al. (Chang et al. 2019). Briefly, kidneys were harvested and immediately frozen, sectioned at 10-μm thickness. According to the ROS Fluorescein Assay Kit (GMS10016.6, Genmed Scientifics, Inc) instructions, the sections were incubated with stains for 20 min at 37℃, then washed with cleaning liquid, incubated with DAPI and observed under a confocal system and a digital camera. The number of ROS-positive cells were calculated by the ImageJ software (10 random fields per section, × 400).
2.16 Statistics analysis
Data were presented as the mean ± standard error of the mean (SEM) and analysed by GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). Continuous variables were analysed by analysis of variance or t-test, and nonparametric test was used if the data did not assume Gaussian distribution. One-way analysis of variance was performed to compare differences when more than two groups. P < 0.05 was considered statistically significant, and two-tailed P-value was used in analyses.
3 Results
3.1 Nitrate suppresses cisplatin-induced renal cell apoptosis in vitro
To assess the role of nitrate (NaNO3) in cisplatin-treated renal cells, cisplatin-treated HK2 or NRK52E cells were incubated with or without nitrate. Firstly, the result of CCK8 assay indicated that HK2 cell viability was descended in a concentration- and time-dependent manner after cisplatin treatment (Fig. 1A). Secondly, we explored the optimal acting concentration of nitrate with sodium chloride as an osmotic control and found a significant decrease in cell viability at 50 mmol/L (Fig. 1B). Therefore, we chose cisplatin for 20 μmol/L, nitrate for 20 mmol/L and action for 48 h as the conditions for the subsequent cell experiments. Subsequently, the cisplatin-treated HK2 cell viability was increased to 0.69 ± 0.04-fold from 0.52 ± 0.05-fold after nitrate incubation (Fig. 1C). Similar results were obtained in NRK52E cell and the cisplatin-treated NRK52E cell viability was ascended to 0.73 ± 0.07-fold from 0.46 ± 0.06-fold under the action of nitrate (Fig. 1D). Moreover, the number of apoptotic cells caused by cisplatin was notably decreased after nitrate treatment (Fig. 1E and F). Consistent with attenuating cell apoptosis, the elevated expression of Bax and decreased expression of Bcl-2 induced by cisplatin treatment were reduced and increased after nitrate incubation, respectively (Fig. 1G-I). These data indicate that nitrate inhibit cisplatin-induced renal cell apoptosis in vitro.

Inorganic nitrate (NaNO3) inhibits cisplatin-induced injury in vitro. Nacl was used as osmotic control of NaNO3. A HK2 cells were exposed to cisplatin (0, 5, 10, 20 μmol/L) for 24, 48 or 72 h, and the cell viability was determined by CCK8. B HK2 cells were incubated with NaNO3 or Nacl (0, 1, 10, 20, 50, 100 mmol/L) for 48 h, and the cell viability was determined. C, D HK2 or NRK52E cells were treated with cisplatin (20 μmol/L) and were incubated with NaNO3 (20 mmol/L) for 48 h, and the cell viability was determined. E, F The effects of NaNO3 on cisplatin-induced apoptosis in HK2 or NRK52E cells were determined by flow cytometry assay. G-I Whole HK2 cell lysates were collected for immunoblot analysis of Bax, Bcl-2 and β-actin (loading control). (G) Representative blots. Three independent experiments to quantify Bax (H) and Bcl-2 (I). Data are shown as the means ± SEM and analyzed by one-way ANOVA with Tukey’s test. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Ctrl; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Cisp; NS, no significance (Ctrl, control; NaNO3, Nitrate; Cisp, cisplatin; Cisp + NaNO3, cisplatin + Nitrate)
3.2 Oral administration of inorganic nitrate attenuates cisplatin-induced AKI
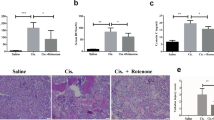
Considering the prospect of nitrate application in clinical practice in the future, we constructed cisplatin-induced AKI mice model to further explore whether oral administration of nitrate could attenuate cisplatin-induced renal injury in vivo. Male C57BL/6N mice orally administered nitrate drinking water (2 mmol/L) one week before intraperitoneally injected with cisplatin (20 mg/kg) and sacrificed after 72 h for related testing (Fig. 2A). As expected, the weight loss of cisplatin-induced AKI mice was remarkable increased compared with the controls. Importantly, nitrate pre-treatment tended to attenuate this cisplatin-induced severe undesirable toxic effects (Fig. 2B). To further validate the protective role of nitrate on cisplatin-induced kidney injury, we measured mice renal function with BUN and Crea. The results indicated that cisplatin treatment caused a significant increase of BUN and Crea compare to controls, and these were prevented by nitrate pre-treatment. Baseline BUN and Crea were similar between controls and nitrate-treated mice (Fig. 2C and D). In addition, severe histological changes of renal injury were observed in the kidney of cisplatin-induced AKI model mice, including cast formation, brush border loss, vacuolisation and loss of tubular epithelial cells, whereas this renal structural damage was improved by dietary nitrate supplementation (Fig. 2E and F).

Oral administration of inorganic nitrate protects the kidney against cisplatin-induced AKI. A Schematic of the experimental design for nitrate administration of cisplatin-induced AKI mice model. B Body weight changes of mice at the end of the experiment. C, D Serum BUN and Crea levels were quantified in each group (5 mice in each group). E Representative histological changes in hematoxylin–eosin (H&E) and periodic acid-schiff (PAS) stained of mice kidney sections at 72 h after cisplatin injection. The yellow arrow indicates normal appearance of tubules; the black arrow indicates cytoplasmic vacuolization and desquamation of tubular epithelium. Scale bars: 50 μm. F Tubular damage score. G Representative images of KIM-1 IHC staining in kidney sections. Scale bars: 50 μm. H Percentage of KIM-1 staining positive cells. I-L The protein levels of Bax, Caspase 3 and Cleaved Caspase 3 in renal cortical tissues of mice were evaluated by western blotting. Representative images (I) of western blotting and three independent experiments to quantify Bax (J), Caspase 3 (K) and Cleaved Caspase 3 (L). M Representative micrographs showing TUNEL staining. N The quantitative analyses of apoptotic cell. Data are shown as the means ± SEM and analyzed by one-way ANOVA with Tukey’s test. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Ctrl; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Cisp; NS, no significance (Ctrl, control; NaNO3, Nitrate; Cisp, cisplatin; Cisp + NaNO3, cisplatin + Nitrate)
To confirm the nephroprotective effect of inorganic nitrate in cisplatin-induced AKI mice, we further measured the level of kidney injury marker-1 (KIM-1). Immunohistochemical results showed that cisplatin treatment caused a significant increase of KIM-1 levels compare to controls, and this was prevented by inorganic nitrate pre-treatment (Fig. 2G and H). The same results were obtained by western blot for detecting KIM-1 (Figure S1). Subsequently, we tested the effect of oral nitrate on apoptosis related proteins in vivo models. In line with the results of in vitro studies, western blot assay indicated that oral administration of nitrate reduced the level of pro-apoptotic Bax and cleaved caspase 3 in cisplatin-induced AKI mice (Fig. 2I-L), which was consistent with the results of immunofluorescence staining of TUNEL (Fig. 2M and N).
3.3 Dietary nitrate improves DNA damage and inhibits ROS release induced by cisplatin
DNA damage and ROS production are considered to critically contribute to kidney injury (Tejchman et al. 2021). Firstly, we compared the levels of phosphorylated histone H2AX (p-H2AX), a marker of early-stage DNA damage, at different cisplatin treatment time points between the groups. After 2 or 4 h of cisplatin treatment, the levels of p-H2AX in the kidney of mice significantly increased compared to the controls, while in cisplatin-induced AKI group given nitrate, it decreased remarkably (Fig. 3A and B). Similar results were obtained in western bolt experiment (Figure S2A and B). However, the level of p-H2AX tended to be reduced in nitrate-treated AKI model mice compared to the AKI group at 6 h of cisplatin treatment, but the differences between the two groups were not statistically significant.

Dietary nitrate inhibits DNA damage and ROS release induced by cisplatin. A Representative images of p-H2AX immunofluorescence staining of kidney tissue from the mice. Nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Bar: 50 μm. B The quantitative analyses of p-H2AX staining positive cells. (C) Representative images of ROS immunofluorescence staining of kidney tissue from the mice. Nuclei were stained with DAPI. Bar: 50 μm. D The quantitative analyses of ROS staining positive cells. E–H The activities of superoxide dismutase (SOD) and catalase (CAT) and together with the contents of glutathione (GSH) and malondialdehyde (MDA) were measured in the mice kidney tissues of each group (5 mice in each group). Data are shown as the means ± SEM and analyzed by one-way ANOVA with Tukey’s test. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Ctrl; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Cisp; NS, no significance (Ctrl, control; NaNO3, Nitrate; Cisp, cisplatin; Cisp + NaNO3, cisplatin + Nitrate)
Next, we detected the ROS changes in the kidney following cisplatin or/and nitrate treatment. The results displayed that the number of ROS-positive cells were significant ascended at 2, 4, 6 and 72 h after cisplatin treatment compare to controls. As expected, the productions of ROS were reduced in the nitrate-treated AKI model mice at 2, 4 and 72 h compare to cisplatin-treated mice, although the decrease of ROS is not statistically significant at 6 h (Fig. 3C and D). In addition, the activities of superoxide dismutase (SOD) and catalase (CAT) and together with the contents of glutathione (GSH) and malondialdehyde (MDA) in the kidney tissues were measured to assess the antioxidant effect of nitrate in cisplatin-treated AKI mice. The results showed that cisplatin significantly decreased the activities of kidney SOD and CAT, reduced the GSH content and increased the MDA content compare to those of controls. Importantly, nitrate treatment in cisplatin-induced AKI model mice markedly increased the levels of CAT, SOD and GSH, and decreased the MDA level compare to cisplatin-treated mice (Fig. 3E-H). These data prove that nitrate has a potential protective effect against cisplatin-induced AKI by improving oxidative stress.
3.4 Dietary nitrate induces autophagy in cisplatin-induced injury
To explore the molecular mechanisms by which nitrate attenuated cisplatin-induced kidney injury, and considering the importance of autophagy in tubular injury and renal function, we investigated the effect of nitrate on autophagy in cisplatin-treated HK2 cell. Consistent with previous studies, our western blot results showed a dramatic increase in LC3B-II and a significant reduction in p62, two biochemical markers of autophagy activation, in HK2 cells treated with cisplatin (Figure S3A-C, 1 vs 2 lane). These data indicated that autophagic flux were induced by cisplatin in HK2 cells. Interestingly, subsequent analysis showed that the level of LC3 II was further ascended and the level of p62 was decreased in cisplatin + NaNO3 group compare to cisplatin-treated group (Figure S3A-C, 2 vs 4 lane). Furthermore, we determined whether autophagy was activated in cisplatin-induced AKI mice model. Renal cortical tissues were harvested for immunoblot assay and the result showed that cisplatin induced significant increases in LC3B-II and reduction in p62 (Fig. 4A-C, 1 vs 2 lane). Expectedly, we observed higher LC3B-II and lower p62 level in the cisplatin + NaNO3 group compared with the cisplatin-induced AKI group (Fig. 4A-C, 2 vs 4 lane). Furthermore, HK2 cells were transfected with a mCherryGFP-LC3 fusion construct tool to determine autophagy flux by estimating the relative numbers of autophagosomes and autolysosomes, as indicated by yellow and red fluorescence due to the expression of mCherry and GFP. The results showed that cisplatin caused an increase in autophagic flux as indicated by the ratio of mCherry to GFP compare to control group. In cisplatin + NaNO3 group, the further elevated ratio of mCherry to GFP suggesting a more active autophagy (Fig. 4D-E). We also monitored changes in autophagy using transmission electron microscope (TME) analysis. As shown in Fig. 4F, electron microscope data indicated that more autolysosomes were observed in cisplatin + NaNO3 treated HK2 cells, suggesting NaNO3 promoted autophagy flux of HK2 cell incubated with cisplatin.

Dietary nitrate promotes autophagy in cisplatin-induced injury. A-C The protein levels of LC3B II and p62 in renal cortical tissues of mice were evaluated by western blotting. A Representative blots. Quantification of LC3BII/ LC3BI (B) and p62 (C), (3 mice in each group). D, E HK2 cells were transfected with a mCherry-GFP-LC3B construct and were cultured with cisplatin and/or NaNO3 for 48 h. Autophagic flux was measured by counting the yellow autophagosomes fluorescence dots and red autolysosomes fluorescence dots under a confocal microscope. The representative images (D) and statistical data were shown (E). DAPI was used for nuclear staining. Scale bar: 10 μm. F, G Transmission electron microscopy observation of autophagic ultra-structure in HK2 cells treated with cisplatin and/or NaNO3 for 48 h, white rectangles are magnified in the right panels. The representative images (F) and statistical data were shown (G). N, nuclei. Red arrows indicate autophagosomes; White arrows for autolysomes; Scale bar: 5 μm. Data are presented as mean ± SEM and analyzed by one-way ANOVA with Tukey’s test. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Ctrl; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Cisp (Ctrl, control; NaNO3, Nitrate; Cisp, cisplatin; Cisp + NaNO3, cisplatin + Nitrate)
To confirm the protective role of autophagy on cisplatin-treated cell damage, an autophagy inhibitor (Hydroxychloroquine, HCQ) or activator (Rapamycin; Rapa) was added to the HK2 cells. The result showed that cell viability was decreased in the cisplatin + HCQ group (Figure S3D), while increased in the cisplatin + Rapa group (Figure S3E). These data confirm that autophagy plays a protective effect on cisplatin-induced cell damage.
3.5 Nitrate induced PINK1 and PRKN/PARK2-mediated mitophagy in HK-2 cells
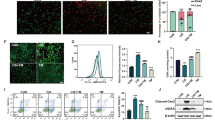
Given the critical role of mitophagy on mitochondrial maintenance, we explored the effect of nitrate on cisplatin-induced mitophagy. The result of western blot showed that the change of autophagy activation in HK2 cells during nitrate treatment was closed related to a marked decrease in the mitochondrial inner membrane protein TIMM23 and TOMM20 (Fig. 5A-C), suggesting activation of mitophagy or mitochondrial clearance. To further confirm mitophagy induction, we tested the colocalization of mitochondria and autophagosomes, as indicated by staining of autophagosome marker LC3B and mitochondrial marker MitoTracker. The result of confocal microscopy displayed a significant colocalization of LC3B and MitoTracker in cisplatin + NaNO3 treated HK2 cells, indicating the formation of mitophagosomes induced by NaNO3. Next, we observed mitochondria morphology in the cortices of the mouse kidneys using transmission electron microscopy. Compared with the normal control group, more swollen and round mitochondria could be observed in the renal tubular cells of AKI model group mice. Subsequently, quantitative morphometric analysis revealed a decrease in the aspect ratio of mitochondria (the long axis of mitochondria/short axis of mitochondria), which is indicative of mitochondrial fragmentation. However, these phenomena were significantly alleviated after NaNO3 administration (Fig. 5F-G). Together, these results demonstrate that nitrate is able to further activate mitophagy in cisplatin-induced AKI models.

Dietary nitrate induced mitophagy cisplatin-induced injury. A-C The protein levels of TOMM20 and TIMM23 in HK2 cells were evaluated by western blotting. A Representative blots. Three independent experiments to quantify TOMM20 (B) and TIMM23 (C). D, E Representative images and quantification of immunofluorescence double-labelling LC3B and mitochondrial marker (MitoTracker), DAPI was used for nuclear staining. Scale bar: 10 μm. F Representative TEM micrographs of mouse renal tubular epithelial cell mitochondria from each group, white rectangles are magnified in the right panels. Scale bar: 1 μm. G The aspect ratio (major axis/minor axis) of mitochondria were evaluated via ImageJ (n = 10 mitochondria). Data are presented as mean ± SEM and analyzed by one-way ANOVA with Tukey’s test. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Ctrl; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Cisp (Ctrl, control; NaNO3, Nitrate; Cisp, cisplatin; Cisp + NaNO3, cisplatin + Nitrate)
Next, to further dissect the molecular mechanism underlying nitrate-induced mitophagy in HK2 cells, we detected the effect of nitrate on PINK1-PRKN/PARK2 pathway closely associated with mitophagy. In response to cisplatin + NaNO3 treatment, HK-2 cells showed notable elevation in PINK1 and PRKN/PARK2 levels compare to cisplatin treatment (Fig. 6A-C). Consistent with the change in HK2 cells, the protein levels of PINK1 and PRKN/PARK2 in kidney tissue from the mice were increased in cisplatin + NaNO3 group compared with the cisplatin group (Fig. 6D-F). To further clarify the role of PINK1-PRKN/PARK2 on mitophagy in HK2 cell, we knocked down their expression with specific siRNAs. Western blot assay showed that compared with control siRNA transfected cells, the cells with either PINK1 or PRKN siRNA showed higher mitochondrial protein TIMM23 and TOMM20 subjected to cisplatin + NaNO3 (Fig. 6G-I). Collectively, these data demonstrated that the PINK1-PRKN/PARK2 pathway is critically to tubular cell mitophagy in response to nitrate treatment under the cisplatin stress.

Dietary nitrate induced mitophagy dependent on PINK1 and PRKN/PARK2 pathway in cisplatin-induced injury. The protein levels of PRKN and PINK1 in HK2 cells were evaluated by western blotting. A Representative blots. Three independent experiments to quantify PINK1 (B) and PRKN (C). D-F The protein levels of PRKN and PINK1 in renal cortical tissues of mice were evaluated by western blotting. D Representative blots. Quantification of PINK1 (E) and PRKN (F), (3 mice in each group). G-I Silencing of PINK1or PRKN restores TIMM23 and TOMM20 levels in HK-2 following cisplatin treatment. HK-2 cells were transfected with PINK1 siRNA, PRKN siRNA or control siRNA. At 48 h after transfection, the cells were subjected to 48 h of cisplatin, and whole cell lysates were collected for immunoblot analysis of TIMM23, TOMM20 and β-actin (loading control). G Representative blots. Three independent experiments to quantify TOMM20 (H) and TIMM23 (I). Data are shown as the means ± SEM and analyzed by one-way ANOVA with Tukey’s test. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Ctrl; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Cisp; NS, no significance (Ctrl, control; NaNO3, Nitrate; Cisp, cisplatin; Cisp + NaNO3, cisplatin + Nitrate)
4 Discussion
Previous reports have shown that dietary nitrate play a renoprotective role in rat model of renal disease induced by high-salt diet and renal ischemia–reperfusion injury (Carlström et al. 2011; Yang et al. 2017). Therefore, we hypothesized that dietary nitrate might have potential as an effective drug to cure cisplatin-induced AKI. In the present study we demonstrated that dietary nitrate notably reduced the tubular and glomerular injuries as well as the loss of renal function following cisplatin-induced AKI. p-H2AX is an early marker of DNA double-strand breaks, which reaches its peak at 3 h after DNA damage. Our results showed that p-H2AX reached the peak after 4 h of cisplatin treatment and decreased at 6 h. Nitrate reduced the p-H2AX levels induced by cisplatin damage at 2 and 4 h, but this reduction was not observed at 6 h, which may be related to the rapid decrease of p-H2AX within 6 h. In addition, our data showed that at the time point of 6 h, nitrate reduced the ROS production caused by cisplatin, although the difference between the two groups did not reach statistical significance. These protective effects are closely related to down-regulated apoptosis and reduced generation of ROS. This is the first description of the salutary effects of inorganic nitrate in protecting the kidney from cisplatin-induced AKI.
Autophagy is a cytoprotective mechanism to boost cellular adaptation by eliminating and recycling of damaged organelles and macromolecules under numerous stresses. Growing studies show that autophagy induction has been proved in various AKI experimental models (Kaushal and Shah 2016; Havasi and Dong 2016). The activation of autophagy alleviates renal injury, whereas inhibition of autophagy shown adverse effect, suggesting the renal protective role of autophagy in AKI disease (Jiang et al. 2010; Yang et al. 2008; Periyasamy-Thandavan et al. 2008). In line with these studies, our research shown autophagy induction in renal tubular cells during cisplatin-induced AKI using both in vitro and in vivo experimental models. Importantly, the data of this study indicate that inorganic nitrate is an autophagic inducer in cisplatin-treated cells and AKI mice models, as shown by western blot, immunofluorescence staining and transmission electron microscopy. However, the mechanism of autophagy protecting renal tubule cells in AKI is not clarified. So far, there are two schools of thought to explicate the cytoprotective effects of autophagy. First, the activation of autophagy may release anti-apoptotic protein BCL2 from BECN1 to protect cells from death (Kang et al. 2011). Second, autophagy is a known process for clearing dysfunctional organelles that are potentially cytotoxic (Mizushima and Komatsu 2011). Our results support that inorganic nitrate function as a renoprotective role through the second possibility.
Mitochondria are known as the main intracellular organelles for ROS production and mitochondria ROS (mtROS) can initiate oxidative stress and apoptosis, which eventually leading to kidney damage (Ishimoto and Inagi 2016; Yang et al. 2014). Timely clearing of impaired mitochondria can reduce ROS production and have a protective effect on cisplatin-induced AKI. Mitophagy is a form of selective autophagy where tagging damaged mitochondria for autophagic identification and degradation. In renal pathophysiology, the mechanism of mitophagy is not clarified. Variation in PINK1 and PRKN/PARK2 expression was observed in tubular cells in diabetic nephropathy experimental models (Zhan et al. 2015). Dong et al. recently reported that silencing of PINK1 or PRKN/PARK2 reduced mitophagosome formation and restored mitochondrial protein TIMM23 during ATP depletion-repletion in HK2 cells, a commonly used in vitro model of renal ischemia–reperfusion (Tang et al. 2018). In vivo, deficiency of Pink1 and Park2 caused the decrease of ubiquitination in mitochondrial proteins and the accumulation of damaged mitochondria in renal tubule cells during renal ischemia–reperfusion (Tang et al. 2018). In present study, we founded the decrease of mitochondrial membrane protein (TIMM23, TOMM20) and the increase of PINK1 and PRKN/PARK2 protein in tubular cells in both in vitro and in vivo models of cisplatin-induced AKI. This observation is not surprising, because, these data suggest that PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal injury induced by cisplatin. Of note, our results showed that after inorganic nitrate treatment, the protein levels of PINK1 and PRKN/PARK2 in cisplatin-treated cells were further elevated, while the levels of TIMM23 and TOMM20 were decreased. Moreover, PINK or PRKN/PARK2 knockdown attenuated the promotion of inorganic nitrate on mitophagy identified by TIMM23 and TOMM20. Together, these results demonstrate that timely removal of impaired mitochondria via mitophagy is a pivotal mechanism of the renoprotective effect supplied by inorganic nitrate in cisplatin-induced AKI models.
In summary, this study has demonstrated that inorganic nitrate plays a key protective role in kidney damage induced by cisplatin in both in vitro and in vivo AKI models. Mechanistically, inorganic nitrate promotes the activation of mitophagy and reduces the level of oxidative stress markers. Mitophagy induced by inorganic nitrate is mainly mediated by the PINK1-PRKN/PARK2 pathway that plays a crucial role in maintaining mitochondrial quality and helping renal tubular cells to survive under cisplatin stress by clearing damaged mitochondria. It is suggested that promoting mitophagy may provide a novel therapeutic strategy for cisplatin-induced AKI. Future clinical study shall illuminate if inorganic nitrate can offer protective effects in patients with renal injury caused by cisplatin treatment.
Availability of data and materials
The datasets and documents generated, analyzed or used during the current study are available from corresponding author upon reasonable request.
Abbreviations
- AKI:
-
Acute kidney injury
- BUN:
-
Blood urea nitrogen
- CAT:
-
Catalase
- Crea:
-
Creatinine
- GSH:
-
Glutathione
- HCQ:
-
Hydroxychloroquine
- H&E:
-
Hematoxylin-eosin
- IR:
-
Ischemia-reperfusion
- KIM-1:
-
Kidney injury marker-1
- MDA:
-
Malondialdehyde
- NO:
-
Nitrogen oxide
- PAS:
-
Periodic acid-schiff
- p-H2AX:
-
Phosphorylated-H2A.X variant histone
- Rapa:
-
Rapamycin
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
References
Carlström M, Persson AEG, Larsson E, Hezel M, Scheffer PG, Teerlink T, et al. Dietary nitrate attenuates oxidative stress, prevents cardiac and renal injuries, and reduces blood pressure in salt-induced hypertension. Cardiovasc Res. 2011;89:574–85.
Chang S, Hu L, Xu Y, Li X, Ma L, Feng X, et al. Inorganic nitrate alleviates total body irradiation-induced systemic damage by decreasing reactive oxygen species levels. Int J Radiation Oncol Biol Phys. 2019;103:945–57.
Duncan C, Dougall H, Johnston P, Green S, Brogan R, Leifert C, et al. Chemical generation of nitric oxide in the mouth from the enterosalivary circulation of dietary nitrate. Nat Med. 1995;1:546–51.
Goldberg M, Manzi A, Birdi A, Laporte B, Conway P, Cantin S, et al. A nanoengineered topical transmucosal cisplatin delivery system induces anti-tumor response in animal models and patients with oral cancer. Nat Commun. 2022;13:1–14.
Gonsalez SR, Cortês AL, Silva RCD, Lowe J, Prieto MC, Silva Lara LD. Acute kidney injury overview: from basic findings to new prevention and therapy strategies. Pharmacol Ther. 2019;200:1–12.
Havasi A, Dong Z. Autophagy and tubular cell death in the kidney. Semin Nephrol. 2016;36:174–88.
Ichinomiya M, Shimada A, Ohta N, Inouchi E, Ogihara K, Naya Y, et al. Demonstration of mitochondrial damage and mitophagy in cisplatin-mediated nephrotoxicity. Tohoku J Exp Med. 2018;246:1–8.
Ishimoto Y, Inagi R. Mitochondria: a therapeutic target in acute kidney injury. Nephrol Dial Transplant. 2016;31:1062–9.
Jia P, Wu X, Pan T, Xu S, Hu J, Ding X. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. EBioMedicine. 2019;49:331–40.
Jiang M, Liu K, Luo J, Dong Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176:1181–92.
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–80.
Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int. 2016;89:779–91.
Klumpers MJ, Witte WD, Gattuso G, Schiavello E, Terenziani M, Massimino M, et al. Genome-wide analyses of nephrotoxicity in platinum-treated cancer patients identify association with genetic variant in RBMS3 and acute kidney injury. J Pers Med. 2022;12:892.
Lewington AJP, Cerdá J, Mehta RL. Raising awareness of acute kidney injury: a global perspective of a silent killer. Kidney Int. 2013;84:457–67.
Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26:101254.
Liu J, Livingston MJ, Dong G, Tang C, Su Y, Wu G, et al. Histone deacetylase inhibitors protect against cisplatin-induced acute kidney injury by activating autophagy in proximal tubular cells. Cell Death Dis. 2018;9:1–15.
Lundberg JO, Weitzberg E, Gladwin MT. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–67.
Ma PA, Xiao H, Yu C, Liu J, Cheng Z, Song H, et al. Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett. 2017;17:928–37.
Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins (basel). 2010;2:2490–518.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41.
Oh CJ, Ha C, Choi Y, Park S, Choe MS, Jeoung NH, et al. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 2017;91:880–95.
Ohtake K, Nakano G, Ehara N, Sonoda K, Ito J, Uchida H, et al. Dietary nitrite supplementation improves insulin resistance in type 2 diabetic KKAy mice. Nitric Oxide. 2015;44:31–8.
Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 2008;74:631–40.
Pickles S, Vigié P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. 2018;28:R170–85.
Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer. 2021;21:37–50.
Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14:880–97.
Tang Chengyuan, Livingston Man J, Safirstein Robert, Dong Z. Cisplatin nephrotoxicity: new insights and therapeutic implications. Nat Rev. 2023;19:53–72.
Tejchman K, Kotfis K, Sieńko J. Biomarkers and mechanisms of oxidative stress—last 20 years of research with an emphasis on kidney damage and renal transplantation. Int J Mol Sci. 2021;22:8010.
Volarevic V, Djokovic B, Jankovic MG, Harrell CR, Fellabaum C, Djonov V, et al. Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor toxicity. J Biomed Sci. 2019;26:1–14.
Wang H, Yang W, Qin Q, Yang X, Yang Y, Liu H, et al. E3 ubiquitin ligase MAGI3 degrades c-Myc and acts as a predictor for chemotherapy response in colorectal cancer. Mol Cancer. 2022;21:1–19.
Wen X, Zhang R, Hu Y, Wu L, Bai H, Song D, et al. Controlled sequential in situ self-assembly and disassembly of a fluorogenic cisplatin prodrug for cancer theranostics. Nat Commun. 2023;14:1–12.
Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol. 2008;294:F777–87.
Yang Y, Liu H, Liu F, Dong Z. Mitochondrial dysregulation and protection in cisplatin nephrotoxicity. Arch Toxicol. 2014;88:1249–56.
Yang T, Zhang X, Tarnawski L, Peleli M, Zhuge Z, Terrando N, et al. Dietary nitrate attenuates renal ischemia-reperfusion injuries by modulation of immune responses and reduction of oxidative stress. Redox Biol. 2017;13:320–30.
Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. 2015;26:1304–21.
Zhang Y, Wang L, Meng L, Cao G, Wu Y. Sirtuin 6 overexpression relieves sepsis-induced acute kidney injury by promoting autophagy. Cell Cycle. 2019;18:425–36.
Zhu L, Yuan Y, Yuan L, Li L, Liu F, Liu J, et al. Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics. 2020;10:5829–44.
Acknowledgements
Not applicable.
Funding
This work was supported by the Beijing Municipal Government Grant (Beijing Laboratory of Oral Health, PXM2021-014226–000041), National Natural Science Foundation of China (No. 81972338), National Natural Science Foundation of China (No. 91649124), Chinese Academy of Medical Sciences Research Unit Capital Medical University (No. 2019RU020) and Beijing Municipality Government (Beijing Scholar Program-PXM2018_014226_000021, PXM2017_014226_000023 and PXM2018_193312 _000006_0028S643_FCG).
Author information
Authors and Affiliations
Contributions
Conceptualisation: W. Li and S. Wang; Data curation: H. Wang and C. Song; Methodology: H. Wang and C. Song; Investigation: F. Chen and L. Hu; Validation: X. Liu; Visualization: H. Wang and X. Liu; Formal analysis: L. Hu and F. Chen; Project administration: W. Li and S. Wang; Resources: C. Zhang; Supervision: S. Wang; Funding acquisition: W. Li and S. Wang; Writing-original draft: H. Wang and C. Song; Writing-review & editing: W. Li and S. Wang.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Use and Care Committee of Capital Medical University (ethical code: AEEI-2018–216).
Consent for publication
Not applicable.
Competing interests
Author Songlin Wang and Wenbin Li are members of the Editorial Board for Current Medicine. The paper was handled by the other Editor and has undergone rigrous peer review process. And they were not involved in the journal's review of, or decisions related to, this manuscript.
The other authors declare that they have no competing interests.
Supplementary Information
Additional file 1:
Supplementary Figure 1. Oral administration of inorganic nitrate protects the kidney against cisplatin-induced AKI mice. The protein levels of KIM and β-actin (loading control) in renal cortical tissues of mice were evaluated by western blotting. Supplementary Figure 2. Dietary nitrate inhibits DNA damage. (A) The protein levels of p-H2AX and β-actin (loading control) in renal cortical tissues of mice at different time points were evaluated by western blotting. (B) Quantification of p-H2AX (3 mice in each group). Supplementary Figure 3. Dietary nitrate promotes autophagy in cisplatin-induced injury and autophagy plays a protective effect on cisplatin-induced cell damage. (A-C) Whole HK2 cell lysates were collected for immunoblot analysis of LC3 II, p62 and β-actin (loading control). (A) Representative blots. Three independent experiments to quantify LC3 II/LC3 I (B) and p62 (C). (D, E) HK2 cells were treated with cisplatin in the presence or absence of hydroxychloroquine (HCQ, 25 μmol/L) or Rapamycin (Rapa, 50 nmol/L), and cell viability was determined by CCK8. Data are shown as the means ± SEM and analyzed by one-way ANOVA with Tukey’s test. * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Ctrl; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. Cisp; (Ctrl, control; NaNO3, Nitrate; Cisp, cisplatin; Cisp + NaNO3, cisplatin + Nitrate).
Additional file 2:
Supplementary Table 1. The antibodies for western blotting, IF and IHC.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Song, C., Chen, F. et al. Nitrate attenuates cisplatin-induced acute kidney injury by promotion of mitophagy and reduction of oxidative stress. Curr Med 2, 8 (2023). https://doi.org/10.1007/s44194-023-00024-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44194-023-00024-3