
Abstract
Background:
Selective platelet release of pro- or anti-angiogenic factors distinctly regulated angiogenesis. We hypothesised that selective release of platelet angiogenic factors could differently regulate tumour growth.
Methods:
Breast cancer cell proliferation, cancer cell-induced endothelial tube formation in vitro, and tumour growth in vivo were studied in the presence of protease-activated receptor 1-stimulated platelet releasate (PAR1-PR; rich in pro-angiogenic factors) or PAR4-PR (rich in anti-angiogenic factors).
Results:
The PAR1-PR and PAR4-PR supplementation (10%) similarly enhanced cell proliferation of MCF-7 and MDA-MB-231 breast cancer cells. The cancer cells triggered capillary-like tube formation of endothelial cells that was further enhanced by pro-angiogenic factor-rich PAR1-PR. The VEGF, but not SDF-1α, receptor blockade abolished PAR1-PR/PAR4-PR-enhanced cancer cell proliferation. Integrin blockade by RGDS had identical effects as VEGF inhibition. The Src and ERK inhibition diminished, whereas PI3K and PKC blockade abolished platelet releasate-enhanced cancer cell proliferation. Using a model of subcutaneous implantation of MDA-MB-231 cells in nude mice, PAR1-PR enhanced tumour growth more markedly than PAR4-PR, and seemed to achieve the exaggeration by promoting more profound tumour angiogenesis.
Conclusions:
Platelet releasate increases breast cancer cell proliferation through VEGF–integrin cooperative signalling. Pro-angiogenic factor-rich platelet releasate enhances cancer cell-induced angiogenesis more markedly, and thus exaggerates tumour growth in vivo.
Similar content being viewed by others


Main
Platelets are the principal cells in thrombosis and haemostasis. Platelets are, however, versatile cells that play important roles in many other physiological and pathophysiological processes, such as inflammation, immunity, angiogenesis, wound healing, and cancer progression (Gay and Felding-Habermann, 2011; Li, 2016). It has been well recognised that platelet dysfunction and thrombotic disorders are important manifestations of cancer progression (Gay and Felding-Habermann, 2011; Lal et al, 2013; Li, 2016). Thus, cancer patients have a higher risk of platelet hyperactivities (Blann et al, 2001) and thrombotic events (Sorensen et al, 1998; Schulman and Lindmarker, 2000). Cancer patients diagnosed together with a thrombotic event or thrombocytosis have a poor prognosis of survival (Sorensen et al, 2000; Digklia and Voutsadakis, 2014; Bottsford-Miller et al, 2015).
There are complex cross-talks between platelets and cancer cells (Gay and Felding-Habermann, 2011; Li, 2016). Cancer cells can induce platelet activation by releasing various platelet-activating mediators, such as adenosine diphosphate (ADP) (Egan et al, 2011) and thromboxane A2 (Steinert et al, 1993), and/or by direct cell-to-cell contact (Lowe et al, 2012). Hence, different cancer cells may activate platelets via different mechanisms (Gay and Felding-Habermann, 2011; Lal et al, 2013; Li, 2016). Moreover, cancer cells may secrete cytokines, including angiogenic regulators, that can be uptaken by platelets (Klement et al, 2009; Kuznetsov et al, 2012). By doing so, cancer cells create a tumour-supportive microenvironment, and cancer-‘educated’ platelets can then promote cancer growth and metastasis (Kuznetsov et al, 2012). Indeed, platelets regulate diverse aspects of cancer progression and/or cancer cell activities. Platelets release a number of growth factors that promote cancer cell proliferation (e.g., by platelet-derived growth factor (PDGF)) (Sabrkhany et al, 2011) and epithelial–mesenchymal transition (EMT) (by transforming growth factor-1β (TGF-1β)) (Labelle et al, 2011). Platelet adhesion/conjugation on cancer cells can also serve the latter as an immune surveillance-protective ‘cloak’ (Nieswandt et al, 1999) and facilitates cancer cell arrest at the distal vasculature (Gay and Felding-Habermann, 2011; Li, 2016). Moreover, platelets have a rich storage of angiogenic regulators that can not only promote angiogenesis but also prevent haemorrhage in the tumour (Ho-Tin-Noe et al, 2008). Others and we have recently shown that different stimuli induced selective release of pro-angiogenic or anti-angiogenic regulators from platelet α-granules (Ma et al, 2005; Italiano et al, 2008; Chatterjee et al, 2011), and that selective platelet release of pro-angiogenic regulators promoted angiogenesis more markedly (Battinelli et al, 2011; Huang et al, 2015). All these platelet–cancer cell interactions may work in concert to promote cancer growth and metastasis.
Human platelets express protease activated receptor-1 (PAR1) and PAR4 that are best known as thrombin receptors. Interestingly, platelet PAR1 and PAR4 stimulation evoke selective releases of pro-angiogeneic and anti-angiogenic regulators (Ma et al, 2005; Italiano et al, 2008; Chatterjee et al, 2011), respectively. Hence, there is a need to investigate how selective releases of platelet angiogenic regulators influence cancer-related angiogenesis and tumour growth. In the present work, we found that PAR1-stimulated platelet releasates (PAR1-PR) and PAR4-stimulated platelet releasates (PAR4-PR) promoted the proliferation of both MCF-7 and MDA-MB-231 breast cancer cells. We also showed that platelet releasates and breast cancer cells collaborated in promoting capillary-like tube formation of endothelial cells. Importantly, we found that PAR1-PR enhanced tumour growth of implanted MDA-MB-231 cells more markedly than PAR4-PR via promoting more intense angiogenesis in the tumour.
Materials and methods
Study subjects and ethical considerations
Sixteen healthy subjects (9 males and 7 females, aged 28–59 years) gave informed consent to participate in the study. The study was approved by the ethics committee of the Karolinska Institutet in accordance with the guidelines of the Declaration of Helsinki.
Reagents
Thrombin receptor PAR1-activating peptide (PAR1-AP; H-Ser-Phe-Leu-Leu-Arg-Asn-Pro-Asn-Asp-Lys-Tyr-Glu-Pro-Phe-OH), the mitogen-activated protein kinase (MAPK) inhibitor U0126, and the p38-MAPK inhibitor SB203580 were from Calbiochem (Darmstadt, Germany). PAR4-AP (Ala-Tyr-Pro-Gly-Lys-Phe-NH2), prostaglandin (PG) E1, aspirin, apyrase grade VII, and cell detach solution (0.01% trypsin/5 mM EDTA) were all purchased from Sigma (St Louis, MO, USA). Recombinant hirudin was from Ciba Geigy (Base, Switzerland). Endothelial culture media (EGM-2 basic medium and the SingleQuots kit) and fetal bovine serum (FBS) were purchased from Lonza (Basel, Switzerland). The VEGFR2 inhibitor Ki8751, the stromal cell-derived factor (SDF)-1α receptor CXCR4 inhibitor AMD3100, and RGDS peptide were all from Tocris Bioscience (Bristol, UK). The cell counting kit-8 was from Dojindo (Munich, Germany). The Src kinase inhibitor PP2 and the PI3K inhibitor LY294002 were purchased from Merck Millipore (Darmstadt, Germany). The pan PKC inhibitor Ro 318220 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Culture of HUVECs and cancer cells
Human umbilical vein endothelial cells (HUVECs) were from Lonza. The HUVECs were cultured using the EGM-2 SingleQuots complete medium containing 10% FBS and the SingleQuots Kit. The HUVECs were cultured with fibronectin-coated 24-well culture plates in an incubator at 37 °C with 5% CO2. Human breast cancer cell lines MDA-MB-231 and MCF-7 were from ATCC (Wesel, Germany), and were cultured with RPMI-1640 containing 10% FBS and 1% penicillin–streptomycin at 37 °C with 5% CO2. Both cell lines were authenticated through polymorphic short tandem repeat testing within the past 2 years at Uppsala Genome Center (Uppsala, Sweden), and the cells between passages 3 and 10 were used in the present study.
Cell proliferation assay
The MCF-7 and MDA-MB-231 cells (1.5 × 103 cells in 100 μl per well) were cultured in a 96-well plate in triplicate with complete medium (RPMI-1640 containing 10% FBS). After 24 h of culture, the medium was replaced by serum-free basic medium without or with platelet releasates. Cell proliferation was determined using a cell counting kit (CCK)-8 assay after 0, 24, 48, 72, and 96 h of incubation, and the absorbance was measured at 450 nm using a microplate reader.
Flow cytometric analysis
Cell proliferation was analysed by labelling cells with carboxyfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen, Carlsbad, CA, USA) as described previously (Zhu et al, 2014). Briefly, cells were incubated with 1 μM CFSE for 10 min at 37 °C in the dark, washed, and then cultured for 96 h. Cell proliferation was examined at 96 h. For cell apoptosis analyses, the cells were washed with PBS, and stained with Annexin V and propidium iodide (PI) using a commercial kit (V13241; Invitrogen). Flow cytometric analyses were performed using an FC500 flow cytometer (Beckman Coulter, Hialeah, FL, USA).
Preparation of platelet releasates
Venous blood was collected using 9 ml vacutainers containing 1 : 10 volume of 3.8% sodium citrate and 100 μ M aspirin. Within 3 min of blood collection, blood samples were spun at 190 g for 20 min at room temperature. Upper two-thirds of platelet-rich plasma (PRP) were collected and further centrifuged at 900 g for 10 min in the presence of PGE1 (1 μ M). The pellet was resuspended in Tyrode’s Hepes buffer containing 1 μ M PGE1, centrifuged again, and then resuspended in Tyrode’s Hepes buffer containing 20 μg ml−1 hirudin and 2 U ml−1 apyrase at the concentration of 2 × 109 platelets per ml. The use of aspirin, hirudin, and apyrase was to block the positive feedback of platelet activation upon PAR1 or PAR4 receptor stimulation.
After the recovery of cell reactivity (30 min, 22 °C), platelets were stimulated with PAR1-AP (10 μ M) or PAR4-AP (100 μ M) for 10 min at 37 °C. Afterwards, the samples were centrifuged at 15 000 g for 10 min at 4 °C, and the supernatant was aliquoted and stored at −80 °C. The platelet releasates prepared with the present conditions have been documented by us previously for a selective pro-angiogenic regulator release by PAR1-AP stimulation (e.g., VEGF 50.2±0.9 11.6±1.3 pg ml−1 with PAR4-AP) and a selective anti-angiogenic regulator release by PAR4-AP stimulation (PF4 10.5±2.6 vs 5.6±0.9 μg ml−1 by PAR1-AP) (Chatterjee et al, 2011).
In vitro tube formation on Matrigel-coated plates
Tube formation assay was performed using 96-well plates coated with Matrigel (Corning Inc., Tewksbury, MA, USA). Matrigel was thawed at 4 °C overnight. The solution was added into the wells of a 96-well plate (50 μl per well), and incubated at 37 °C for 1 h to allow matrix gel solidification.
The HUVECs were resuspended in EBM-2 basic medium at 105 per ml. Cell suspensions of MDA-MB-231 and MCF-7 cancer cells (106 per ml for both) were also prepared using EBM-2 basic medium. Afterwards, 104 HUVECs (100 μl cell suspension), 104 cancer cells (10 μl MDA-MB-231 or MCF-7 cell suspension), or/and 10% platelet releasates were added into each matrix gel-coated well and incubated at 37 °C for 6 h. Capillary-like tubular structures were observed using an Olympus CKX41 inverted light microscope (Olympus Life Science, Tokyo, Japan) equipped with a Nikon D5100 camera (Tokyo, Japan). The branch points of capillary-like structure were assessed using the Wimasis software (Munich, Germany).
In vivo experiments of tumour growth
The protocol of in vivo tumour growth using a murine model of Matrigel implantation was approved by the institutional animal care and use committee of the Second Hospital of Shandong University. The study was performed in accordance to the national guidelines of China on ‘Laboratory animals-requirements of environment and housing facilities’ (GB 14925-2001) and to the European Directive 2010/63/EU on the protection of animals used for scientific purposes. Twenty-five female nude mice (8–12 weeks) were obtained from the Model Animal Research Center of Nanjing University (Nanjing, China).
Matrigel implantation cocktails (final volume at 250 μl per implant site) were composed immediately before use. The cocktails contained no cells or 106 MDA-MB-231 breast cancer cells. The cocktails were also supplemented with 10% (final concentration) of either vehicle (Tyrode’s Hepes buffer containing 20 μg ml−1 hirudin and 2 U ml−1 apyrase without or with 1 μ M PAR1-AP or 10 μ M PAR4-AP), PAR1-PR, or PAR4-PR. Two pairs of Matrigel implantations were made by subcutaneous injection, and were symmetrically implanted on the front or back parts of the flanks of each mouse. Analyses of the results were restricted to symmetrical, that is, within pair, comparisons. The tumours were allowed to grow for 4 weeks before the mice were killed and subjected to surgical removal of the tumours. Tumours/Matrigel implants were immediately photographed and weighed. Afterwards, the tumours were divided for further analyses of tumour histology and tumour haemoglobin content.
Tumour histology
One part of each tumour was fixed with 10% neutralised formalin overnight, embedded in paraffin, and sectioned at the thickness of 4 μm. For histological analysis, the sections were subjected to haematoxylin and eosin (HE) staining. The sections were observed, and the images were recorded using a Nikon Eclipse 90i microscope (Nikon Corporation, Tokyo, Japan).
Measurement of haemoglobin in tumour plugs
A portion of each tumour was rinsed with saline, placed on dry ice, and air dried for ⩾12 h in a hood. Dehydration of tumour plugs was employed as a haemoglobin concentrating procedure that enabled us to analyse the plugs with minor angiogenesis. Each dried tumour plug was weighed, and then lysed in 1 ml 0.1% Triton-100 for 2 h at room temperature. Afterwards, the lysates were thoroughly mixed by repeated pipetting, and then centrifuged at 15 000 g for 15 min. The supernatants were collected and stored at 4 °C. Upon measurements, 150 μl supernatant was mixed with 2500 μl colourimetric reagent of haemoglobin analysis kit A071 (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and incubated in a 37 °C water bath for 20 min. When the colourimetric reaction was completed, absorbance of the samples was measured at 510 nm using a Multiskan MK3 microplate spectrophotometer (Thermo Scientific, Waltham, MA, USA). The results are reported as angiogenesis index that is the product of tumour plug weight and haemoglobin content in mg per g of dried tumour plug. Hence, the angiogenesis index reflects both tumour size and blood content/angiogenesis in the tumour plugs.
Statistics
Data are presented as means±s.e.m. Comparisons between treatments were analysed by one-way ANOVA followed by Newman–Keuls test or two-way ANOVA where appropriate using GraphPad 5 (GraphPad Software, San Diego, CA, USA). P<0.05 was deemed statistically significant.
Results
Platelet releasates enhanced the proliferation of breast cancer cells under a serum-free condition
We and others have previously demonstrated that PAR1 and PAR4 stimuli evoke distinct pro- and anti-angiogenic factor release of platelets (Chatterjee et al, 2011), and that PAR1-PR and PAR4-PR regulate angiogenic activities of endothelial cells with different potentials (Huang et al, 2015). To determine whether distinct platelet release of angiogenic factors also differently regulates the behaviours of cancer cells, we first investigated the effects of PAR1-PR and PAR4-PR on breast cancer cell proliferation. The MCF-7 and MDA-MB-231 cells were incubated with supernatant from resting platelets (control), PAR1-PR, or PAR4-PR for 96 h under a serum-free condition, and cell proliferation was monitored using the colourimetric assay CCK-8. We found that supplementation of PAR1-PR and PAR4-PR, but not PAR1-AP or PAR4-AP, significantly increased the proliferation of both MCF-7 (Figure 1A, upper panel) and MDA-MB-231 cells (Figure 1A, lower panel), and that the enhancements led to faster and more intensive cell proliferation. To further characterise this effect, we examined cell division using CFSE staining. As shown in Figure 1B, CFSE fluorescence signals of both MCF-7 and MDA-MB-231 cells were dramatically reduced after 96 h of culture, indicating marked cancer cell proliferation. In the presence of PAR1-PR and PAR4-PR, the CFSE signals of both cell types were further reduced in a similar manner, and the percentages of CFSE-dim cells at 96 h were significantly higher (Figure 1B and C), suggesting that PAR1-PR and PAR4-PR accelerated cell division of MCF-7 and MDA-MB-231 cells similarly. However, supplementation of cultured MCF-7 cells with control vehicle, PAR1-PR, or PAR4-PR did not alter Annexin V+ cells (Figure 1D, upper panels), with the percentages of Annexin V+ MCF-7 at 5.1±1.6%, 5.6±1.5%, and 5.9±1.6%, respectively. The same was also true for MDA-MB-231 cells (Figure 1D, lower panels). The results suggested that either PAR1-PR or PAR4-PR had no influence on apoptosis of these breast cancer cells during 72 h of cell culture.

Platelet releasates enhance cell proliferation without affecting apoptosis of breast cancer cells. Breast cancer cell lines MCF-7 and MDA-MB-231 were treated with or without 10% (final concentration) of PAR1-PR or PAR4-PR under a serum-free culture condition. (A) Cell proliferation rate was detected at indicated time points using a CCK assay. The fold increases of OD values, proportional to the increase of cell numbers, were referred to those at time 0. Data are presented as mean±s.e.m. from at least five independent experiments. #P<0.05, †P<0.001 vs control at corresponding time points. (B) CFSE fluorescence intensities of breast cancer cells were determined by flow cytometry after MCF-7 and MDA-MB-231 cells were cultured without (control; black solid lines) or with PAR1-PR (blue dash lines) or PAR4-PR (red dot lines) for 96 h. (C) Quantification of CFSE dilution. #P<0.05 vs control. (D) The MCF-7 and MDA-MB-231 cells were stained with FITC-conjugated Annexin V and PE-conjugated PI after 72 h of culture, and were examined using the Beckman Coulter F500 flow cytometer. Percentages of total apoptotic cells, that is, all Annexin-V-positive cells, were plotted. Images in (B and D) are the representatives from three independent experiments. A full colour version of this figure is available at the British Journal of Cancer journal online.
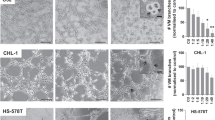
Platelet releasates promoted cancer cell-induced endothelial cell tube formation
Angiogenesis is critical for cancer growth and metastasis. Effects of platelet releasates on breast cancer cell-regulated angiogenic activities of cultured endothelial cells were thus studied. The HUVECs incubated on Matrigel over 6 h had limited capillary-like tube formation (Figure 2A, up-left panel). Supplementation of platelet releasates enhanced endothelial tube formation, and in line with our previous studies (Huang et al, 2015), PAR1-PR promoted capillary-like tube formation of HUVECs more potently than PAR4-PR (Figure 2A and B). Interestingly, co-incubation of HUVECs with breast cancer cells also facilitated tube formation. The enhancement was much more marked with MDA-MB-231 cells than with MCF-7 cells, probably indicating a more aggressive nature of MDA-MB-231 cells. Moreover, the combination of platelet releasate with either MDA-MB-231 or MCF-7 cells further promoted endothelial tube formation (Figure 2A). When endothelial angiogenic activities were quantified and assessed as branch points of capillary network, it was found that MDA-MB-231 cells increased the branch points of HUVEC capillary-like network from 4.8±1.3 of HUVECs alone to 66.8±20.5, and that supplementation of PAR1-PR further increased the branch points to 90.4±21.3 (P<0.05, as compared with MDA-MB-231 cell co-incubation only; Figure 2B). Supplementation of PAR4-PR only showed a weak trend to enhance MDA-MB-231 cell-induced tube formation, with a slightly higher branch points at 75.0±17.7. Similar collaborative enhancements on endothelial tube formation were also seen with MCF-7 cells and platelet releasates, although the enhancements were less potent as compared with those with MDA-MB-231 cells (Figure 2B). Together, these results indicate that platelet releasates can enhance angiogenic potentials of breast cancer cells.

Platelet releasates and cancer cells collaborate to potentiate the tube formation of endothelial cells. Endothelial cells (ECs) were supplemented with vehicle (EC only), PAR1-PR (10%), PAR4-PR (10%), MCF-7 cells, or/and MDA-MB-231 cells at the EC/cancer cell ratio of 1 : 1. The mixtures (100 μl) were added into Matrigel-coated wells and incubated at 37 °C for 6 h. (A) The EC tube formation was photographed using an inverted light microscope. (B) The branch point numbers of capillary-like structures were assessed using the Wimasis software. Data are presented as mean±s.e.m. from five independent experiments. *P<0.001, #P<0.05 vs EC only; φP<0.05 vs EC+PAR4-PR; †P<0.05 vs EC+MCF-7; ‡P<0.05 vs EC+MDA-MB-231. A full colour version of this figure is available at the British Journal of Cancer journal online.
VEGFR-2 and integrin mediated platelet releasate-enhanced breast cancer cell proliferation
To determine the mechanism by which platelet releasates increased the proliferation of breast cancer cells, we investigated the possible roles of multiple platelet-derived angiogenic factors. In the absence of platelet releasates, the proliferation of MCF-7 (Figure 3A) or MDA-MB-231 cells (Figure 3B) was not affected by the VEGFR-2 inhibitor Ki8751, the CXCR4 inhibitor ADM3100, or the integrin blocking peptide RGDS. In the presence of either PAR1-PR or PAR4-PR, the proliferation of MCF-7 and MDA-MB-231 cells was markedly increased. The enhancements were totally inhibited by VEGFR-2 inhibition. Interestingly, integrin blockage by the pan integrin inhibitor RGDS peptide also abolished PAR1-PR- and PAR4-PR-enhanced proliferation in MCF-7 and MDA-MB-231 cells. However, CXCR4 blockade had no effects on platelet releasate-dependent breast cancer cell proliferation. Thus, these data demonstrated that the proliferation-enhancing effects of PAR1-PR and PAR4-PR on MCF-7 and MDA-MB-231 cells were mediated by VEGFR-2 and integrins.

Blockade of vascular endothelial growth factor receptor (VEGFR) and integrin inhibits platelet releasate-enhanced breast cancer cell proliferation. The breast cancer cell line MCF-7 (A) and MDA-MB-231 cells (B) were treated without (control) or with 10% (final concentration) PAR1-PR or PAR4-PR in the absence or presence of the VEGFR-2 inhibitor Ki8751 (0.5 μ M), the CXCR4 inhibitor AMD3100 (10 μ M), or the integrin blocking peptide RGDS (10 μg ml−1). After 96 h of incubation, cell proliferation was assessed using a CCK assay. The percentage changes of cell proliferation were referred to the cells cultured with vehicle and without platelet releasate. Data are presented as mean±s.e.m. from four independent experiments. #P<0.05, *P<0.01, and †P<0.001 compared with corresponding treatments without inhibitors.
Platelet releasates promoted breast cancer cell proliferation via PI3K/PKC pathway
To clarify signalling pathways downstream of platelet releasate-enhanced proliferation of breast cancer cells, several kinase-specific small molecular inhibitors, including the PI3K inhibitor LY294002, the ERK inhibitor U0126, the p38 inhibitor SB203580, the PKC inhibitor Ro318220, and the Src kinase inhibitor PP2, were employed during 96 h cultures of breast cancer cells. As expected, supplementation of platelet releasates markedly increased breast cancer cell proliferation, and the stimulatory effects of PAR1-PR and PAR4-PR on either MCF-7 or MDA-MB-231 cells were similar. Among MCF-7 cells (Figure 4A), PI3K and PKC blockade abolished platelet releasate-dependent enhancements, and reduced cell proliferation to the same levels of MCF-7 cells cultured without platelet releasates, indicating a critical role of PI3K and PKC signalling in cell proliferation. The Src and ERK inhibition also diminished PAR1-PR- and PAR4-PR-enhanced breast cancer proliferation, whereas MAPK p38 intervention had little impact. Among MDA-MB-231 cells (Figure 4B), more clearcut inhibitions were seen, and all the inhibitors diminished PAR1-PR- and PAR4-PR-dependent enhancements of MDA-MB-231 cell proliferation.

Platelet releasates promote breast cancer cell proliferation through PI3K/PKC signalling. Breast cancer cell lines MCF-7 (A) and MDA-MB-231 (B) were treated with or without 10% (final concentration) of PAR1-PR or PAR4-PR in the absence or presence of the Src kinase inhibitor PP2 (10 μ M), the PI3K inhibitor LY294002 (25 μ M), the PKC inhibitor Ro318220 (10 μ M), the ERK inhibitor U0126 (10 μ M), or the p38 inhibitor SB203580 (10 μ M). After 96 h of incubation, cell proliferation was detected using a CCK assay. Impacts of platelet releasates and signalling interventions on breast cancer cell proliferation were assessed as the percentage changes compared with the cells cultured without platelet releasate and treated with vehicle. Data are presented as mean±s.e.m. from four independent experiments. #P<0.05, *P<0.01, and †P<0.001 compared with corresponding treatments without inhibitors.
Impact of platelet releasates on MDA-MB-231 tumour growth in vivo
The presence of PAR1-PR or PAR4-PR significantly enhanced tumour growth of subcutaneously implanted MDA-MB-231 cells in nude mice. This was evidenced by our observation showing that supplementation of either PAR1-PR or PAR4-PR markedly increased MDA-MB-231 tumour growth ratios as compared with MDA-MB-231 cancer cell implants with vehicle (Figure 5A). Moreover, the tumour growth ratio of MDA-MB-231 tumour with PAR1-PR was higher than that with PAR4-PR. Light microscopic observation of histological sections (Figure 5B–D) showed that there was significant breast cancer cell proliferation in the tumour plugs, and that cancer cells grew into lobular cell masses. In the tumour implanted with vehicle (Figure 5B), there were considerable amounts of noncancer cell tissues between lobular cancer cell mass, and the blood vessels were sparse. In contrast, higher densities of breast cancer cells and blood vessels were observed in the tumours that were implanted with PAR1-PR (Figure 5C) and PAR4-PR (Figure 5D).

Platelet releasates enhance tumour growth in vivo. MDA-MB-231 breast cancer cells in Matrigel (106 cells in 250 μl) were subcutaneously implanted without or with 10% (final concentration) PAR1-PR or PAR4-PR in female nude mice. After 4 weeks, tumours were surgically removed, weighted, and sectioned for HE staining. (A) Tumour growth ratios were calculated using the following formula: tumour growth ratio=(tumour weight with vehicle (or PAR1-PR or PAR4-PR))/tumour weight with vehicle. Mean±s.e.m. from eight tumour pairs are plotted. *P<0.05 vs vehicle, †P<0.05 vs PAR1-PR. Representative images of MDA-MB-231 cell tumour sections with vehicle (B), PAR1-PR (C), and PAR4-PR (D) are shown. Black asterisks indicate cancer tissues, and white arrows indicate vessels in the tumour plugs. A full colour version of this figure is available at the British Journal of Cancer journal online.
Five pairs of contralateral implanted MDA-MB-231 tumour with PAR1-PR or PAR4-PR are shown in Figure 6A. It can be clearly seen that most of the tumours implanted with PAR1-PR were larger (mean tumour weight 0.44±0.07 g) than the ones with PAR4-PR (0.31±0.06 g; P<0.05). The only exception, the upper PAR4-PR plug, appeared to be larger, but was actually elongated/thin and not heavier than the countering PAR1-PR plug. Using tumour weight of PAR1-PR-containing tumour as the standard, the tumour growth ratio of PAR1-PR-containing tumour was significantly higher than that of PAR4-PR-containing tumour (Figure 6B). Furthermore, when haemoglobin contents in the tumours were measured and the angiogenesis indexes were calculated, the index of PAR1-PR-containing tumours was significantly higher than that of PAR4-PR-containing tumours (Figure 6C), indicating more profound angiogenesis in the presence of PAR1-PR.

The PAR1-PR enhances tumour growth more potently than PAR4-PR. The MDA-MB-231 breast cancer cells in Matrigel (106 cells in 250 μl, containing 10% PAR1-PR or PAR4-PR) were subcutaneously implanted in female nude mice. After 4 weeks, tumours were removed, weighted, and photographed. A portion of each tumour plug was used for measurement of haemoglobin content. (A) Five pairs of symmetrically implanted tumour plugs with PAR1-PR or PAR4-PR are shown. (B) Tumour growth ratios were calculated using the following formula: tumour growth ratio=(tumour weight with PAR1-PR (or PAR4-PR))/tumour weight with PAR1-PR. Mean±s.e.m. from five tumour pairs are plotted. *P<0.05 vs PAR1-PR. (C) Mean±s.e.m. of angiogenesis index of five tumour pairs are plotted. *P<0.05 vs PAR1-PR. A full colour version of this figure is available at the British Journal of Cancer journal online.
Discussion
The present work demonstrated that platelet releasates promoted the proliferation of the breast cancer cell lines MCF-7 and MDA-MB-231 through VEGF–integrin cooperative signalling. Platelet releasates and breast cancer cells each enhanced capillary-like tube formation of HUVECs, and the combination of platelet releasates and breast cancer cells further enhanced the tube formation. Moreover, selective release of platelet pro-angiogenic regulators exaggerated breast cancer growth in vivo, and the distinct effect was associated with intensified angiogenesis in the tumours.
A series of recent reports from several laboratories indicate that platelets store pro-angiogenic and anti-angiogenic regulators in separate α-granules, and that platelets selectively release pro- or anti-angiogenic regulators upon different stimuli (Ma et al, 2005; Italiano et al, 2008; Chatterjee et al, 2011). Although some concerns have also been raised that selective release of platelet angiogenic regulators may be activation intensity and/or secretion kinetics dependent (Jonnalagadda et al, 2012; Van Holten et al, 2014), selective release of counteracting angiogenic regulators remains a tempting intervention target for the development of a platelet-targeted cancer intervening approach. It has been reported that selective platelet release of pro-angiogenic regulators by ADP stimulation promoted whereas selective platelet release of anti-angiogenic regulators by TXA2 stimulation inhibited angiogenic responses of endothelial cells (Battinelli et al, 2011). We have, however, shown that both PAR1-PR and PAR4-PR enhanced angiogenic activities of endothelial progenitor cells, and that the enhancement by pro-angiogenic regulator-rich PAR1-PR was more potent (Huang et al, 2015). Supporting and expanding our earlier findings, the present work demonstrated that both PAR1-PR and PAR4-PR enhanced the proliferation of breast cancer cells in vitro, and that both of them promoted tumour growth in vivo of implanted MDA-MB-231 cells. Although there was no major difference between PAR1-PR- and PAR4-PR-enhanced breast cancer cell proliferation in vitro, PAR1-PR enhanced tumour growth in vivo more profoundly. The favourable enhancement of tumour growth by PAR1-PR was likely caused by increased angiogenesis. The latter was supported by the evidence showing that PAR1-PR, alone or in combination with cancer cells, promoted more profound endothelial cell tube formation in vitro, and that angiogenesis index of tumours implanted with PAR1-PR was higher than that with PAR4-PR. Hence, our data suggest that selective release of platelet anti-angiogenic regulators upon certain stimuli (e.g., by PAR4 stimulation) may not be used as a simple platelet-targeted approach for the intervention of tumour growth.
The VEGF is a potent inducer of angiogenesis that is required for a wide variety of physiological and pathological processes, including tumour progression. The present study showed that VEGFR-2 blockade abolished platelet releasate-enhanced breast cancer cell proliferation, implying that VEGF is the prominent mediator of the platelet releasates promoting MCF-7 and MDA-MB-231 cell proliferation. Our findings also support the notion that VEGF is not only a potent angiogenic regulator but also a versatile actor in cancer cell biology, for example, cancer cell survival and proliferation, as well as resistance to chemotherapy (Dias et al, 2002). Interestingly, our data showed that integrin inhibition by RGDS strongly diminished platelet releasate-promoted proliferation of breast cancer cells, and to the same inhibitory extent as by VEGFR-2 blockade. The observation highlighted the close and important collaboration between integrin and growth factor receptors (Moreno-Layseca and Streuli, 2014). It has previously been shown that VEGF directly bound to α9β1 integrin, and that VEGF-induced adhesion, migration, and angiogenesis of endothelial cells depended on cooperative signal inputs of α9β1 integrin and VEGFR-2 (Vlahakis et al, 2007). Of note, both VEGFR-2 signalling and α9β1 outside-in signalling operated through PI3K–PKC–MAPK pathway (Moreno-Layseca and Streuli, 2014; Olsson et al, 2006). Indeed, the present study showed that either PI3K or PKC inhibition completely blocked PAR1-PR- and PAR4-PR-enhanced cancer cell proliferation. Hence, our findings suggest that VEGF from the platelet releasates is the key growth factor stimulating breast cancer cell proliferation via VEGFR2–integrin cooperative signalling through PI3K/PKC signalling axis.
In conclusion, platelet releasates promote cancer cell proliferation mainly through VEGF triggered VEGFR2–integrin cooperative signalling via PI3K/PKC signalling pathway. Platelet releasates and cancer cells also cooperate to promote angiogenic activities of endothelial cells. Pro-angiogenic factor-rich PAR1-PR promotes breast cancer growth more markedly than anti-angiogenic factor-rich PAR4-PR by promoting more intense angiogenesis in the tumour.
References
Battinelli EM, Markens BA, Italiano Jr JE (2011) Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood 118: 1359–1369.
Blann AD, Gurney D, Wadley M, Bareford D, Stonelake P, Lip GY (2001) Increased soluble P-selectin in patients with haematological and breast cancer: a comparison with fibrinogen, plasminogen activator inhibitor and von Willebrand factor. Blood Coagul Fibrinolysis 12: 43–50.
Bottsford-Miller J, Choi HJ, Dalton HJ, Stone RL, Cho MS, Haemmerle M, Nick AM, Pradeep S, Zand B, Previs RA, Pecot CV, Crane EK, Hu W, Lutgendorf SK, Afshar-Kharghan V, Sood AK (2015) Differential platelet levels affect response to taxane-based therapy in ovarian cancer. Clin Cancer Res 21: 602–610.
Chatterjee M, Huang Z, Zhang W, Jiang L, Hultenby K, Zhu L, Hu H, Nilsson GP, Li N (2011) Distinct platelet release and surface expression of proangiogenicand antiangiogenic factors upon different platelet stimuli. Blood 117: 3907–3911.
Dias S, Choy M, Alitalo K, Rafii S (2002) Vascular endothelial growth factor (VEGF)-C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood 99: 2179–2184.
Digklia A, Voutsadakis IA (2014) Thrombocytosis as a prognostic marker in stage III and IV serous ovarian cancer. Obstet Gynecol Sci 57: 457–463.
Egan K, Crowley D, Smyth P, O'toole S, Spillane C, Martin C, Gallagher M, Canney A, Norris L, Conlon N, Mcevoy L, Ffrench B, Stordal B, Keegan H, Finn S, Mceneaney V, Laios A, Ducree J, Dunne E, Smith L, Berndt M, Sheils O, Kenny D, O'leary J (2011) Platelet adhesion and degranulation induce pro-survival and pro-angiogenic signalling in ovarian cancer cells. PLoS One 6: e26125.
Gay LJ, Felding-Habermann B (2011) Contribution of platelets to tumour metastasis. Nat Rev Cancer 11: 123–134.
Ho-Tin-Noe B, Goerge T, Cifuni SM, Duerschmied D, Wagner DD (2008) Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res 68: 6851–6858.
Huang Z, Miao X, Luan Y, Zhu L, Kong F, Lu Q, Pernow J, Nilsson G, Li N (2015) PAR1-stimulated platelet releasate promotes angiogenic activities of endothelial progenitor cells more potently than PAR4-stimulated platelet releasate. J Thromb Haemost 13: 465–476.
Italiano JE Jr, Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, Ryeom S, Folkman J, Klement GL (2008) Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 111: 1227–1233.
Jonnalagadda D, Izu LT, Whiteheart SW (2012) Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood 120: 5209–5216.
Klement GL, Yip TT, Cassiola F, Kikuchi L, Cervi D, Podust V, Italiano JE, Wheatley E, Abou-Slaybi A, Bender E, Almog N, Kieran MW, Folkman J (2009) Platelets actively sequester angiogenesis regulators. Blood 113: 2835–2842.
Kuznetsov HS, Marsh T, Markens BA, Castano Z, Greene-Colozzi A, Hay SA, Brown VE, Richardson AL, Signoretti S, Battinelli EM, Mcallister SS (2012) Identification of luminal breast cancers that establish a tumor-supportive macroenvironment defined by proangiogenic platelets and bone marrow-derived cells. Cancer Discov 2: 1150–1165.
Labelle M, Begum S, Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20: 576–590.
Lal I, Dittus K, Holmes CE (2013) Platelets, coagulation and fibrinolysis in breast cancer progression. Breast Cancer Res 15: 207.
Li N (2016) Platelets in cancer metastasis: to help the ‘villain’ to do evil. Int J Cancer 138: 2078–2087.
Lowe KL, Navarro-Nunez L, Watson SP (2012) Platelet CLEC-2 and podoplanin in cancer metastasis. Thromb Res 129 (Suppl 1): S30–S37.
Ma L, Perini R, Mcknight W, Dicay M, Klein A, Hollenberg MD, Wallace JL (2005) Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc Natl Acad Sci USA 102: 216–220.
Moreno-Layseca P, Streuli CH (2014) Signalling pathways linking integrins with cell cycle progression. Matrix Biol 34: 144–153.
Nieswandt B, Hafner M, Echtenacher B, Mannel DN (1999) Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res 59: 1295–1300.
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L (2006) VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol 7: 359–371.
Sabrkhany S, Griffioen AW, Oude Egbrink MG (2011) The role of blood platelets in tumor angiogenesis. Biochim Biophys Acta 1815: 189–196.
Schulman S, Lindmarker P (2000) Incidence of cancer after prophylaxis with warfarin against recurrent venous thromboembolism. Duration of Anticoagulation Trial. N Engl J Med 342: 1953–1958.
Sorensen HT, Mellemkjaer L, Olsen JH, Baron JA (2000) Prognosis of cancers associated with venous thromboembolism. N Engl J Med 343: 1846–1850.
Sorensen HT, Mellemkjaer L, Steffensen FH, Olsen JH, Nielsen GL (1998) The risk of a diagnosis of cancer after primary deep venous thrombosis or pulmonary embolism. N Engl J Med 338: 1169–1173.
Steinert BW, Tang DG, Grossi IM, Umbarger LA, Honn KV (1993) Studies on the role of platelet eicosanoid metabolism and integrin alpha IIb beta 3 in tumor-cell-induced platelet aggregation. Int J Cancer 54: 92–101.
Van Holten TC, Bleijerveld OB, Wijten P, De Groot PG, Heck AJ, Barendrecht AD, Merkx TH, Scholten A, Roest M (2014) Quantitative proteomics analysis reveals similar release profiles following specific PAR-1 or PAR-4 stimulation of platelets. Cardiovasc Res 103: 140–146.
Vlahakis NE, Young BA, Atakilit A, Hawkridge AE, Issaka RB, Boudreau N, Sheppard D (2007) Integrin alpha9beta1 directly binds to vascular endothelial growth factor (VEGF)-A and contributes to VEGF-A-induced angiogenesis. J Biol Chem 282: 15187–15196.
Zhu L, Huang Z, Stålesen R, Hansson GK, Li N (2014) Platelets provoke distinct dynamics of immune responses by differentially regulating CD4+ T cell proliferation. J Thromb Haemost 12: 1156–1165.
Acknowledgements
The present work was supported by the Swedish Heart-Lung Foundation, the Swedish Research Council, the Swedish Cancer Foundation, the National Natural Science Foundation of China (81400098), the KC Wong Magna Fund in Ningbo University, the China Scholarship Council, the Stockholm County Council, and Karolinska Institutet.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Jiang, L., Luan, Y., Miao, X. et al. Platelet releasate promotes breast cancer growth and angiogenesis via VEGF–integrin cooperative signalling. Br J Cancer 117, 695–703 (2017). https://doi.org/10.1038/bjc.2017.214
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.214
- Springer Nature Limited
Keywords
This article is cited by
-
Role and research progress of hematological markers in laryngeal squamous cell carcinoma
Diagnostic Pathology (2023)
-
The critical role of platelet in cancer progression and metastasis
European Journal of Medical Research (2023)
-
The role of tumor-platelet interplay and micro tumor thrombi during hematogenous tumor metastasis
Cellular Oncology (2023)
-
Predictive value of NLR and PLR for immune-related adverse events: a systematic review and meta-analysis
Clinical and Translational Oncology (2023)
-
Tissue factor: a neglected role in cancer biology
Journal of Thrombosis and Thrombolysis (2022)