
Abstract
Runt-related transcription factor 2 (RUNX2) has been considered to be one of master regulators for osteoblast differentiation and bone formation. Recently, we have described that RUNX2 attenuates p53/TAp73-dependent cell death of human osteosarcoma U2OS cells bearing wild-type p53 in response to adriamycin. In this study, we have asked whether RUNX2 silencing could enhance gemcitabine (GEM) sensitivity of p53-deficient human pancreatic cancer AsPC-1 cells. Under our experimental conditions, GEM treatment increased the expression level of p53 family TAp63, whereas RUNX2 was reduced following GEM exposure, indicating that there exists an inverse relationship between the expression level of TAp63 and RUNX2 following GEM exposure. To assess whether TAp63 could be involved in the regulation of GEM sensitivity of AsPC-1 cells, small interfering RNA-mediated knockdown of TAp63 was performed. As expected, silencing of TAp63 significantly prohibited GEM-dependent cell death as compared with GEM-treated non-silencing cells. As TAp63 was negatively regulated by RUNX2, we sought to examine whether RUNX2 knockdown could enhance the sensitivity to GEM. Expression analysis demonstrated that depletion of RUNX2 apparently stimulates the expression of TAp63, as well as proteolytic cleavage of poly ADP ribose polymerase (PARP) after GEM exposure, and further augmented GEM-mediated induction of p53/TAp63-target genes, such as p21WAF1, PUMA and NOXA, relative to GEM-treated control-transfected cells, implying that RUNX2 has a critical role in the regulation of GEM resistance through the downregulation of TAp63. Notably, ablation of TAp63 gave a decrease in number of γH2AX-positive cells in response to GEM relative to control-transfected cells following GEM exposure. Consistently, GEM-dependent phosphorylation of ataxia telangiectasia-mutated protein was remarkably impaired in TAp63 knockdown cells. Collectively, our present findings strongly suggest that RUNX2-mediated repression of TAp63 contributes at least in part to GEM resistance of AsPC-1 cells, and thus silencing of RUNX2 may be a novel strategy to enhance the efficacy of GEM in p53-deficient pancreatic cancer cells.
Similar content being viewed by others


Introduction
Human pancreatic cancer is a highly aggressive, as well as metastatic tumor, and represents the fourth and fifth leading causes of cancer-related death in the United States and Japan, respectively, whose incidence is increasing.1,2 Although the best chance of long-term survival is the complete surgical resection, most patients (over 80%) are not amenable to surgery at the time of diagnosis because of its difficulty in early detection.3 Unfortunately, even in patients who underwent surgery, a high rate of relapse with high local recurrence rates up to 50% is detectable, which leads to a 5-year survival rate of under 5%.4 Therefore, chemotherapy and/or radiotherapy is the only option.
For chemotherapy, the antimetabolite deoxycytidine nucleoside analog gemcitabine (GEM) is the current first line of standard treatment given to most patients with advanced pancreatic cancer.5,6 To achieve its anticancer activity, GEM requires phosphorylation within tumor cells to become an active form. The active one then accumulates within cancer cells and its incorporation into genome DNA correlates with its cytotoxicity.7 However, GEM treatment provides limited clinical benefits, especially in advanced and metastatic disease.8 Furthermore, a lot of clinical studies demonstrated that the combination of GEM with other cytotoxic drugs does not remarkably improve the poor prognosis of pancreatic cancer patients.9–12 Thus, a novel therapeutic option(s) based on biology of pancreatic cancer and also the understanding of the precise molecular mechanism(s) behind its resistance to GEM should be necessary to improve outcomes in patients with pancreatic cancer.
Mammalian RUNX family of transcription factors including runt-related transcription factor 1 (RUNX1), RUNX2 and RUNX3, is an evolutionarily conserved regulator of cell fate. Each of RUNX family members contains a highly conserved ‘runt’ domain, which recognizes and directly binds to the consensus sequences (TGTGGT or ACCACA), and mediates transcriptional activation or repression of their target genes.13,14 In spite of their structural similarities, RUNX family members have divergent physiological roles. It has been well established that RUNX1, RUNX2 and RUNX3 have a critical role in the regulation of hematopoiesis,15 bone formation16,17 and gastrointestinal/neuronal development,18,19 respectively. Meanwhile, all of these RUNX family members are also implicated in carcinogenesis. For example, RUNX1 is a frequent target of chromosomal translocations in hematopoietic malignancies,20 and the loss or reduction of RUNX3 expression can be detected in over 80% of gastric cancers.21,22 These observations strongly suggest that RUNX1, as well as RUNX3, acts as a putative tumor suppressor.
In a sharp contrast to RUNX1 and RUNX3, RUNX2 may have a pro-oncogenic potential. A growing body of evidence demonstrated that RUNX2 is aberrantly expressed in several human cancers including pancreatic,23 thyroid,24 breast,25,26 prostate,27 lung,28 colon,29 ovarian cancers30 and osteosarcoma.31,32 Consistent with these observations, it has been shown that RUNX2 has an ability to transactivate genes implicated in cancer cell migration and invasion.33–38 Indeed, Tandon et al.39 revealed that knockdown of RUNX2 in invasive breast cancer cells promotes cell death in response to glucose- and growth factor-deprivation. Similarly, Akech et al.40 described that depletion of RUNX2 in prostate cancer cells inhibits cell migration and invasion in vitro and RUNX2 expression in prostate cancer tissues is associated with metastasis. In addition, it has been found that there exists a positive correlation between RUNX2 gene amplification and poor chemo-response in osteosarcoma patients.32 Unfortunately, the precise molecular mechanism(s) how RUNX2 could contribute to the development and progression of the above-mentioned cancers remains elusive.
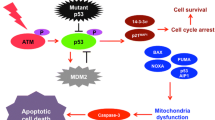
The representative tumor-suppressor p53 protects normal cells from onocogenic transformation by prohibiting undesirable propagation of damaged cells. As expected from its structural property, p53 acts as a nuclear transcription factor, which transactivates numerous of its target genes implicated in the induction of cell cycle arrest, cellular senescence and/or cell death following DNA damage.41 Accumulating evidence strongly suggests that p53-mediated cellular processes are tightly linked to its transcriptional activity. Although extensive mutation searches revealed that p53 is mutated in over 50% of human cancers. Among them, p53 mutation has been detectable in approximately 75% of pancreatic cancer.42 As most of p53 mutations are found within the genomic region encoding its DNA-binding domain, mutant forms of p53 lack sequence-specific transactivation ability and thereby act as dominant-negative inhibitors against wild-type p53.41,43 Unlike p53, p73 and p63, which belong to p53 family, are rarely mutated in human cancers.44 Owing to the alternative C-terminal splicing and promoter usage, p73 and p63 encode multiple isoforms such as transactivating isoforms (TAp73 and TAp63) and N-terminally truncated isoforms lacking transactivation domain (ΔNp73 and ΔNp63).45,46 As expected from their structural similarity to p53, TAp73 and TAp63 have a fundamental role in the regulation of DNA damage response.41
Recently, we have demonstrated for the first time that RUNX2 attenuates p53 and/or TAp73-dependent cell death in p53-proficient osteosarcoma U2OS cells following DNA damage inducer adriamycin (ADR) exposure.47,48 In this study, we have focused on p53-deficient pancreatic cancer AsPC-1 cells and found that depletion of RUNX2 enhances the sensitivity to GEM of AsPC-1 cells in association with a significant stimulation of TAp63-dependent cell death pathway.
Results
AsPC-1 cells are much more resistant to GEM than SW1990 cells
As described,49 human pancreatic cancer-derived AsPC-1 cells lacking p53 were resistant to GEM. Here, we compared the effects of GEM between AsPC-1 and human pancreatic cancer SW1990 cells carrying wild-type p53. Toward this end, AsPC-1 and SW1990 cells were treated with the indicated concentrations of GEM. Forty-eight hours after treatment, cells were observed under phase-contrast microscope. As shown in Supplementary Figure S1, number of attached cells was apparently decreased in GEM-treated SW1990 cells but not significantly in AsPC-1 cells following GEM exposure. Under these experimental conditions, floating and attached cells were harvested and their DNA content was analyzed by fluorescence-activate cell sorting (FACS). As seen in Figure 1a, number of cells with sub-G1 DNA content was smaller in AsPC-1 cells than SW1990 cells after GEM treatment. Consistent with these observations, Trypan blue exclusion assays demonstrated that number of Trypan blue-positive cells (dead cells) is much more smaller in AsPC-1 cells than SW1990 cells in response to GEM (Figure 1b). Thus, our present results indicate that AsPC-1 cells display GEM resistance as compared with SW1990 cells.

AsPC-1 cells are much more resistant to GEM than SW1990 cells. (a) FACS analysis. SW1990 (upper panels) and AsPC-1 (lower panels) cells were exposed to the indicated concentrations of GEM. Forty-eight hours after treatment, floating and attached cells were collected and their cell cycle distribution was analyzed by FACS. (b) Trypan blue exclusion assay. Cells were treated as in a. Twenty-four and 48 hours after treatment, floating and attached cells were combined, cell suspensions were mixed with equal volume of 0.4% Trypan blue solution, and number of Trypan blue-positive cells (dead cells) was scored.
Expression pattern of p53 family-related gene products in response to GEM
To shed light on the molecular mechanism(s) behind GEM-resistant phenotype of AsPC-1 cells, we have examined the expression pattern of p53 family-related gene products following GEM exposure. In these experiments, cleavage of PARP and accumulation of γH2AX were also checked as cell death and DNA damage markers, respectively. As shown in Figure 2a, GEM-dependent robust accumulation of γH2AX and cleavage of PARP were detectable in SW1990 cells, implying that DNA damage-mediated cell death takes place. As expected, GEM exposure resulted in an induction of p53, as well as phosphorylation of p53, at Ser-15 accompanied by a stimulation of a subset of p53 family-target gene products such as p21WAF1 and pro-apoptotic BAX, suggesting that GEM-mediated induction of cell death of SW1990 cells is regulated at least in part in a p53-dependent manner. In addition, the expression level of p53 family member TAp63 and RUNX2 was reduced and increased in response to GEM, respectively. As RUNX2 inhibited pro-apoptotic activity of p53 following DNA damage,47 it is possible that GEM-mediated upregulation of RUNX2 attenuates the inappropriate cell death caused by overactive p53.

Expression analysis of p53/TAp63-related gene products in response to GEM. SW1990 (a) and AsPC-1 (b) cells were exposed to the indicated concentrations of GEM. Forty-eight hours after treatment, whole-cell lysates were prepared and subjected to immunoblotting with the indicated antibodies. Actin was used as a loading control.
In contrast to SW1990 cells, GEM treatment led to an accumulation of γH2AX but not a promotion of PARP cleavage in AsPC-1 cells (Figure 2b). Instead of p53, GEM-dependent induction of TAp63 was observed in association with an upregulation of p21WAF1. Thus, it is likely that GEM-mediated induction of p21WAF1 may be due to TAp63. On the other hand, the expression level of BAX remained unchanged regardless of GEM treatment, whereas RUNX2 level declined in response to GEM, raising a possibility that there exists an inverse relationship between the expression level of TAp63 and RUNX2 following GEM exposure. We then focused on GEM-resistant AsPC-1 cells in our further experiments.
TAp63 is implicated in the regulation of GEM sensitivity in AsPC-1 cells
As the expression level of TAp63 was increased in AsPC-1 cells exposed to GEM, it is plausible that TAp63 may be involved in the regulation of their GEM sensitivity. To test this possibility, we sought to deplete the endogenous TAp63. AsPC-1 cells were transfected with control small interfering RNA (siRNA) or with siRNA against TAp63. These experiments were performed in the presence or in the absence of GEM. As shown in Figure 3 and Supplementary Figure S2A, FACS analysis and Trypan blue exclusion assay demonstrated that GEM sensitivity is markedly decreased in TAp63 knockdown cells relative to non-silencing cells. These results were also supported by WST cell survival assay (Supplementary Figure S2B).

Silencing of TAp63 lowers the sensitivity to GEM. AsPC-1 cells were transfected with control siRNA or with siRNA against TAp63. Twenty-four hours after transfection, cells were maintained in the presence or absence of GEM (1 μ M). Forty-eight hours after treatment, pictures were taken (upper panels). Under the same experimental conditions, floating and adherent cells were collected and their DNA contents were measured by FACS analysis (lower panels).
These findings prompted us to examine the effects of TAp63 silencing on GEM-dependent upregulation of p53/TAp63-target genes. For this purpose, AsPC-1 cells were transfected with control siRNA or with siRNA targeting TAp63. Twenty-four hours after transfection, cells were exposed to GEM or left untreated for 48 h. As shown in Figure 4a, knockdown of TAp63 attenuated GEM-mediated induction of p21WAF1 and NOXA. Consistent with these observations, immunoblotting revealed that GEM-stimulated expression of p21WAF1, as well as NOXA, and cleavage of PARP are remarkably prohibited by TAp63 depletion (Figure 4b). Together, our present results strongly suggest that TAp63-driven cell death pathway is tightly linked to GEM sensitivity of AsPC-1 cells.

Knockdown of TAp63 attenuates GEM-mediated induction of certain p53/TAp63-target genes. AsPC-1 cells were transfected as in Figure 3. Twenty-four hours after transfection, cells were incubated in the presence or absence of GEM (1 μ M) for 48 h. Total RNA and whole-cell lysates were then prepared and analyzed by RT-PCR (a) and immunoblotting (b), respectively.
Knockdown of RUNX2 enhances GEM sensitivity of AsPC-1 cells through the stimulation of TAp63-dependent cell death pathway
As shown in Figure 2b, there existed an inverse relationship between the expression level of TAp63 and RUNX2 in GEM-treated AsPC-1 cells, raising a possibility that RUNX2 could negatively regulate TAp63 expression. To address this issue, AsPC-1 cells were transfected with the empty plasmid or with the expression plasmid for RUNX2. As clearly seen in Supplementary Figure S3, forced expression of RUNX2 in AsPC-1 cells resulted in a marked decrease in the expression of TAp63 at mRNA and protein levels, indicating that RUNX2 has an ability to trans-repress TAp63.
Considering that silencing of RUNX2 enhances ADR sensitivity of osteosarcoma U2OS cells through the stimulation of p53/TAp73-dependent cell death pathway,47,48 we asked whether depletion of the endogenous RUNX2 could augment the sensitivity to GEM in AsPC-1 cells. To this end, AsPC-1 cells were transfected with control siRNA or with siRNA targeting RUNX2. Transfected cells were then treated with GEM or left untreated. As shown in Figure 5 and Supplementary Figure S4A, GEM-mediated cell death was further promoted in RUNX2 knockdown cells as compared with non-silencing control cells. These observations were supported by WST cell survival assay (Supplementary Figure S4B). Similar results were also obtained in p53-mutated human pancreatic cancer MiaPaCa-2 cells (Nakamura et al., manuscript in preparation), indicating that RUNX2 depletion-mediated enhancement of GEM sensitivity may not be restricted to p53-deficient AsPC-1 cells. Together, these results suggest that silencing of RUNX2 augments GEM-mediated cell death in AsPC-1 cells.

siRNA-mediated knockdown of RUNX2 enhances GEM sensitivity of AsPC-1 cells. AsPC-1 cells were transfected with control siRNA or with siRNA against RUNX2. Twenty-four hours after transfection, cells were treated with GEM (1 μ M) or left untreated. Forty-eight hours after treatment, images were obtained with phase-contrast microscopy (upper panels). Under the same experimental conditions, floating and attached cells were collected and number of cells with sub-G1 DNA content was measured by FACS analysis (lower panels).
We next sought to investigate the influence of RUNX2 silencing on p53/TAp63-related gene expression under these experimental conditions. As expected, GEM-dependent cleavage of PARP was increased in RUNX2 knockdown cells as compared with non-targeting siRNA-transfected cells (Figure 6a). Of note, TAp63 was strongly induced in RUNX2 knockdown cells exposed to GEM. Not surprisingly, these observations were also supported by RT-PCR analysis. As seen in Figure 6b, depletion of RUNX2 markedly stimulated GEM-mediated upregulation of TAp63, as well as a subset of p53/TAp63-target genes such as p21WAF1, NOXA and PUMA, indicating that silencing of RUNX2 activates TAp63-dependent cell death pathway in response to GEM.

Knockdown of RUNX2 further stimulates GEM-mediated induction of TAp63. AsPC-1 cells were transfected with control siRNA or with siRNA against RUNX2. Twenty-four hours after transfection, cells were treated with 1.0 μ M of GEM or left untreated. Forty-eight hours after treatment, whole-cell lysates and total RNA were prepared and analyzed by immunoblotting (a) and RT-PCR (b), respectively. Actin and GAPDH were used as a loading and an internal control, respectively.
To further verify whether TAp63 could suppress cell proliferation and/or promote cell death in AsPC-1 cells, AsPC-1 cells were transfected with the empty plasmid or with the expression plasmid for TAp63α. As shown in Supplementary Figures S5A and B, forced expression of TAp63α induced a subset of p53/TAp63-target genes, whereas the expression of RUNX2 remained unchanged at mRNA and protein levels regardless of the exogenous TAp63α. In addition, AsPC-1 cells were transfected as described above and transfected cells were maintained in the presence of G418. Two weeks after selection, cells were observed under phase-contrast microscope. Three weeks after the selection, G418-resistant colonies were stained with Giemsa’s solution. As shown in Supplementary Figure S5C, number of drug-resistant colonies was markedly reduced upon transfection of TAp63α as compared with control transfection. Thus, it is conceivable that TAp63 alone is capable to suppress cell proliferation and/or induce cell death in AsPC-1 cells.
TAp63 is involved in the regulation of GEM-dependent activation of ataxia telangiectasia-mutated protein (ATM)
Close inspection of the immunoblots as shown in Figures 4b and 6a revealed that silencing of TAp63 or RUNX2 results in a decrease or increase in GEM-dependent accumulation of γH2AX relative to GEM-treated non-silencing cells, respectively. To further confirm these observations, AsPC-1 cells were transfected with control siRNA or with siRNA against TAp63 followed by the incubation with or without GEM for 48 h. Cells were then fixed and stained with anti- γH2AX antibody. As clearly shown in Figure 7 and Supplementary Figure S6, knockdown of TAp63 plus GEM treatment caused the reduction in number of γH2AX-positive cells as compared with non-silencing cells exposed to GEM. As it has been well documented that histone H2AX is phosphorylated by phospho-ATM (p-ATM) in response to DNA damage,50 we have assessed the effects of TAp63 silencing on the amount of p-ATM in response to GEM. For this purpose, AsPC-1 cells were treated as in Figure 7. As expected, ATM became phosphorylated at Ser-1981 after GEM exposure (Figure 8). Intriguingly, GEM-dependent phosphorylation of ATM was markedly prohibited in TAp63 knockdown cells. Meanwhile, total amount of ATM was basically unaffected. Collectively, these results indicate that TAp63 is required for GEM-mediated activation of ATM and/or the maintenance of the activated form of ATM.

Knockdown of TAp63 results in decreased γH2AX-positive cells in response to GEM. AsPC-1 cells were transfected as in Figure 5. Twenty-four hours after transfection, cells were treated with or without GEM (1 μ M) for 48 h. Cells were then incubated with anti-γH2AX antibody (green). Cell nuclei were stained with DAPI (blue).

Silencing of TAp63 prohibits GEM-dependent phosphorylation of ATM. AsPC-1 cells were transfected as in Figure 7. Twenty-four hours after transfection, cells were incubated with or without GEM (1 μ M) for 48 h. Total RNA and whole-cell lysates were then extracted and analyzed by RT-PCR and immunoblotting, respectively.
Discussion
As described,51 transcriptionally active TAp63 has an overlapping function of the other p53 family members such as TAp73 and p53. For example, TAp63 can promote cell cycle arrest and/or cell death in response to genotoxic insults. In this study, we have found for the first time that silencing of pro-oncogenic RUNX2 in p53-deficient pancreatic cancer AsPC-1 cells helps to amplify TAp63 response to GEM and bolsters its tumor-suppressive function. As GEM-mediated accumulation of TAp63 in RUNX2 knockdown cells remarkably stimulated the expression of a subset of p53/TAp63-target gene products including cell cycle-related p21WAF1 and pro-apoptotic NOXA, it is highly likely that RUNX2 contributes at least in part to the acquisition and/or maintenance of GEM-resistant phenotype of p53-deficient pancreatic cancer cells through the downregulation of TAp63-dependent cell death pathway.
It has been well documented that, unlike p73 and p63, p53 is frequently mutated in human cancer tissues.43,44 Especially, around 75% of pancreatic cancers carry p53 mutations.42 Hamed et al.52 showed that AsPC-1 cells display GEM-resistant phenotype.51 According to our present results, AsPC-1 cells exhibited a much more higher resistance to GEM relative to p53-proficient pancreatic cancer SW1990 cells, indicating that SW1990 cells but not AsPC-1 cells undergo cell death following GEM exposure in a p53-dependent manner. Of note, GEM-dependent accumulation of γH2AX was also observed in AsPC-1 cells to an extent similar to SW1990 cells as revealed by immunoblotting and immunostaining (data not shown), implying that AsPC-1 cells receive DNA damage in the presence of GEM. These observations may rule out the possibility that the transporter function, which protects cells from the accumulation of undesirable chemotherapeutic agents such as GEM,53 is specifically augmented in AsPC-1 cells. Therefore, it is conceivable that γH2AX-dependent downstream cell death signaling pathway(s) is suppressed in AsPC-1 cells. Although AsPC-1 cells lack functional p53, the efforts should be made to improve GEM sensitivity of p53-defective and/or p53-mutated pancreatic cancer cells.
Intriguingly, siRNA-mediated silencing of RUNX2 positively impacted on GEM sensitivity of AsPC-1 cells, which was accompanied by a marked upregulation of TAp63 and a subset of p53/TAp63-target genes, indicating that RUNX2 suppresses TAp63-driven cell death pathway following GEM exposure. Under our experimental conditions, our siRNA against TAp63 did not affect the expression level of pro-oncogenic ΔNp63 (data not shown). In accordance with these observations, forced expression of RUNX2 declined the expression of TAp63 at mRNA and protein levels, whereas TAp63 had an undetectable effect on RUNX2 expression. Consistent with these results, knockdown of TAp63 lowered GEM sensitivity of AsPC-1 cells and overexpression of TAp63α gave a significant decrease in number of G418-resistant colonies as compared with control plasmid-transfected cells. These results further support the notion that TAp63 contributes to the enhancement of GEM sensitivity in AsPC-1 cells. In a good agreement with this notion, it has been shown that p53-mediated cell death is severely impaired in the absence of TAp63 or TAp73 in response to DNA damage, whereas TAp63 or TAp73 alone is able to induce cell death following DNA damage.54
As knockdown and overexpression of RUNX2 increased and decreased the expression level of TAp63, respectively, it is likely that RUNX2 serves as a negative transcription regulator for TAp63. From the close inspection of 5’-upstream region of human TAp63 gene, we could find out a putative RUNX2-binding sequence ACCACA (from –553 to –548). At present, it remains unclear whether this canonical sequence could be functional or not. It has been described that RUNX2 represses the transcription of its target genes by recruiting histone deacetylases to their promoters.55 Alternatively, Ng et al.56 found that transcription factor OCT4 has an ability to stimulate TAp63 but not ΔNp63 expression at mRNA level. According to their results, the upstream region of human TAp63 gene contained a functional OCT4-binding site (from –3044 to –3037). In addition, Park et al.57 found that mono-ubiquitinated FANCD2 together with its binding partner FANCP suppresses squamous cell cancers through the activation of TAp63 transcription. Thus, it is possible that RUNX2 may abrogate OCT4- and/or FANCD2/FANCP-mediated transcriptional activation of TAp63. Further studies should be required to adequately address this issue.
Another finding coming out of this study was that RUNX2/TAp63 regulatory axis is strongly involved in the regulation of GEM-mediated activation of ATM. According to our present results, knockdown of RUNX2 or TAp63 resulted in GEM-dependent increase or decrease in the amounts of γH2AX, respectively. Moreover, GEM-mediated phosphorylation (activation) of ATM was robustly reduced in TAp63-depleted cells as compared with non-silencing cells. Consistently, GEM-mediated cell death was prohibited in TAp63 knockdown cells relative to non-silencing cells. Wilhelm et al.58 described that ΔNp73 binds to DNA damage sensor protein 53BP1 and thereby inhibits ATM activation without direct interaction with ATM. Based on their results, cisplatin-mediated phosphorylation of H2AX was enhanced in ΔNp73-deficient mouse embryonic fibroblasts as compared with wild-type ones. As ΔNp73 displays a dominant-negative behavior against TAp73,41 it is plausible that TAp73 may have an ability to stimulate ATM activity in response to DNA damage. However, the precise molecular mechanism(s) how TAp73 could enhance ATM activity remains elusive. Under our experimental conditions, p-ATM was induced following GEM exposure in association with upregulation of TAp63, whereas ΔNp63 remained unchanged regardless of GEM treatment (data not shown). These observations indicate that the intracellular balance between TAp63 and ΔNp63 may affect GEM-dependent activation of ATM. Further studies should be required to elucidate the exact role of TAp63 in the modulation of ATM phosphorylation in response to DNA damage.
Taken together, our present findings strongly suggest that silencing of pro-oncogenic RUNX2 enhances GEM sensitivity of p53-deficient AsPC-1 cells through the upregulation of pro-apoptotic TAp63 and may be an attractive therapeutic strategy for the treatment of the patients with GEM-resistant malignant pancreatic cancer.
Materials and Methods
Cells and transfection
Human pancreatic cancer-derived SW1990 and AsPC-1 cells (ATCC, Manassas, VA, USA) were maintained in Dulbecco’s modified Eagle’s medium (Wako, Osaka, Japan) supplemented with heat-inactivated 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and penicillin–streptomycin at 37 °C in 5% CO2. For transfection, cells were transfected with the indicated expression plasmids using Lipofectamine 2000 transfection reagent according to the manufacturer’s instructions (Invitrogen).
Immunoblotting
GEM-treated cells were harvested in lysis buffer (25 mM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl and 1% Triton X-100) supplemented with a commercial protease inhibitor mixture (Sigma, St. Louis, MO, USA). Whole-cell lysates were analyzed by immunoblotting as described,47 using the following antibodies: anti-p53 (DO-1, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phospho-p53 at Ser-15 (Cell Signaling, Beverly, MA, USA), anti-p63 (4A4, NeoMarkers, Fremont, CA, USA), anti-p21WAF1 (H-164, Santa Cruz Biotechnology), anti-BAX (Cell Signaling), anti-NOXA (114C307, Abcam, Cambridge, MA, USA), anti-RUNX2 (Cell Signaling), anti-PARP (Cell Signaling), anti-γH2AX (2F3, BioLegend, San Diego, CA, USA), anti-ATM (5C2, Santa Cruz Biotechnology), anti-phospho-ATM at Ser-1981 (Cell Signaling) and anti-actin (20-33, Sigma).
WST assay
Cell were seeded at a density of 1×103 cells/96-well plates and allowed to attach overnight. Cells were then treated with the indicated concentrations of GEM. Forty-eight hours after treatment, 10 μl of a modified 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyl-tetrazolium bromide solution (Dojindo, Kumamoto, Japan) were added to the culture and reaction mixtures were incubated at 37°C for 2 h. The absorbance readings for each well were carried out at 570 nm using the microplate reader (Model 450, Bio-Rad, Hercules, CA, USA).
FACS analysis
For flow cytometric analysis, cells were fixed in 70% ethanol at –20°C overnight, then incubated with 1 mg/ml of RNase A at 37°C for 30 min and stained with 50μg/ml of propidium iodide. The DNA contents were analyzed by a FACScan flow cytometer (Becton Dickinson, Lincoln Park, NJ, USA).
Trypan blue exclusion assay
Floating and trypsinized attached cells were collected and suspended in fresh medium at room temperature. Twenty microliters of cell suspension were mixed with 20 μl of 0.4% Trypan blue solution (Bio-Rad), and 10 μl of this mixture were used for counting blue (dead) and white (alive) cells using TC20 automated cell counter (Bio-Rad).
RT-PCR
Total RNA extracted from the indicated cells using RNeasy mini kit (Qiagen, Valencia, CA, USA) was subjected to semiquantitative RT-PCR using SuperScript II reverse transcriptase and random primers (Invitrogen) according to the manufacturer’s instructions. Oligonucleotide primer sets used in this study were as follows: p53, 5′-CTGCCCTCAACAAGATGTTTTG-3′ (forward) and 5′-CTATCTGAGCAGCGCTCATGG-3′ (reverse); TAp63, 5′-GACCTGAGTGACCCCATGTG-3′ (forward) and 5′-CGGGTGATGGAGAGAGAGCA-3′ (reverse); RUNX2, 5′-TCTGGCCTTCCACTCTCAGT-3′ (forward) and 5′-GACTGGCGGGGTGTAAGTAA-3′ (reverse); p21WAF1, 5′-ATGAAATTCACCCCCTTTCC-3′ (forward) and 5′-CCCTAGGCTGTGCTCACTTC-3′ (reverse); 14-3-3σ, 5′-GAGCGAAACCTGCTCTCAGT-3′ (forward) and 5′-CTCCTTGATGAGGTGGCTGT-3′ (reverse); NOXA, 5′-CTGGAAGTCGAGTGTGCTACT-3′ (forward) and 5′-TCAGGTTCCTGAGCAGAAGAG-3′ (reverse); PUMA, 5′-GCCCAGACTGTGAATCCTGT-3′ (forward) and 5′-TCCTCCCTCTTCCGAGATTT-3′ (reverse); BAX, 5′-AGAGGATGATTGCCGCCGT-3′ (forward) and 5′-CAACCACCCTGGTCTTGGAT-3′ (reverse); glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′-ACCTGACCTGCCGTCTAGAA-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse).
Colony formation assay
AsPC-1 cells were transfected with the empty plasmid (pcDNA3) or with the expression plasmid for TAp63α. Forty-eight hours after transfection, cells were transferred into fresh medium containing G418 (400 μg/ml). Two weeks after the selection, cells were observed under phase-contrast microscope. Three weeks after the selection, G418-resistant colonies were fixed and stained with Giemsa’s solution.
Immunostaining
Immunostaining was performed by fixing cells in 3.7% formaldehyde at room temperature for 30 min followed by permeabilization with 0.1% Triton X-100 in PBS for 5 min. Cells were then blocked with 3% BSA in PBS at room temperature for 1 h. After blocking, cells were incubated with anti-γH2AX antibody at room temperature for 1 h. Cells were washed in PBS and incubated with fluorescein isothiocyanide-conjugated goat anti-mouse IgG (Invitrogen) at room temperature for 1 h. After incubation, cells were extensively washed in PBS and the coverslips were mounted on glass slides with VectaShield (Vector Laboratories, Peterborough, UK). Images were captured using a confocal laser scanning microscope (Leica, Wetzlar, Germany). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI).
Statistical analysis
All cell-based assays described in this study were performed independently at least three times. The data in all graphs were analyzed with Microsoft Excel (Microsoft Co., Redmond, WA, USA) and represented means±S.D. from two independent experiments performed in triplicates. Statistical significance was determined with Student’s t-test, and P-values <0.05 were considered statistically significant.
Abbreviations
- ADR:
-
adriamycin
- ATM:
-
ataxia telangiectasia-mutated protein
- DAPI:
-
4′,6-diamidino-2-phenylindole
- FACS:
-
fluorescence-activate cell sorting
- GAPDH:
-
glyceraldehyde 3-phosphate dehydrogenase
- GEM:
-
gemcitabine
- PARP:
-
poly ADP ribose polymerase
- RUNX2:
-
runt-related transcription factor 2
- siRNA:
-
small interfering RNA.
References
Siegel R, Ma J, Zou Z, Jemal A . Cancer statistics. CA Cancer J Clin 2014; 64: 9–29.
Kuroda T, Kumagi T, Yokota T, Seike H, Nishiyama M, Imai Y et al. Improvement of long-term outcomes in pancreatic cancer and its associated factors within the gemcitabine era: a collaborative retrospective multicenter clinical review of 1,082 patients. BMC Gastroenterol 2013; 13: 134.
Smeenk HG, Tran TC, Erdmann J, van Eijck CH, Jeekel J . Survival after surgical management of pancreatic adenocarcinoma: does curative and radical surgery truly exist? Langenbecks Arch Surg 2005; 390: 94–103.
Janes Jr RH, Niederhuber JE, Chmiel JS, Winchester DP, Ocwieja KC, Karnell JH et al. National patterns of care for pancreatic cancer. Results of a survey by the Commission on Cancer. Ann Surg 1996; 223: 261–272.
Burris 3rd HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997; 15: 2403–2413.
Moss RA, Lee C . Current and emerging therapies for the treatment of pancreatic cancer. Onco Targets Ther 2010; 3: 111–127.
Huang P, Chubb S, Hertel LW, Grindey GB, Plunkett W . Action of 2',2'-difluorodeoxycytidine on DNA synthesis. Cancer Res 1991; 51: 6110–6117.
Li Q, Yan H, Liu W, Zhen H, Yang Y, Cao B . Efficacy and safety of gemcitabine-fluorouracil combination therapy in the management of advanced pancreatic cancer: a meta-analysis of randomized controlled trials. PLoS One 2014; 9: e104346.
Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, André T et al. GISCAD: gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a ERCOR and GISCAD phase III trial. J Clin Oncol 2005; 23: 3509–3516.
Cunningham D, Chau I, Stocken DD, Valle JW, Smith D, Steward W et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J Clin Oncol 2009; 27: 5513–5518.
Heinemann V, Quietzsch D, Gieseler F, Gonnermann M, Schönekäs H, Rost A et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J Clin Oncol 2006; 24: 3946–3952.
Van Cutsem E, Vervenne WL, Bennouna J, Humblet Y, Gill S, Van Laethem JL et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol 2009; 27: 2231–2237.
Chuang LS, Ito K, Ito Y . RUNX family: regulation and diversification of roles through interacting proteins. Int J Cancer 2013; 132: 1260–1271.
Ito Y, Bae SC, Chuang LS . The RUNX family: developmental regulators in cancer. Nat Rev Cancer 2015; 15: 81–95.
Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR . AML1 the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996; 84: 321–330.
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997; 89: 755–764.
Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997; 89: 765–771.
Levanon D, Bettoun D, Harris-Cerruti C, Woolf E, Negreanu V, Eilam R et al. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. EMBO J 2002; 21: 3454–3463.
Fukamachi H, Ito K . Growth regulation of gastric epithelial cells by Runx3. Oncogene 2004; 23: 4330–4335.
Peterson LF, Zhang DE . The 8;21 translocation in leukemogenesis. Oncogene 2004; 23: 4255–4262.
Li QL, Ito K, Sakakura C, Fukamachi H, Inoue Ki, Chi XZ et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002; 109: 113–124.
Hsu PI, Hsieh HL, Lee J, Lin LF, Chen HC, Lu PJ et al. Loss of RUNX3 expression correlates with differentiation, nodal metastasis, and poor prognosis of gastric cancer. Ann Surg Oncol 2009; 16: 1686–1694.
Kayed H, Jiang X, Keleg S, Jesnowski R, Giese T, Berger MR et al. Regulation and functional role of the Runt-related transcription factor-2 in pancreatic cancer. Br J Cancer 2007; 97: 1106–1115.
Endo T, Ohta K, Kobayashi T . Expression and function of Cbf a-1/Runx2 in thyroid papillary carcinoma cells. J Clin Endocrinol Metab 2008; 93: 2409–2412.
Pratap J, Imbalzano KM, Underwood JM, Cohet N, Gokul K, Akech J et al. Ectopic runx2 expression in mammary epithelial cells disrupts formation of normal acini structure: implications for breast cancer progression. Cancer Res 2009; 69: 6807–6814.
Onodera Y, Miki Y, Suzuki T, Takagi K, Akahira J, Sakyu T et al. Runx2 in human breast carcinoma: its potential roles in cancer progression. Cancer Sci 2010; 101: 2670–2675.
Brubaker KD, Vessella RL, Brown LG, Corey E . Prostate cancer expression of runt-domain transcription factor Runx2, a key regulator of osteoblast differentiation and function. Prostate 2003; 56: 13–22.
Li H, Zhou RJ, Zhang GQ, Xu JP . Clinical significance of RUNX2 expression in patients with nonsmall cell lung cancer: a 5-year follow-up study. Tumour Biol 2013; 34: 1807–1812.
Sase T, Suzuki T, Miura K, Shiiba K, Sato I, Nakamura Y et al. Runt-related transcription factor 2 in human colon carcinoma: a potent prognostic factor associated with estrogen receptor. Int J Cancer 2012; 131: 2284–2293.
Li W, Xu S, Lin S, Zhao W . Overexpression of runt-related transcription factor-2 is associated with advanced tumor progression and poor prognosis in epithelial ovarian cancer. J Biomed Biotechnol 2012; 2012: 456534.
Nathan SS, Pereira BP, Zhou YF, Gupta A, Dombrowski C, Soong R et al. Elevated expression of Runx2 as a key parameter in the etiology of osteosarcoma. Mol Biol Rep 2009; 36: 153–158.
Martin JW, Zielenska M, Stein GS, van Wijnen AJ, Squire JA . The role of RUNX2 in osteosarcoma oncogenesis. Sarcoma 2011; 2011: 282745.
Barnes GL, Hebert KE, Kamal M, Javed A, Einhorn TA, Lian JB et al. Fidelity of Runx2 activity in breast cancer cells is required for the generation of metastases-associated osteolytic disease. Cancer Res 2004; 64: 4506–4513.
Pratap J, Javed A, Languino LR, van Wijnen AJ, Stein JL, Stein GS et al. The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Mol Cell Biol 2005; 25: 8581–8891.
Pratap J, Lian JB, Javed A, Barnes GL, van Wijnen AJ, Stein JL et al. Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev 2006; 25: 589–600.
Pratap J, Wixted JJ, Gaur T, Zaidi SK, Dobson J, Gokul KD et al. Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res 2008; 68: 7795–7802.
Mendoza-Villanueva D, Deng W, Lopez-Camacho C, Shore P . The Runx transcriptional co-activator, CBFbeta, is essential for invasion of breast cancer cells. Mol Cancer 2010; 9: 171.
Chimge NO, Frenkel B . The RUNX family in breast cancer: relationships with estrogen signaling. Oncogene 2013; 32: 2121–2130.
Tandon M, Chen Z, Pratap J . Runx2 activates PI3K/Akt signaling via mTORC2 regulation in invasive breast cancer cells. Breast Cancer Res 2014; 16: R16.
Akech J, Wixted JJ, Bedard K, van der Deen M, Hussain S, Guise TA et al. Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene 2010; 29: 811–821.
Vousden KH, Lu X . Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604.
Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005; 7: 469–483.
Hollstein M, Sidransky D, Vogelstein B, Harris CC . p53 mutations in human cancers. Science 1991; 253: 49–53.
Ikawa S, Nakagawara A, Ikawa Y . p53 family genes: structural comparison, expression and mutation. Cell Death Differ 1999; 6: 1154–1161.
Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997; 90: 809–819.
Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 1998; 2: 305–316.
Ozaki T, Wu D, Sugimoto H, Nagase H, Nakagawara A . Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis 2013; 4: e610.
Ozaki T, Sugimoto H, Nakamura M, Hiraoka K, Yoda H, Sang M et al. Runt-related transcription factor 2 attenuates the transcriptional activity as well as DNA damage-mediated induction of pro-apoptotic TAp73 to regulate chemosensitivity. FEBS J 2015; 282: 114–128.
Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE . BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol Cancer Ther 2012; 11: 2644–2253.
Falck J, Coates J, Jackson SP . Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005; 434: 605–611.
Melino G . p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ 2011; 18: 1487–1499.
Hamed SS, Straubinger RM, Jusko WJ . Pharmacodynamic modeling of cell cycle and apoptotic effects of gemcitabine on pancreatic adenocarcinoma cells. Cancer Chemother Pharmacol 2013; 72: 553–563.
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG . Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013; 13: 714–726.
Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002; 416: 560–564.
McGee-Lawrence ME, Bradley EW, Dudakovic A, Carlson SW, Ryan ZC, Kumar R et al. Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone 2013; 52: 296–307.
Ng WL, Chen G, Wang M, Wang H, Story M, Shay JW et al. OCT4 as a target of miR-34a stimulates p63 but inhibits p53 to promote human cell transformation. Cell Death Dis 2014; 5: e1024.
Park E, Kim H, Kim JM, Primack B, Vidal-Cardenas S, Xu Y et al. FANCD2 activates transcription of TAp63 and suppresses tumorigenesis. Mol Cell 2013; 50: 908–918.
Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev 2010; 24: 549–560.
Acknowledgements
This research was supported by a research grant (grant no. 23501278) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to TO).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Sugimoto, H., Nakamura, M., Yoda, H. et al. Silencing of RUNX2 enhances gemcitabine sensitivity of p53-deficient human pancreatic cancer AsPC-1 cells through the stimulation of TAp63-mediated cell death. Cell Death Discovery 1, 15010 (2015). https://doi.org/10.1038/cddiscovery.2015.10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/cddiscovery.2015.10
- Springer Nature Limited
This article is cited by
-
Impact of RUNX2 on drug-resistant human pancreatic cancer cells with p53 mutations
BMC Cancer (2018)
-
Improvement of gemcitabine sensitivity of p53-mutated pancreatic cancer MiaPaCa-2 cells by RUNX2 depletion-mediated augmentation of TAp73-dependent cell death
Oncogenesis (2016)
-
Silencing of RUNX2 enhances gemcitabine sensitivity of p53-deficient human pancreatic cancer AsPC-1 cells through the stimulation of TAp63-mediated cell death
Cell Death & Disease (2015)