
Abstract
Novel PLA2G6 mutations associated with p.Asp283Asn and a unique intragenic deletion of exons 4 and 5 due to non-allelic homologous recombination were identified in a Japanese female patient with typical infantile neuroaxonal dystrophy. The patient showed progressive tetraplegia beginning at 9 months. An electroencephalogram showed a diffuse increase in fast waves, and brain magnetic resonance imaging showed progressive brain atrophy and T2 hypointensity in the globus pallidus.
Similar content being viewed by others


Infantile neuroaxonal dystrophy (INAD; MIM #256600) is a rare autosomal-recessive neurodegenerative disorder involving both the central and peripheral nervous system. INAD is diagnosed when patients show several clinical features, including the onset of symptoms before 3 years of age, psychomotor regression, hypotonia, symmetrical pyramidal tract signs, a relentlessly progressive course leading to spastic tetraplegia, visual impairment with nystagmus due to optic atrophy, and dementia.1,2 Brain pathological examinations demonstrated characteristic findings of axonal spheroids.3 This disorder typically occurs because of mutations in the phospholipase A2 group VI gene (PLA2G6) located on the 22q13.1 region, which encodes a calcium-independent phospholipase A2 enzyme involved in phospholipid remodeling and catalyzes the hydrolysis of glycerophospholipids.4 Recent molecular investigations identified nearly 100 PLA2G6 mutations not only in patients with INAD but also in patients with parkinsonism.1,5–25 Thus, the disease concept has been expanded to include all of these disorders into a group called neurodegeneration with brain iron accumulation.2,26 Currently, these broad clinical features due to PLA2G6 mutations are recognized as a phenotypic spectrum of PLA2G6-associated neurodegeneration (PLAN). Recently, we encountered a Japanese female patient with a typical form of INAD and identified compound heterozygous mutations.
At present, the patient is of 2 years and 8 months old. She is the only child of healthy non-consanguineous parents. Her family history was unremarkable. She was born after a 40-week-long uneventful gestation with a weight of 2,840 g without asphyxia. Her initial development was normal during early infancy: she attained head control at 3 months and sat without support at 7 months. Her development, however, slowed after she began to crawl at 9 months. Although she stood with a support at 14 months, her developmental deterioration gradually emerged at ~17 months. At 19 months, hypotonia was noted, particularly in the lower extremities. She lost the ability to sit and roll, and showed decreased voluntary movements at 23 months.
Neurological examination revealed generalized hypotonia, hyperreflexia in all extremities and bilateral Babinski reflexes. Electroencephalogram (EEG) showed a diffuse increase in fast waves without apparent rhythmic activity (Figures 1a and b). Brain magnetic resonance imaging (MRI) showed atrophy of the cerebellum (Figures 1d–g). Auditory brainstem responses were almost lacking and the cortical component of short-latency sensory-evoked potentials were not detected. Electromyogram showed signs of denervation. Other laboratory examinations revealed unremarkable results.

Clinical examinations of the present patient. Electroencephalogram (EEG) of the patient (a–c). Diffuse increase of low-amplitude 15–30 Hz fast waves are demonstrated during wakefulness at 23 months of age (a). At 2 years and 8 months, extremely fast waves during wakefulness (b) and multifocal spike-waves during sleep (c, arrowheads) are recorded. Brain magnetic resonance imaging (MRI) of the patient (d–i). Axial T1 (d) and T2 (e) images and a sagittal T1 (f) and a coronal T2 (g) images examined at 23 months. Mild enlargement of the lateral ventricles (d, e) and cerebellar atrophy (f, g) are demonstrated. These findings are remarkable in axial T1 (h) and T2 (i) images examined at 2 years and 8 months. Progressive T2 hypointensity in the globus pallidus is noted (i). ECG, electrocardiogram.
She gradually lost her visual ability to follow objects with emergence of strabismus and transient nystagmus at 2 years and 8 months. Her optic nerves showed initial signs of atrophy. Tetraplegia was progressive and the bilateral upper extremities were rigid. The EEG revealed a further increase in fast waves and multifocal spike-waves, particularly in the bilateral temporal regions (Figure 1c). She had no apparent seizures, although she often showed episodic stiffening and extension of the bilateral upper extremities associated with no ictal epileptic patterns in EEG. Brain MRI revealed a slight progression of diffuse cerebral atrophy and T2 hypointensity in the globus pallidus (Figures 1h and i). Based on these findings, she was clinically diagnosed with INAD.
Although PLA2G6 was listed as a candidate target of molecular diagnosis, the contribution of other genes could not be excluded. Thus, targeted re-sequencing was performed as the first step to screen single-nucleotide variants using TruSight One v1.0 sequencing panel (Illumina, San Diego, CA, USA), which includes 125,396 probes aimed to capture 11,946,514-bp targeted exon regions consisting of 4,813 genes associated with known clinical phenotypes. This study was performed in accordance with the Declaration of Helsinki Principles, and the ethics committee of Tokyo Women’s Medical University approved this study. Blood samples were obtained from the patient and her parents after receiving written informed consent thorough careful genetic counseling regarding the appropriate dealing of genetic information and possible incidental findings. Genomic DNA was extracted from blood samples and used for further examination. After constructing the sequence library using 50 ng of genomic DNA, the MiSeq next-generation sequencer (Illumina) was used to sequence 151-bp paired-end reads according to the manufacturer’s instructions. The extracted data were mapped to a reference genome (GRCh37/hg19) using BWA Enrichment v1.0 cloud software (Illumina).
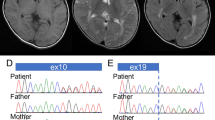
The total aligned bases were 2.08-Gb with a mean coverage depth of 83.7 (the target coverage at 20× was 95.0%). A total of 7,919 variants were extracted. The extracted variants were annotated and filtered using Variant Studio software (Illumina) as described previously.27 As a result, 20 variants were considered as candidates (Supplementary Table S1). Among them, a missense mutation, NM_003560.2(PLA2G6):c.847G>A (NP_003551.2(PLA2G6):p.Asp283Asn), was included (Figure 2a). Although the same variant has never been reported, this codon is located on the ankyrin repeat region (which is crucial for protein–protein interaction)4,28 and was affected by a previously reported disease-causing mutation, p.Asp283Gly.4 The prediction scores suggested pathogenesis, i.e., SIFT (0; deleterious) and PolyPhen-2 (0.999; probably damaging). From these findings, we considered this variant to be a disease-causing mutation. Subsequent Sanger sequencing confirmed that this variant was inherited from the patient’s father (Figure 2b). However, a maternally inherited variant was not detected through targeted re-sequencing.

Molecular analysis results. (a) Image view using Integrative Genomics Viewer (IGV) demonstrates an altered ‘T’ nucleotide in approximately half of the reads. The affected amino-acid ‘D’ is conserved among species. (b) Electropherograms of Sanger sequencing. The present patient and her father show heterozygous for c.847G>A. Thus, her father is an obligate carrier of this mutation. (c) Agarose gel electrophoresis of RT-PCR amplicons. Compared with the normal control (N), the patient (P) shows an aberrant short band (arrow). M4; phi-X HaeIII digest. (d) An electropherogram of Sanger sequencing for an aberrant short band demonstrates a skipping of exons 4 and 5. (e) Agarose gel electrophoresis of long PCR products. In addition to the normal bands with an expected size of 4,949 bp, the patient (P) and her mother (Mo) show aberrant short bands (arrow). Thus, the mother is an obligate carrier of this large deletion. (f) An electropherogram of Sanger sequence for the short band extracted from the agarose gel. A 12-bp homologous region is identified in the breakpoint. The genomic positions are indicated on the sequences. Because PLA2G6 is encoded in an anti-sense direction, the locations of genomic numbers are inverted. (g) The result of homology searching between two breakpoints. “Query” and “Sbjct” indicate the nucleotide sequences around the breakpoints in intron 3 and 5, respectively. An identity of 86% is calculated between them. The box indicates the homologous region in the breakpoint. Fa, patient’s father; M1, lambda/Hind III digest; N, normal control sample.
To identify an undiagnosed mutation in the homologous allele, PLA2G6 mRNA was analyzed by reverse transcription (RT)-PCR amplification. Total RNA was extracted from Epstein-Barr virus-transfected immortalized lymphocytes established from the patient and transcribed into complementary DNA. The RT-PCR product using the primer set (5ʹ-TGCTACCCTTCTATGAGAGC-3ʹ and 5ʹ-TGGTGCTGTTCACGTTGCAG-3ʹ) showed an aberrant shorter band in addition to a normal band (Figure 2c). DNA extracted from the short band was analyzed by Sanger sequencing and the result revealed deletions in exons 4 and 5 (Figure 2d), indicating a 372-bp in-flame deletion, c.426_797del372 (p.Ser142_Gly266delinsArg). Because no splicing mutations located at the exon–intron boundaries nearby this region were detected, we suspected exonic deletions of this region. Next, the genomic region encompassing exons 4 and 5 was amplified by PCR amplification using the primer set (5ʹ-ACCAGTTGGCCATCTTGTGC-3ʹ and 5ʹ-AAGGAGCACTGAAGCCATCG-3ʹ), which was designed to amplify a 4,949-bp PCR product, and an abnormal short band was identified (Figure 2e). DNA extracted from the short band was analyzed by Sanger sequencing and a genomic deletion of exons 4 and 5 was confirmed (Figure 2f). The size of aberrant short band was 640 bp, indicating a 4,309-bp genomic deletion. This deletion was also identified in the sample from the mother (Figure 2e). Thus, compound heterozygous mutations of PLA2G6 inherited from both parents were confirmed and a molecular diagnosis of PLAN was established in this patient.
A similar exonic deletion was previously reported by Tonelli et al. in exons 5 and 6.11 The breakpoints of the reported deletion were analyzed and the homologous sequence was identified in the breakpoints. Based on their findings, they considered Alu-repeats as the genomic characteristic of this region. In this study, the exonic deletion involved exons 4 and 5 was identified, and the breakpoint sequence showed a 12-bp homologous sequence (Figure 2f). Using Genetyx software (Genetyx Corporation, Tokyo, Japan), we further analyzed the homology of the neighboring the 237-bp genomic regions encompassing the 12-bp homologous sequence, and 86% identity was confirmed (Figure 2g). This indicates that the mechanism of the identified exonic deletion is quite similar to that reported by Tonelli et al.11 The genomic deletion between the similar genomic sequences suggests predisposition of the genomic rearrangements in this region.
Previously, nearly 100 PLA2G6 mutations were reported in patients with PLAN and/or parkinsonism (Supplementary Table S2). The identified mutations are distributed throughout the PLA2G6 complementary DNA sequence and there is no clear genotype–phenotype correlation. Particularly, Arg632Trp was identified in patients with both of INAD and parkinsonism.4,10 Thus, phenotypic consequence of PLA2G6 mutations may be related to the combinations of the heterozygous mutations.
References
References
Carrilho I, Santos M, Guimaraes A, Teixeira J, Chorao R, Martins M et al. Infantile neuroaxonal dystrophy: what's most important for the diagnosis? Eur J Paediatr Neurol 2008; 12: 491–500.
Kurian MA, McNeill A, Lin JP, Maher ER . Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev Med Child Neurol 2011; 53: 394–404.
Aicardi J, Castelein P . Infantile neuroaxonal dystrophy. Brain 1979; 102: 727–748.
Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 2006; 38: 752–754.
Arber CE, Li A, Houlden H, Wray S . Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol Appl Neurobiol (e-pub ahead of print 14 April 2015; doi: 10.1111/nan.12242).
Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am J Hum Genet 2006; 79: 942–948.
Biancheri R, Rossi A, Alpigiani G, Filocamo M, Gandolfo C, Lorini R et al. Cerebellar atrophy without cerebellar cortex hyperintensity in infantile neuroaxonal dystrophy (INAD) due to PLA2G6 mutation. Eur J Paediatr Neurol 2007; 11: 175–177.
Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW et al. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 2009; 65: 19–23.
Yoshino H, Tomiyama H, Tachibana N, Ogaki K, Li Y, Funayama M et al. Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 2010; 75: 1356–1361.
Sina F, Shojaee S, Elahi E, Paisan-Ruiz C . R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol 2009; 16: 101–104.
Tonelli A, Romaniello R, Grasso R, Cavallini A, Righini A, Bresolin N et al. Novel splice-site mutations and a large intragenic deletion in PLA2G6 associated with a severe and rapidly progressive form of infantile neuroaxonal dystrophy. Clin Genet 2010; 78: 432–440.
Tan EK, Ho P, Tan L, Prakash KM, Zhao Y . PLA2G6 mutations and Parkinson's disease. Ann Neurol 2010; 67: 148.
Tomiyama H, Yoshino H, Ogaki K, Li L, Yamashita C, Li Y et al. PLA2G6 variant in Parkinson's disease. J Hum Genet 2011; 56: 401–403.
Shi CH, Tang BS, Wang L, Lv ZY, Wang J, Luo LZ et al. PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology 2011; 77: 75–81.
Lu CS, Lai SC, Wu RM, Weng YH, Huang CL, Chen RS et al. PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 2012; 159B: 183–191.
Bower MA, Bushara K, Dempsey MA, Das S, Tuite PJ . Novel mutations in siblings with later-onset PLA2G6-associated neurodegeneration (PLAN). Mov Disord 2011; 26: 1768–1769.
Gui YX, Xu ZP, Wen L, Liu HM, Zhao JJ, Hu XY . Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkinsonism Relat Disord 2013; 19: 21–26.
Tian JY, Tang BS, Shi CH, Lv ZY, Li K, Yu RL et al. Analysis of PLA2G6 gene mutation in sporadic early-onset parkinsonism patients from Chinese population. Neurosci Lett 2012; 514: 156–158.
Romani M, Kraoua I, Micalizzi A, Klaa H, Benrhouma H, Drissi C et al. Infantile and childhood onset PLA2G6-associated neurodegeneration in a large North African cohort. Eur J Neurol 2015; 22: 178–186.
Illingworth MA, Meyer E, Chong WK, Manzur AY, Carr LJ, Younis R et al. PLA2G6-associated neurodegeneration (PLAN): further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab 2014; 112: 183–189.
Malaguti MC, Melzi V, Di Giacopo R, Monfrini E, Di Biase E, Franco G et al. A novel homozygous PLA2G6 mutation causes dystonia-parkinsonism. Parkinsonism Relat Disord 2015; 21: 337–339.
Kim YJ, Lyoo CH, Hong S, Kim NY, Lee MS . Neuroimaging studies and whole exome sequencing of PLA2G6-associated neurodegeneration in a family with intrafamilial phenotypic heterogeneity. Parkinsonism Relat Disord 2015; 21: 402–406.
Salih MA, Mundwiller E, Khan AO, AlDrees A, Elmalik SA, Hassan HH et al. New findings in a global approach to dissect the whole phenotype of PLA2G6 gene mutations. PLoS ONE 2013; 8: e76831.
Karkheiran S, Shahidi GA, Walker RH, Paisan-Ruiz C . PLA2G6-associated Dystonia-Parkinsonism: Case Report and Literature Review. Tremor Other Hyperkinet Mov (NY) 2015; 5: 317.
Wu Y, Jiang Y, Gao Z, Wang J, Yuan Y, Xiong H et al. Clinical study and PLA2G6 mutation screening analysis in Chinese patients with infantile neuroaxonal dystrophy. Eur J Neurol 2009; 16: 240–245.
Schneider SA, Hardy J, Bhatia KP . Syndromes of neurodegeneration with brain iron accumulation (NBIA): an update on clinical presentations, histological and genetic underpinnings, and treatment considerations. Mov Disord 2012; 27: 42–53.
Yamamoto T, Igarashi N, Shimojima K, Sangu N, Sakamoto Y, Shimoji K et al. Use of targeted next-generation sequencing for molecular diagnosis of craniosynostosis: identification of a novel de novo mutation of EFNB1. Congenit Anom (Kyoto) (e-pub ahead of print 24 July 2015; doi: 10.1111/cga.12123).
Li J, Mahajan A, Tsai MD . Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 2006; 45: 15168–15178.
Data Citations
Yamamoto, Toshiyuki HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.735
Yamamoto, Toshiyuki HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.738
Acknowledgements
We would like to express our gratitude to the patients and their families for their cooperation. We are grateful to the technicians from our laboratories including Yumiko Ondo for their skillful help. This research was supported by the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and Development (AMED) and Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 15K09631.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yamamoto, T., Shimojima, K., Shibata, T. et al. Novel PLA2G6 mutations associated with an exonic deletion due to non-allelic homologous recombination in a patient with infantile neuroaxonal dystrophy. Hum Genome Var 2, 15048 (2015). https://doi.org/10.1038/hgv.2015.48
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.48
- Springer Nature Limited