
Abstract
A pilot study of newborn screening for Fabry disease was performed in Okinawa, Japan. A total of 2,443 neonates were screened using dried blood spot samples over 7 years starting in 2007. Of 13 neonates determined to have low α-galactosidase A (GLA) activity, one boy had a new missense mutation, p.G144D of the GLA gene. This mutation was considered to be a late-onset type, as evaluated based on plasma globotriaosylsphingosine levels and family history.
Similar content being viewed by others


Fabry disease (FD; MIM# 301500) is an X-linked lysosomal storage disorder resulting from a deficiency in α-galactosidase A (GLA) activity. This enzyme defect leads to the systemic lysosomal accumulation of globotriaosylceramide (Gb3) in multiple organs, such as the skin, eyes, kidneys, ears, lungs, heart and brain.1 Affected males with the classic phenotype of FD present with acroparesthesias, angiokeratomas and hypohidrosis in early childhood or adolescence, progressing to renal insufficiency, cardiomyopathy and cerebrovascular disease in adulthood.2 Patients with late-onset phenotypes of FD lack the classic symptoms of FD, and their symptoms primarily involve the heart, kidneys, or cerebrovascular systems in adulthood.3 Clinical manifestations in heterozygous females vary from no symptoms to abnormalities as severe as those in affected males.4 Enzyme replacement therapy is effective against the progression of FD. Therefore, newborn mass screening is considered important for early treatment before the onset of irreversible organ defects, including renal failure, cardiac disease and early-onset stroke.5,6 The plasma or urinary levels of Gb3 in neither hemizygotes nor heterozygotes correlate with the severity of disease manifestations.7 Plasma Gb3 levels are not useful for the secondary screening of FD, but plasma globotriaosylsphingosine (lyso-Gb3) levels are greatly increased in patients with classic FD. Therefore, lyso-Gb3 levels might be helpful for the secondary screening and severity of FD.7–9
In this pilot study, we performed newborn screening for FD. We measured GLA activity using dried blood spot samples and then analyzed GLA and lyso-Gb3 concentrations.
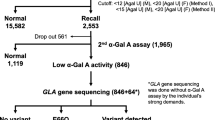
A pilot study of newborn screening for FD was performed in the hospital of University of the Ryukyus in Okinawa Prefecture, Japan. The study (approval number: H18.12–9) was performed in accordance with the standards of the Ethics Committee in the Ryukyus Graduate School of Medicine (Okinawa, Japan). Dried blood spot samples collected from the heel at 5 days of age were used, and GLA activity was assayed using a fluorescent substrate as previously described.6,10,11 One unit (AgalU) of enzymatic activity was equal to 0.34 pmol of 4-methylumbelliferyl-d-galactopyranoside cleaved/hour/disc. The results from this assay were repeatedly <17.0 AgalU in males and <20.0 AgalU in females. We analyzed all seven exons of GLA by direct sequencing from the peripheral blood.12 Plasma GLA activity was measured as described previously.8 Plasma lyso-Gb3 concentrations were analyzed by LC–MS/MS using the lyso-Gb3-Gly internal standard.13 A normal assay result for plasma GLA activity was >4.0 nmol/hour/ml and that for plasma lyso-Gb3 was <2.0 ng/ml. The average value and s.d. of the normal control plasma lyso-Gb3 of 1,000 males and females was 0.4±0.3 ng/ml.
A total of 2,443 neonates with informed consent from their guardians were included in the study for 7 years, from May 2007 to September 2014. Thirteen neonates, including two boys and 11 girls, were found to have low GLA activity. The GLA activity in the 11 girls was normal when measured a second time. Because the girls had no family history with clinical information regarding FD, at their request, the analysis of GLA was not performed. One boy without a family history of FD had GLA activity of 13.6 AgalU (normal GLA activity: <17.0) and p.E66Q of GLA, previously suggested to be a functional mutation.14,15 The other boy showed GLA activity of 6.1 AgalU (normal GLA activity: <17.0) and p.G144D, which was not previously described. This finding indicated that asparaginic acid was substituted for glycine at the 144th amino acid in GLA. His plasma GLA activity was low (0.8 nmol/hour/ml, normal: >4.0 nmol/hour/ml). Plasma lyso-Gb3 concentrations (normal: <2.0 ng/ml, mean±s.d.: 0.4±0.3) were 0.9 ng/ml (+1.7 s.d.) at 11 months old and 1.4 ng/ml (+3.3 s.d.) at 21 months old. He was born as the second child to a healthy father and a mother with hypothyroidism. The pregnancy and delivery at 39 weeks of gestation were uneventful. Birth weight was 2,874 g (−0.3 s.d.), length 50.0 cm (+0.47 s.d.) and occipitofrontal circumference 33.0 cm (−0.2 s.d.). At 21 months old, he did not have acroparesthesias, angiokeratomas, or hypohidrosis. Urinary mulberry cells were not observed in a spot of urine. In pedigree analysis, his mother and maternal grandmother also had p.G144D in GLA (Figure 1). Both healthy female relatives showed a normal range of plasma lyso-Gb3 concentrations. The boy’s male relatives (Figure 1, II-2 and III-1) did not participate in the study. The mutation p.G144D was predicted to be disease-causing, as the probable damaging score according to the software PolyPhen-2 (http://genetics.bwh.harvard.edu/pph/) was 1.000 and the probability value score according to Mutation Taster (http://www.mutationtaster.org) was 0.999. When valine is substituted for glycine at the 144th amino acid in GLA, the p.G144V mutation of GLA is a disease-causing mutation, previously described as classic FD.16 Known pathological missense mutations at the same position cause different clinical types of FD (e.g., R112H results in the late-onset type and R112C or R112S in the classic type).16–18 The levels of lyso-Gb3 gradually increase with age in individuals carrying a GLA mutation of the late-onset type.19 The rate of disease onset increases with the elevation of lyso-Gb3 levels in individuals carrying a GLA mutation of the late-onset type.19 We speculate that the p.G144D causes FD as a late-onset type because of the patient’s low plasma GLA activity levels, his slightly high plasma lyso-Gb3 levels and the results in his family analysis. Further careful investigation of the boy’s male relatives is warranted.

Pedigree analysis with results for the GLA gene, GLA activity and lyso-Gb3 levels. p.G144D: II-3 and III-2 (heterozygous), IV-3 (hemizygous); GLA activity (normal: >4.0 nmol/hour per ml): II-3 (3.1), III-2 (4.3) and IV-3 (0.8); lyso-Gb3 (normal: <2.0 ng/ml): II-3 (0.3), III-2 (0.2) and IV-3 (0.9, 1.4). Neither persons II-2 nor III-1 participated in the study. CA, cerebral aneurysm; CC, carcinoma of the colon; Gb3, globotriaosylceramide; GLA, α-galactosidase A; MI, myocardial infarction; SH, subarachnoid hemorrhage; Y, years of age.
References
References
Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease, 8th edn. McGraw-Hill: New York, NY, USA, 2001, pp 3733–3774.
Desnick RJ, Brady RO . Fabry disease in childhood. J Pediatr 2004; 144: 20–26.
Lin HY, Huang CH, Yu HC, Chong KW, Hsu JH, Lee PC et al. Enzyme assay and clinical assessment in subjects with a Chinese hotspot late-onset Fabry mutation (IVS4 + 919G→A). J Inherit Metab Dis 2010; 33: 619–624.
Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U et al. Fabry Registry. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab 2008; 93: 112–128.
Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U et al. Fabry disease: a review of current management strategies. QJM 2010; 103: 641–659.
Nakamura K, Hattori K, Endo F . Newborn screening for lysosomal storage disorders. Am J Med Genet C Semin Med Genet 2011; 157C: 63–71.
Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci USA 2008; 105: 2812–2817.
Maruyama H, Takata T, Tsubata Y, Tazawa R, Goto K, Tohyama J et al. Screening of male dialysis patients for Fabry disease by plasma globotriaosylsphingosine. Clin J Am Soc Nephrol 2013; 8: 629–636.
Niemann M, Rolfs A, Störk S, Bijnens B, Breunig F, Beer M et al. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet 2014; 7: 8–16.
Chamoles NA, Blanco M, Gaggioli D . Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta 2001; 308: 195–196.
Inoue T, Hattori K, Ihara K, Ishii A, Nakamura K, Hirose S . Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J Hum Genet 2013; 58: 548–552.
Nakamura K, Sekijima Y, Nakamura K, Hattori K, Nagamatsu K, Shimizu Y et al. Cerebral hemorrhage in Fabry’s disease. J Hum Genet 2010; 55: 259–261.
Krüger R, Tholey A, Jakoby T, Vogelsberger R, Mönnikes R, Rossmann H et al. Quantification of the Fabry marker lysoGb3 in human plasma by tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 2012; 883–884: 128–135.
Gal A, Hughes DA, Winchester B . Toward a consensus in the laboratory diagnostics of Fabry disease—recommendations of a European expert group. J Inherit Metab Dis 2011; 34: 509–514.
Togawa T, Tsukimura T, Kodama T, Tanaka T, Kawashima I, Saito S et al. Fabry disease: biochemical, pathological and structural studies of the α-galactosidase A with E66Q amino acid substitution. Mol Genet Metab 2012; 105: 615–620.
Eng CM, Niehaus DJ, Enriquez AL, Burgert TS, Ludman MD, Desnick RJ . Fabry disease: twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase A gene. Hum Mol Genet 1994; 3: 1795–1799.
Ashton-Prolla P, Tong B, Shabbeer J, Astrin KH, Eng CM, Desnick RJ . Fabry disease: twenty-two novel mutations in the alpha-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes. J Investig Med 2000; 48: 227–235.
Shabbeer J, Robinson M, Desnick RJ . Detection of alpha-galactosidase a mutations causing Fabry disease by denaturing high performance liquid chromatography. Hum Mutat 2005; 25: 299–305.
Liao HC, Huang YH, Chen YJ, Kao SM, Lin HY, Huang CK et al. Plasma globotriaosylsphingosine (lysoGb3) could be a biomarker for Fabry disease with a Chinese hotspot late-onset mutation (IVS4+919G>A). Clin Chim Acta 2013; 426: 114–120.
Data Citations
Chinen, Yasutsugu HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.932
Acknowledgements
We thank Ryuji Tasaki of the Chemo-Sero-therapeutic Research Institute (Kumamoto, Japan) for laboratory assistance and Satoshi Ishii of GlycoPharma Corporation (Oita, Japan) for determining plasma lyso-Gb3 concentrations. We are indebted to the participants, their parents, and the nurses and physicians who supported this study. This work was supported by a grant-in-aid for research on rare and intractable diseases, Health and Labour Sciences Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Chinen, Y., Nakamura, S., Yoshida, T. et al. A new mutation found in newborn screening for Fabry disease evaluated by plasma globotriaosylsphingosine levels. Hum Genome Var 4, 17002 (2017). https://doi.org/10.1038/hgv.2017.2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.2
- Springer Nature Limited