

Abstract
Hookworms infect over 400 million people, stunting and impoverishing them1,2,3. Sequencing hookworm genomes and finding which genes they express during infection should help in devising new drugs or vaccines against hookworms4,5. Unlike other hookworms, Ancylostoma ceylanicum infects both humans and other mammals, providing a laboratory model for hookworm disease6,7. We determined an A. ceylanicum genome sequence of 313 Mb, with transcriptomic data throughout infection showing expression of 30,738 genes. Approximately 900 genes were upregulated during early infection in vivo, including ASPRs, a cryptic subfamily of activation-associated secreted proteins (ASPs)8. Genes downregulated during early infection included ion channels and G protein–coupled receptors; this downregulation was observed in both parasitic and free-living nematodes. Later, at the onset of heavy blood feeding, C-lectin genes were upregulated along with genes for secreted clade V proteins (SCVPs), encoding a previously undescribed protein family. These findings provide new drug and vaccine targets and should help elucidate hookworm pathogenesis.
Similar content being viewed by others
Main
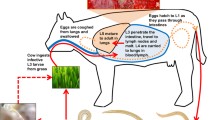
The two hookworm species causing the most infections are Necator americanus and Ancylostoma duodenale, which are generally restricted to human hosts1,9. Hookworms are free living during part of their life cycle, with eggs hatching in soil and larvae feeding on bacteria through the first and second larval stages. At the infectious third-stage larval phase (L3i), hookworms cease feeding and wait until they encounter a human host. They generally enter their host by burrowing into skin, although Ancylostoma can alternatively enter by being swallowed. Hookworms then pass through the bloodstream, lungs and digestive tract to the small intestine, where they affix themselves, mature to adulthood, mate and lay eggs that are excreted by the host1. The ability to culture A. ceylanicum in golden hamster allows it to be used as a model system for the human-specific hookworms N. americanus and A. duodenale, upon which new drug and vaccine candidates can be tested (Fig. 1)6,10,11. Human-specific hookworms belong to a class of parasitic nematodes, strongylids, that are more closely related to the free-living Caenorhabditis elegans than is the free-living Pristionchus pacificus (Fig. 2)12,13,14,15. Treatments effective against A. ceylanicum might thus also prove useful against other strongylids, such as Haemonchus contortus, that infect farm animals and depress agricultural productivity16. Characterizing the genome and transcriptome of A. ceylanicum is a key step toward such comparative analysis.

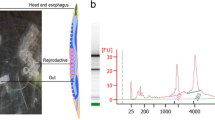
A. ceylanicum hatch in feces and grow as free-living first- to third-stage (L1–L3) larvae. Before exiting the third larval stage, they mature into infectious third-stage (L3i) larvae, arresting further development until they are inside a host. In 24 h after gavage into golden hamsters, A. ceylanicum are still in the stomach but have exited the L3i stage (24.PI). A standard model for parasite infection is to incubate L3i larvae for 24 h in hookworm culture medium (24.HCM), which evokes changes in larval shape and behavior thought to mimic those of 24.PI larvae in vivo. By 5 d after infection, larvae have migrated further to the intestine, affixed themselves there and grown into early fourth-stage (L4) larvae (5.D; female shown) with visible sexual differentiation. By 12 d (12.D; female shown), they start heavy blood feeding and become young adults with mature males and a few gravid females, with little or no egg laying. By 17 d (17.D; male shown), they are fully mature adults. They begin laying many eggs that are deposited outside the host during defecation, renewing the life cycle. From 19 d onward (19.D; male shown), they remain fertile adults for weeks in hamsters. Scale bars: 100 μm (L3i through 5.D), 500 μm (12.D) and 1 mm (17.D and 19.D).

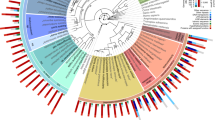
The phylogeny is derived from van Megen et al.13 and Kiontke et al.14. N. americanus and H. contortus are strongylid parasites15 and the closest relatives of A. ceylanicum. C. elegans, C. briggsae and P. pacificus are free-living, non-parasitic nematodes. Nematodes from distinct groups (clades)12 within the phylum are color-coded: black, A. ceylanicum and close relatives, clade V; green, plant parasites, clade IV; pink, ascarid and filarial animal parasites, clade III; orange, Trichinella, an animal parasite from clade I. To the right are the numbers of strictly orthologous genes for A. ceylanicum or C. elegans and other species. Self-comparisons (bold) list all strictly defined orthologs within a genome. A. ceylanicum and C. elegans have similar orthology to diverse nematode species.
We assembled an initial A. ceylanicum genome sequence of 313 Mb and a scaffold N50 of 668 kb, estimated to cover ∼95% of the genome, with Illumina sequencing and RNA scaffolding17,18 (Supplementary Tables 1, 2, 3). The genome size was comparable to those of Ancylostoma caninum (347 Mb)19 and H. contortus (320–370 Mb)20,21 but larger than those of N. americanus, C. elegans and P. pacificus (100–244 Mb)22,23,24. We found that 40.5% of the genomic DNA was repetitive, twice as much as in N. americanus, C. elegans or P. pacificus (17–24%). We predicted 26,966 protein-coding genes25 with products of ≥100 residues (Supplementary Table 4). We also predicted 10,050 genes with products of 30–99 residues, to uncover smaller proteins that might aid in parasitism26. With RNA sequencing (RNA-seq), we detected expression of 23,855 (88.5%) and 6,883 (68.5%) of these genes, respectively (Fig. 3).

Gene activity during infection is shown in log2-transformed transcripts per million (TPM), with k partitioning of the genes into 20 groups. Genes in yellow and blue are up- and downregulated, respectively; TPM values are shown ranging from ≤2−3 to ≥23. Developmental stages are as in Figure 1. Changes in gene expression after 24 h of growth in HCM (24.HCM) are relatively minor, as opposed to the far-reaching changes in gene expression seen after 24 h of infection in vivo (24.PI).
The genomes of plant-parasitic, necromenic and animal-parasitic nematodes have all acquired bacterial genes through horizontal gene transfer (HGT)27,28. We detected one instance of bacterial HGT in A. ceylanicum: Acey_s0012.g1873, a homolog of the N-acetylmuramoyl-L-alanine amidase amiD, which encodes a protein that may help bacteria recycle their murein29. Acey_s0012.g1873 was strongly expressed in L3i and then downregulated in all later stages of infection. It has nine predicted introns, presumably acquired after HGT; it has only one homolog in the entire nematode phylum (NECAME_15163 from N. americanus) but many bacterial homologs (Supplementary Fig. 1 and Supplementary Table 5). The sap-feeding insects Acyrthosiphon pisum and Planococcus citri also have amiD genes, acquired by HGT, that may promote bacterial lysis30,31.
To find genes acting at specific points of infection, we carried out RNA-seq on specimens collected at developmental stages spanning the onset and establishment of infection by A. ceylanicum in golden hamster (Figs. 1 and 3, and Supplementary Table 6), beginning at L3i and followed by 24 h either of incubation in hookworm culture medium (24.HCM), a standard model for early hookworm infection32, or infection in the hamster stomach (24.PI). We found 942 genes to be significantly upregulated from L3i after 24 h of infection in vivo (Supplementary Table 7). In contrast, we observed only 240 genes significantly upregulated from L3i after 24 h of incubation in HCM, of which 141 were also upregulated with in vivo infection. This lower number matches previous observations32 and shows that infection in vivo has stronger effects on gene activity than its in vitro model.
We linked known or probable gene functions to steps of infection by assigning gene ontology (GO) terms to A. ceylanicum genes33 and computing which GO terms were over-represented among genes upregulated or downregulated in developmental transitions (Supplementary Tables 8 and 9)34. We also analyzed homologous gene families for disproportionate upregulation or downregulation; in particular, gene families identified by orthology of A. ceylanicum with N. americanus or other nematodes might encode previously undescribed components of infection (Supplementary Table 10).
Proteases, protease inhibitors, nucleases and protein synthesis were upregulated during early infection (L3i to 24.PI; Supplementary Tables 9a and 11a); proteases and protease inhibitors were also upregulated after L3i in N. americanus24, as were proteases in H. contortus21. Secreted proteases could allow hookworms to digest host proteins in blood and intestinal mucosa6,11,35,36,37. Secreted proteases might also digest and inactivate proteins of the host's immune system37,38. Conversely, secreted protease inhibitors could also suppress host immunity39,40,41.
G protein–coupled receptors (GPCRs), receptor-gated ion channels and neurotransmission-related functions in general were downregulated during early infection (L3i to 24.PI), along with transcription factors (Supplementary Tables 9b and 11b). We observed the same pattern among genes downregulated in the transition from L3 to fourth-stage (L4) larvae both in H. contortus21 and C. elegans42 (Supplementary Table 8). This finding is consistent with downregulation after L3 of sensory perception and transcription genes in both C. elegans43 and N. americanus24 and of ion channel genes in A. caninum and Brugia malayi32,44. Such downregulation might thus be conserved in both parasitic and free-living nematodes.
Among gene families upregulated during early infection, we found some already known from other parasitic nematodes, such as ASPs (Supplementary Table 12a)21,24,45,46. ASP genes encode a diverse set of secreted cysteine-rich proteins, whose functions probably include blocking immune responses and blood clotting8. However, we also found a family of 92 genes collectively upregulated during early infection in vivo (24.PI; q value = 0.003) that had no obvious similarity to known gene families (Supplementary Tables 4 and 12a). By contrast, upregulation of these genes after 24 h of simulated infection in vitro was insignificant (24.HCM; q value = 0.93). These homologs were distantly related to ASPs, so we termed them ASP-related genes (ASPRs; Fig. 4 and Supplementary Fig. 2). We found other ASPRs in some strongylids (for example, N. americanus; Supplementary Tables 13 and 14) but not all (for example, H. contortus). Most ASPR proteins were predicted to be secreted (Supplementary Table 4), and one ASPR in Heligmosomoides bakeri is secreted by parasitic adults46. Thus, like ASPs, ASPRs might comprise an important element of hookworm infection in vivo.

The tree shows a maximum-likelihood phylogeny of protein domains rather than full-length proteins at the tips (as ASP genes sometimes encode two or more tandem ASP domains). All ASP domains and most ASPR domains are from A. ceylanicum or N. americanus. Almost all domains from ASPRs fall within a single branch, labeled in blue. ASPR genes are labeled blue (A. ceylanicum), green (N. americanus), purple (Oesophagostomum dentatum) or magenta (Heligmosomoides bakeri). ASP domains from orthologs of known ASP genes are labeled in gold, with their branches. N- and C-terminal domains from two-domain proteins are noted as “N” or “C.” Domains from other, less familiar ASP genes are labeled in gray. Confidence values are given as decimal fractions (Supplementary Fig. 2). Identities of the corresponding genes and domains are given in Supplementary Tables 4, 13 and 14.
A. ceylanicum had 432 ASP genes, noticeably more than the related parasites N. americanus (128 genes) and H. contortus (161 genes) and remarkably more than the non-parasitic C. elegans and P. pacificus (35 and 33 genes, respectively). A. ceylanicum and N. americanus also had 92 and 25 ASPR genes, respectively, which were missing entirely from the other species. One explanation for this diversity is the 'gray pawn' hypothesis: members of a large gene family might have little individual effect on phenotypic fitness yet be collectively needed for robust fitness under variable conditions47. For parasites, a relevant variable condition might be diverse host immune systems, which might favor continually diversifying sequences and expression profiles of ASPs and ASPRs.
For development from 24 h to 5 d after infection (24.PI to 5.D), genes encoding structural components of cuticle and genes whose products bind cytoskeletal proteins such as actin were prominently upregulated (Supplementary Table 11e). This period in the life cycle corresponds with the start of parasitic feeding, molting into L4 larvae and overt sexual differentiation (Fig. 1)6,10. We also observed a new protein family upregulated at this stage, with homologs in the strongylids A. ceylanicum, N. americanus, H. contortus and Angiostrongylus cantonensis (Supplementary Fig. 3 and Supplementary Tables 4, 12b and 15); the corresponding genes in A. cantonensis are expressed in L4 larvae infecting brain tissue48. We thus named this family strongylid L4 proteins (SL4Ps). In A. ceylanicum, 24 SL4P genes encoded proteins of ∼200 residues, of which 21 were predicted to be non-classically secreted49 without a leader sequence (Supplementary Table 16); notably, parasitic nematodes often use non-classical rather than classical secretion to export proteins into their hosts50.
From 5 to 12 d after infection (5.D to 12.D), genes encoding protein tyrosine phosphatases, serine/threonine kinases and C-lectins were prominently upregulated (Supplementary Tables 11g and 12c). This period in the life cycle corresponds with maturation from late-L4 larvae to young adults with incipient fertility and the onset of heavy blood feeding, which exposes A. ceylanicum to the host's immune system (Fig. 1)10,11. Among 22 C-lectin genes upregulated by 12 d, we detected 6 whose products had greater apparent similarity to mammalian than to nematode lectins (Supplementary Tables 4 and 17). Two of these genes encoded structural mimics of mammalian mannose receptor51, with five tandem C-lectin domains that had arisen through intragenic duplication (Supplementary Fig. 4a). The other four C-lectin genes resembled mammalian asialoglycoprotein receptors and neurocans51 but arose phylogenetically from nematode lectins (Supplementary Fig. 4b,c). Lectin genes with similarities to mammalian rather than nematode lectins have also been observed in the parasitic nematodes Ascaris suum and Toxocara canis and might help suppress host immune responses52,53.
We also observed a previously undescribed gene family upregulated at 12 d after infection, with members not only in strongylid parasites (A. ceylanicum, N. americanus, H. contortus and Heterorhabditis bacteriophora) but also in related non-parasitic clade V species (C. elegans, Caenorhabditis briggsae and P. pacificus; Fig. 5, Supplementary Fig. 5 and Supplementary Tables 12c and 18). We thus named this family secreted clade V proteins (SCVPs). In A. ceylanicum, 53 SCVP genes encoded ∼150-residue proteins, of which 48 were predicted to be classically secreted (Supplementary Table 4). Whereas N. americanus and H. contortus had 11 to 101 SCVP genes, other nematodes had only 1 to 6, suggesting an expansion of SCVP genes in mammalian-parasitic nematodes analogous to those observed for ASP and ASPR genes.

A maximum-likelihood phylogeny of SCVPs (Supplementary Fig. 5 and Supplementary Tables 4 and 18) is shown. Species are indicated by color: the hookworms A. ceylanicum and N. americanus are shown in green and olive green, respectively; H. contortus is shown in orange; the free-living Caenorhabditis nematodes (C. elegans and C. briggsae) and P. pacificus are shown in blue and light blue; and H. bacteriophora, an insect parasite, is shown in purple. Confidence values are given as decimal fractions (Supplementary Fig. 5b). The SCVP phylogeny falls into five branches: two large, independent gene expansions in hookworms (green); two more branches in H. contortus (orange); and one small branch for non-parasitic nematodes (blue). Like ASPs, SCVPs appear to have existed as a small gene family in free-living nematodes but then to have expanded greatly in both hookworms and other mammalian parasites.
A key motivation for parasite genomics is to identify targets for drugs or vaccines. Because drug development often fails54, it is essential to identify as many targets as possible. Four drug targets (adenylosuccinate lyase, carnitine O-palmitoyltransferase, dTDP-4-dehydrorhamnose 3,5-epimerase and trehalose-6-phosphatase) have recently been identified in H. contortus and N. americanus20,24,55,56,57,58,59. All four are encoded by genes with A. ceylanicum orthologs (Supplementary Table 4). To identify additional drug targets across the genome, we searched for genes that were conserved by diverse parasites but absent from mammals, might be essential for survival in the host (determined on the basis of C. elegans loss-of-function phenotypes), had homologs with known three-dimensional protein structures and had at least one homolog bound by a known small molecule (Supplementary Fig. 6). This screen yielded 72 genes in A. ceylanicum, one of which (Acey_s0015.g2804) encoded trehalose-6-phosphatase (Table 1 and Supplementary Tables 4, 19 and 20).
Vaccine targets should be both immunologically accessible and crucial for survival. Proteases meet these requirements, as they are expressed in the intestine (and thus exposed to the host's immune system) and because, without them, hookworms cannot digest host proteins such as hemoglobin36. We thus selected genes encoding proteases that were permanently upregulated by 5 d after infection and that lacked mammalian orthologs but had H. contortus homologs that are also upregulated during infection21. This screen yielded 12 cathepsin B–like protease genes, with 4 orthologs in H. contortus; by 19 d after infection, 5 of these 12 genes generated 1% of all transcripts (Supplementary Table 4). Because protease inhibitors were also upregulated during early infection, we searched for ones meeting our criteria; this screen yielded a previously undescribed protease inhibitor predicted to be a 79-residue secreted protein with consistently strong expression (∼0.1% of all adult transcripts) and one H. contortus homolog upregulated during infection.
The sequencing of A. ceylanicum adds to a growing number of genomes for parasitic nematodes that, collectively, infect over 1 billion humans60. Practically, these genomes will be crucial for inventing new drugs and vaccines against nematodes that rapidly evolve drug resistance61 and that have been parasitizing vertebrates since the Cretaceous62. Understanding immunosuppression by parasitic nematodes might also help alleviate autoimmune disorders, which may be partly due to improved hygiene ridding humans of chronic worm infections63. Intellectually, understanding these genomes may illuminate remarkable evolutionary changes. Parasitism allows adult nematodes to grow larger and live longer than their free-living relatives (N. americanus adults are ∼1 cm long and live for 3–10 years, whereas C. elegans adults are ∼1 mm long and live for 3 weeks), but the genomic changes underlying these adaptations are essentially unknown1,64,65,66. The genome and transcriptome of A. ceylanicum should provide lasting benefits for biology and medicine.
Methods
General summary.
Culture and infection of A. ceylanicum in golden hamster (Mesocricetus auratus) were carried out as described69. All housing and care of laboratory animals used in this study conformed with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals in Research (see 18-F22) and all requirements and all regulations issued by the US Department of Agriculture (USDA), including regulations implementing the Animal Welfare Act (Public Law 89-544, US Statutes at Large) as amended (see 18-F23). Stages of A. ceylanicum selected for developmental RNA-seq are shown in Figure 1 and listed in Supplementary Table 6; they are based on previously described stages of growth in golden hamster10.
Genomic sequencing and RNA-seq were carried out largely as described70. The numbers of A. ceylanicum and hamsters used for A. ceylanicum RNA-seq are listed in Supplementary Table 21. The A. ceylanicum genomic sequence was assembled from paired Illumina 100-nt reads (550 nt and 6 kb apart) with Velvet (1.2.05)18, gaps were closed after assembly with BGI GapCloser 1.12 (release_2011)71 and the sequence was reduced in possible heterozygosity72 with HaploMerger (20111230)73. Genomic RNA scaffolding was performed by filtering RNA-seq reads with khmer74 and then scaffolding with ERANGE (3.2)17. RNA-seq reads were assembled into cDNA with Oases (0.2.07)75. Assembled cDNAs (Supplementary Table 2) were used both to assess genome completeness and to aid in the prediction of protein-coding genes. The true genomic size of A. ceylanicum was estimated by counting 31-mer frequencies with SOAPdenovo (V1.05)71, by CEGMA (v2.4.010312) (Supplementary Table 3)76 and by mapping cDNAs to genomic DNA with BLAT (v. 34)77. Repetitive DNA elements in the final genome assembly were identified with RepeatScout (1.0.5)78.
We predicted protein-coding genes for our final genomic assembly with AUGUSTUS (2.6.1)25, after generating species-specific parameters with one round of MAKER2 (2.26-beta)79 (see URLs for the protocol by S. Kumar for running MAKER2) and using hints from cDNA that had been mapped to the genome assembly with BLAT. For predicted A. ceylanicum proteins, we annotated signal and transmembrane sequences with Phobius80, low-complexity regions with SEG81, coiled-coil domains with NCoils82, Pfam 26.0 domains (from both Pfam-A and Pfam-B)83 with HMMER 3.0/hmmsearch84, InterPro domains with InterProScan (4.8)85 and GO terms with Blast2GO 2.5 (build 23092011)33 (see URLs for protocols for running Blast2GO and InterProScan). We also assigned GO terms to C. elegans and H. contortus genes with Blast2GO so that comparisons of GO terms between different nematode species would be based on equivalent GO term assignments. We performed InterProScan and Blast2GO for both A. ceylanicum and C. elegans. We computed orthologies with OrthoMCL (1.3)86. Strict orthologies between genes from two or more species were defined as those orthology groups that contained only one predicted gene for each of the species. Annotations for protein-coding genes are listed in Supplementary Table 4.
For RNA-seq analysis of C. elegans, we used published developmental data from the modENCODE consortium (Supplementary Table 22)42. For H. contortus, we used published developmental RNA-seq data21.
We mapped RNA-seq reads to genes with Bowtie 2 (ref. 87) and quantified gene expression with RSEM (1.2.0)88. For individual genes, we computed the significance for changes in gene activity between stages or biological conditions (Supplementary Tables 7 and 23) with NOISeq-sim (2.13)89. Because we had only one biological replicate per condition, we sampled five random subsets of RNA-seq data per condition to estimate the significance of changes in gene activity. For A. ceylanicum, C. elegans and H. contortus, we used FUNC 0.4.5 with Wilcoxon rank-sum statistics34 to compute which GO terms were significantly associated with genes up- or downregulated between developmental stages or environmental conditions (for example, changes of drug treatment). For A. ceylanicum, we also used rank-sum statistics to compute such associations for protein families.
For phylogenetic analyses, sequences homologous to a protein or single domain were extracted with psi-BLAST90 or HMMER/jackhmmer. Protein sequences were aligned with MUSCLE (3.8.31)91 or MAFFT (v7.158b)92; alignments were edited with Trimal (v1.4.rev15)93 and visualized with JalView (2.8)94. Protein maximum-likelihood phylogenies and their branch confidence levels were computed with FastTree (2.1.7)95 and visualized with FigTree 1.4 (see URLs).
Some details of these methods are provided below; considerably more extensive details are provided in the Supplementary Note.
Assessing the completeness of genomic DNA.
We estimated the assembly's completeness as 98% by computing the frequencies of 31-mers71, as 91–99% by searching for conserved eukaryotic genes (Supplementary Table 3)76 and as 93% by mapping cDNA (assembled independently from RNA-seq reads) to genomic DNA: these calculations supported a consensus value of 95%. The average number of orthologs observed for full-length core eukaryotic genes76 was 1.13, which matched averages of 1.11–1.15 in C. elegans, C. briggsae and Caenorhabditis tropicalis (all of which are hermaphrodites and thus are completely homozygous), suggesting that the assembly was largely free of unresolved heterozygosity. We searched the genome for tRNA genes with tRNAscan-SE-1.3.1 (ref. 96); this analysis detected a full complement of 426 tRNAs decoding all 20 standard amino acids and one selenocysteine tRNA (Supplementary Table 24).
Examining repetitive elements for possible horizontal gene transfer.
In A. caninum, the repetitive element bandit resembles the HSMAR1 mariner-like transposon of humans and has been postulated to arise from a mammalian host by HGT97. To determine whether a bandit homolog also existed in A. ceylanicum, we searched our library of A. ceylanicum repetitive elements with the DNA sequence for bandit via BLASTN (2.2.26+)90 (arguments: “-task blastn -evalue 1e-03”). This analysis yielded two hits, with E values of 0.0 and 7 × 10−170 (Supplementary Table 25). Phylogenetic analysis (Supplementary Fig. 7) and domain analysis with HMMER/hmmsearch indicated that the higher-scoring hit represented an A. ceylanicum homolog of bandit, whereas the lower-scoring hit represented a partial homolog of bandit that did not encode a transposase domain (Transposase_1/PF01359.13 in Pfam).
To examine whether more evidence for lateral acquisition of repetitive elements existed in human hookworms, we used the DFAM database98 to identify repetitive DNA elements in A. ceylanicum and N. americanus with similarity to human repetitive elements. This analysis identified two classes of elements with mammalian similarities, L3/Plat_L3-like retrotransposons and HSMAR1/2-like mariner elements (Supplementary Table 25). To determine whether these similarities were adventitious or real, we computed maximum-likelihood phylogenies for reverse-transcriptase domains (for L3/Plat_L3-like elements) and transposase domains (for HSMAR1/2-like elements). These phylogenies included all of the L3/Plat_L3-like and HSMAR1/2-like repetitive elements that we could detect in A. ceylanicum and N. americanus, in a diverse set of other published genome sequences from nematodes, vertebrates, arthropods, lophotrochozoans and deuterostomes (Supplementary Table 26) and in a curated collection of eukaryotic elements from RepBase99 (see URLs for source). We extracted well-aligned, full-length protein domains from repetitive elements by requiring that they match the Pfam domains Transposase_1/PF01359.13 (for HSMAR1/2-like elements) or RVT_1 (reverse transcriptase)/PF00078.22 (for L3/Plat_L3-like elements) and also by excluding the shortest 10% of domain matches. These criteria led us to select 988 Plat_L3/L3-like RVT_1/PF00078.22 peptides (Supplementary Table 27a) and 168 HSMAR1/2-like Transposase_1/PF01359.13 peptides (Supplementary Table 27b), which we subjected to multiple-sequence alignment and phylogenetic analysis.
Analyzing protein-coding genes.
For motif searches or OrthoMCL analyses of protein sequences, we used nematode and mammalian proteomes from genomic sequences and partial nematode proteomes from translated ESTs. All proteomes and their sources are listed in Supplementary Table 28. We classified A. ceylanicum, H. contortus and C. elegans genes both by known protein motifs (through HMMER 3.0/Pfam-A 26 and InterProScan 4.8)83,84,85 and evolutionary relationships to genes in different species (through OrthoMCL 1.3)86. Pfam-A domains were detected at a threshold of E ≤ 1 × 10−5; InterProScan and OrthoMCL were run with default parameters. We used Pfam-A and InterPro motifs, in turn, to assign GO terms to each gene with Blast2GO 2.5 (build 23092011)33. We performed InterProScan and Blast2GO according to available protocols (see URLs); for Blast2GO, we used both InterProScan predictions and BLASTP results against an animal-specific subset of the NCBI nr database (NCBI-nr)100. We computed orthology groups for our A. ceylanicum genes with OrthoMCL (1.3)86, for numbers of species ranging from 4 to 14 (Supplementary Tables 4 and 28). Strict orthologies between genes of two or more species were defined as those orthology groups that contained only one predicted gene for each of those species (Fig. 2). Strict orthologies allowed us to compare transcriptional profiles between A. ceylanicum and C. elegans and to thereby identify a set of 406 A. ceylanicum genes that were strongly expressed under all conditions for which we had RNA-seq data from either A. ceylanicum or C. elegans.
Searching for horizontal gene transfer of protein-coding genes.
To find possible cases of HGT of protein-coding genes from non-nematodes to A. ceylanicum, we used both orthologies (strict and non-strict) and Pfam-A domains (computed for all proteomes as with A. ceylanicum). Orthologies were considered to represent possible instances of HGT if they included A. ceylanicum, Homo sapiens and Mus musculus but did not include C. elegans, C. briggsae, P. pacificus, Bursaphelenchus xylophilus or Meloidogyne hapla. Sets of genes encoding a shared Pfam-A domain were likewise considered to contain possible instances of HGT if the domains were present in A. ceylanicum and mammals (at E ≤ 1 × 10−6) but absent in C. elegans, C. briggsae, P. pacificus, B. xylophilus and M. hapla (at E ≤ 1 × 10−5). Out of 33,243 orthology groups and 3,545 Pfam-A domains, we found 52 and 15 (respectively) that were instances of possible HGT. Each possible instance of HGT in A. ceylanicum was individually checked by BLASTP searches of NCBI-nr. In most cases, BLASTP showed similarities to C. elegans and other nematodes, which marked the putative HGTs as false positives. However, we also identified (through Pfam-A domains) one A. ceylanicum gene with strong similarity to bacterial amiD, Acey_s0012.g1873. To search for other such homologs, we reran our motif searches without the requirement for mammalian hits, but, on further testing with BLASTP against NCBI-nr, no other bacterial sequences were found.
Phylogenetic analysis of lectin homologs from metazoa.
In addition to the amiD homolog Acey_s0012.g1873, we also observed eight A. ceylanicum genes that were more similar to vertebrate lectins than to nematode ones (Supplementary Table 17): these fell into three classes, showing similarity to mannose receptor (MRC), asialoglycoprotein receptor (ASGR) or neurocan (NCAN). We phylogenetically compared their domains to nematode, arthropod, deuterostome and lophotrochozoan proteomes, along with a small number of added individual nematode lectins that had been characterized because of their previously reported similarities to mammalian proteins (species listed in Supplementary Table 29; sources of proteome sequences listed in Supplementary Table 28). To avoid misalignments and spurious similarities between multidomain proteins, we analyzed individual C-lectin domains rather than full-length lectin proteins; to identify coherent sets of homologs, we searched the custom proteome database with single-domain query sequences via psi-BLAST (2.2.26+)90,101, run for either three or four rounds at an inclusion threshold of E ≤ 1 × 10−20. The query sequences used, with the corresponding numbers of psi-BLAST rounds, are listed in Supplementary Table 30. The resulting single-domain matches were realigned with MUSCLE (3.8.31) and phylogenetically analyzed as above. For each lectin class, the sequences in each resulting phylogeny are listed in Supplementary Table 31.
Phylogenetic analysis of amiD homologs from metazoa and bacteria.
We first characterized non-bacterial and bacterial homologs of Acey_s0012.g1873 with BLASTP of NCBI-nr. This analysis yielded matches to sequences from the hookworms A. ceylanicum (our own data, deposited into GenBank) and N. americanus; it also gave nine matches to non-bacterial sequences from arthropods and basal animals (Supplementary Table 32). To more rigorously determine the phylogenetic origin of the amiD genes in the hookworms A. ceylanicum and N. americanus, we generated a phylogeny for the entire Amidase_2 superfamily (N-acetylmuramoyl-L-alanine amidase; PFAM 27.0 motif PF01510.20), of which bacterial amiD genes represent one of four major subdivisions102. We searched all of the proteomes listed in Supplementary Table 29, along with all of the individual metazoan amiD homologs listed in Supplementary Table 32 and more proteomes from arthropods, two different metagenomes from human stool and cow rumen and the entire 9 July 2014 release of UniProt103. Species and data sources for additional proteomes are listed in Supplementary Table 33; source files are listed in Supplementary Table 28. We extracted subsequences matching the Amidase_2/PF01510.20 domain, realigned them with MAFFT v7.158b and phylogenetically analyzed them as above.
URLs.
FigTree, http://tree.bio.ed.ac.uk/software/figtree/; Gene Ontology term tables, http://archive.geneontology.org/full/; modENCODE, http://www.modencode.org/; NCoils, http://www.russell.embl-heidelberg.de/coils/coils.tar.gz; protocols by S. Kumar for running Blast2GO, InterProScan and MAKER2, https://github.com/sujaikumar/assemblage/blob/master/README-annotation.md; RepBase, http://www.girinst.org/server/RepBase/protected/RepBase19.02.fasta.tar.gz.
Accession codes.
All assemblies and raw reads have been deposited at NCBI. The corresponding NCBI accession numbers are as follows: A. ceylanicum genome assembly, JARK00000000; A. ceylanicum cDNA assembly, GASF00000000; A. ceylanicum genomic DNA reads, SRR1124846 and SRR1124848; A. ceylanicum RNA-seq reads, SRR1124849, SRR1124850, SRR1124900, SRR1124905, SRR1124906, SRR1124907, SRR1124908, SRR1124909, SRR1124910, SRR1124911, SRR1124912, SRR1124913, SRR1124914, SRR1124985 and SRR1124986; C. elegans RNA-seq reads, SRR1125007 and SRR1125008. In addition, we have deposited the genome sequence and gene predictions at WormBase (in archival release WS245).
Accession codes
Primary accessions
NCBI Reference Sequence
Sequence Read Archive
Change history
05 May 2015
In the version of this article initially published, the following two sentences were omitted from the Acknowledgments: "Sequencing was carried out at the Millard and Muriel Jacobs Genome Facility at the California Institute of Technology. This work was supported by US National Institutes of Health grants to P.W.S. (GM084389) and to R.V.A. (AI056189), by Cornell University salary and start-up funds to E.M.S. and by the Howard Hughes Medical Institute to P.W.S." The error has been corrected in the HTML and PDF versions of the article.
References
Brooker, S., Bethony, J. & Hotez, P.J. Human hookworm infection in the 21st century. Adv. Parasitol. 58, 197–288 (2004).
Vos, T. et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2163–2196 (2012).
Pullan, R.L., Smith, J.L., Jasrasaria, R. & Brooker, S.J. Global numbers of infection and disease burden of soil transmitted helminth infections in 2010. Parasit. Vectors 7, 37 (2014).
Keiser, J. & Utzinger, J. The drugs we have and the drugs we need against major helminth infections. Adv. Parasitol. 73, 197–230 (2010).
Schneider, B. et al. A history of hookworm vaccine development. Hum. Vaccin. 7, 1234–1244 (2011).
Garside, P. & Behnke, J.M. Ancylostoma ceylanicum in the hamster: observations on the host-parasite relationship during primary infection. Parasitology 98, 283–289 (1989).
Traub, R.J. Ancylostoma ceylanicum, a re-emerging but neglected parasitic zoonosis. Int. J. Parasitol. 43, 1009–1015 (2013).
Cantacessi, C. et al. A portrait of the “SCP/TAPS” proteins of eukaryotes—developing a framework for fundamental research and biotechnological outcomes. Biotechnol. Adv. 27, 376–388 (2009).
Jian, X. et al. Necator americanus: maintenance through one hundred generations in golden hamsters (Mesocricetus auratus). II. Morphological development of the adult and its comparison with humans. Exp. Parasitol. 105, 192–200 (2003).
Ray, D.K., Bhopale, K.K. & Shrivastava, V.B. Migration and growth of Ancylostoma ceylanicum in golden hamsters Mesocricetus auratus. J. Helminthol. 46, 357–362 (1972).
Menon, S. & Bhopale, M.K. Ancylostoma ceylanicum (Looss, 1911) in golden hamsters (Mesocricetus auratus): pathogenicity and humoral immune response to a primary infection. J. Helminthol. 59, 143–146 (1985).
Blaxter, M. Nematodes: the worm and its relatives. PLoS Biol. 9, e1001050 (2011).
van Megen, H. et al. A phylogenetic tree of nematodes based on about 1200 full-length small subunit ribosomal DNA sequences. Nematology 11, 927–950 (2009).
Kiontke, K.C. et al. A phylogeny and molecular barcodes for Caenorhabditis, with numerous new species from rotting fruits. BMC Evol. Biol. 11, 339 (2011).
Chilton, N.B., Huby-Chilton, F., Gasser, R.B. & Beveridge, I. The evolutionary origins of nematodes within the order Strongylida are related to predilection sites within hosts. Mol. Phylogenet. Evol. 40, 118–128 (2006).
Kaplan, R.M. Drug resistance in nematodes of veterinary importance: a status report. Trends Parasitol. 20, 477–481 (2004).
Mortazavi, A. et al. Scaffolding a Caenorhabditis nematode genome with RNA-seq. Genome Res. 20, 1740–1747 (2010).
Zerbino, D.R. & Birney, E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Abubucker, S. et al. The canine hookworm genome: analysis and classification of Ancylostoma caninum survey sequences. Mol. Biochem. Parasitol. 157, 187–192 (2008).
Laing, R. et al. The genome and transcriptome of Haemonchus contortus, a key model parasite for drug and vaccine discovery. Genome Biol. 14, R88 (2013).
Schwarz, E.M. et al. The genome and developmental transcriptome of the strongylid nematode Haemonchus contortus. Genome Biol. 14, R89 (2013).
Stein, L.D. et al. The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol. 1, E45 (2003).
Dieterich, C. et al. The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nat. Genet. 40, 1193–1198 (2008).
Tang, Y.T. et al. Genome of the human hookworm Necator americanus. Nat. Genet. 46, 261–269 (2014).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Raffaele, S. & Kamoun, S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat. Rev. Microbiol. 10, 417–430 (2012).
Danchin, E.G. & Rosso, M.N. Lateral gene transfers have polished animal genomes: lessons from nematodes. Front. Cell. Infect. Microbiol. 2, 27 (2012).
Wu, B. et al. Interdomain lateral gene transfer of an essential ferrochelatase gene in human parasitic nematodes. Proc. Natl. Acad. Sci. USA 110, 7748–7753 (2013).
Uehara, T. & Park, J.T. An anhydro-N-acetylmuramyl-L-alanine amidase with broad specificity tethered to the outer membrane of Escherichia coli. J. Bacteriol. 189, 5634–5641 (2007).
Nikoh, N. et al. Bacterial genes in the aphid genome: absence of functional gene transfer from Buchnera to its host. PLoS Genet. 6, e1000827 (2010).
Husnik, F. et al. Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153, 1567–1578 (2013).
Wang, Z. et al. Characterizing Ancylostoma caninum transcriptome and exploring nematode parasitic adaptation. BMC Genomics 11, 307 (2010).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 (2008).
Prüfer, K. et al. FUNC: a package for detecting significant associations between gene sets and ontological annotations. BMC Bioinformatics 8, 41 (2007).
Bansemir, A.D. & Sukhdeo, M.V. The food resource of adult Heligmosomoides polygyrus in the small intestine. J. Parasitol. 80, 24–28 (1994).
Ranjit, N. et al. Proteolytic degradation of hemoglobin in the intestine of the human hookworm Necator americanus. J. Infect. Dis. 199, 904–912 (2009).
Knox, D. in Parasitic Helminths: Targets, Screens, Drugs, and Vaccines (ed. Caffrey, C.R.) 399–420 (Wiley-VCH Verlag & Co., 2012).
Pearson, M.S. et al. Molecular mechanisms of hookworm disease: stealth, virulence, and vaccines. J. Allergy Clin. Immunol. 130, 13–21 (2012).
Klotz, C. et al. A helminth immunomodulator exploits host signaling events to regulate cytokine production in macrophages. PLoS Pathog. 7, e1001248 (2011).
Hartmann, S. & Lucius, R. Modulation of host immune responses by nematode cystatins. Int. J. Parasitol. 33, 1291–1302 (2003).
Manoury, B., Gregory, W.F., Maizels, R.M. & Watts, C. Bm-CPI-2, a cystatin homolog secreted by the filarial parasite Brugia malayi, inhibits class II MHC–restricted antigen processing. Curr. Biol. 11, 447–451 (2001).
Gerstein, M.B. et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330, 1775–1787 (2010).
Kim, D., Grun, D. & van Oudenaarden, A. Dampening of expression oscillations by synchronous regulation of a microRNA and its target. Nat. Genet. 45, 1337–1344 (2013).
Choi, Y.J. et al. A deep sequencing approach to comparatively analyze the transcriptome of lifecycle stages of the filarial worm, Brugia malayi. PLoS Negl. Trop. Dis. 5, e1409 (2011).
Osman, A. et al. Hookworm SCP/TAPS protein structure—a key to understanding host-parasite interactions and developing new interventions. Biotechnol. Adv. 30, 652–657 (2012).
Hewitson, J.P. et al. Proteomic analysis of secretory products from the model gastrointestinal nematode Heligmosomoides polygyrus reveals dominance of venom allergen-like (VAL) proteins. J. Proteomics 74, 1573–1594 (2011).
Thomas, J.H. & Robertson, H.M. The Caenorhabditis chemoreceptor gene families. BMC Biol. 6, 42 (2008).
He, H. et al. Preliminary molecular characterization of the human pathogen Angiostrongylus cantonensis. BMC Mol. Biol. 10, 97 (2009).
Bendtsen, J.D., Jensen, L.J., Blom, N., Von Heijne, G. & Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 17, 349–356 (2004).
Borloo, J. et al. In-depth proteomic and glycomic analysis of the adult-stage Cooperia oncophora excretome/secretome. J. Proteome Res. 12, 3900–3911 (2013).
Zelensky, A.N. & Gready, J.E. The C-type lectin–like domain superfamily. FEBS J. 272, 6179–6217 (2005).
Yoshida, A., Nagayasu, E., Horii, Y. & Maruyama, H. A novel C-type lectin identified by EST analysis in tissue migratory larvae of Ascaris suum. Parasitol. Res. 110, 1583–1586 (2012).
Loukas, A., Doedens, A., Hintz, M. & Maizels, R.M. Identification of a new C-type lectin, TES-70, secreted by infective larvae of Toxocara canis, which binds to host ligands. Parasitology 121, 545–554 (2000).
Scannell, J.W., Blanckley, A., Boldon, H. & Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 11, 191–200 (2012).
Taylor, C.M. et al. Discovery of anthelmintic drug targets and drugs using chokepoints in nematode metabolic pathways. PLoS Pathog. 9, e1003505 (2013).
Fyfe, P.K., Dawson, A., Hutchison, M.T., Cameron, S. & Hunter, W.N. Structure of Staphylococcus aureus adenylosuccinate lyase (PurB) and assessment of its potential as a target for structure-based inhibitor discovery. Acta Crystallogr. D Biol. Crystallogr. 66, 881–888 (2010).
Ashrafian, H., Horowitz, J.D. & Frenneaux, M.P. Perhexiline. Cardiovasc. Drug Rev. 25, 76–97 (2007).
Sivendran, S. et al. Identification of triazinoindol-benzimidazolones as nanomolar inhibitors of the Mycobacterium tuberculosis enzyme TDP-6-deoxy-D-xylo-4-hexopyranosid-4-ulose 3,5-epimerase (RmlC). Bioorg. Med. Chem. 18, 896–908 (2010).
Farelli, J.D. et al. Structure of the trehalose-6-phosphate phosphatase from Brugia malayi reveals key design principles for anthelmintic drugs. PLoS Pathog. 10, e1004245 (2014).
Zarowiecki, M. & Berriman, M. What helminth genomes have taught us about parasite evolution. Parasitology 42 (suppl. 1), S85–S97 (2015).
Gilleard, J.S. Haemonchus contortus as a paradigm and model to study anthelmintic drug resistance. Parasitology 140, 1506–1522 (2013).
Durette-Desset, M.C., Beveridge, I. & Spratt, D.M. The origins and evolutionary expansion of the strongylida (Nematoda). Int. J. Parasitol. 24, 1139–1165 (1994).
McSorley, H.J. & Maizels, R.M. Helminth infections and host immune regulation. Clin. Microbiol. Rev. 25, 585–608 (2012).
Yeates, G.W. & Boag, B. Female size shows similar trends in all clades of the phylum Nematoda. Nematology 8, 111–127 (2006).
Gems, D. Longevity and ageing in parasitic and free-living nematodes. Biogerontology 1, 289–307 (2000).
Desjardins, C.A. et al. Genomics of Loa loa, a Wolbachia-free filarial parasite of humans. Nat. Genet. 45, 495–500 (2013).
Somvanshi, V.S., Ellis, B.L., Hu, Y. & Aroian, R.V. Nitazoxanide: nematicidal mode of action and drug combination studies. Mol. Biochem. Parasitol. 193, 1–8 (2014).
Crowther, G.J. et al. Cofactor-independent phosphoglycerate mutase from nematodes has limited druggability, as revealed by two high-throughput screens. PLoS Negl. Trop. Dis. 8, e2628 (2014).
Hu, Y. et al. Mechanistic and single-dose in vivo therapeutic studies of Cry5B anthelmintic action against hookworms. PLoS Negl. Trop. Dis. 6, e1900 (2012).
Srinivasan, J. et al. The draft genome and transcriptome of Panagrellus redivivus are shaped by the harsh demands of a free-living lifestyle. Genetics 193, 1279–1295 (2013).
Li, R. et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272 (2010).
Barrière, A. et al. Detecting heterozygosity in shotgun genome assemblies: lessons from obligately outcrossing nematodes. Genome Res. 19, 470–480 (2009).
Huang, S. et al. HaploMerger: reconstructing allelic relationships for polymorphic diploid genome assemblies. Genome Res. 22, 1581–1588 (2012).
Brown, C.T., Howe, A., Zhang, Q., Pyrkosz, A.B. & Brom, T.H. A single pass approach to reducing sampling variation, removing errors, and scaling de novo assembly of shotgun sequences. arXiv. http://arxiv.org/abs/1203.4802 (2012).
Schulz, M.H., Zerbino, D.R., Vingron, M. & Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092 (2012).
Parra, G., Bradnam, K., Ning, Z., Keane, T. & Korf, I. Assessing the gene space in draft genomes. Nucleic Acids Res. 37, 289–297 (2009).
Kent, W.J. BLAT—the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Price, A.L., Jones, N.C. & Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 21 (suppl. 1), i351–i358 (2005).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Käll, L., Krogh, A. & Sonnhammer, E.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 (2004).
Wootton, J.C. Non-globular domains in protein sequences: automated segmentation using complexity measures. Comput. Chem. 18, 269–285 (1994).
Lupas, A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 266, 513–525 (1996).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301 (2012).
Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009).
McDowall, J. & Hunter, S. InterPro protein classification. Methods Mol. Biol. 694, 37–47 (2011).
Li, L., Stoeckert, C.J. Jr. & Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Langmead, B. & Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, B. & Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Tarazona, S., Garcia-Alcalde, F., Dopazo, J., Ferrer, A. & Conesa, A. Differential expression in RNA-seq: a matter of depth. Genome Res. 21, 2213–2223 (2011).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Katoh, K. & Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutiérrez, S., Silla-Martinez, J.M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Waterhouse, A.M., Procter, J.B., Martin, D.M., Clamp, M. & Barton, G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Price, M.N., Dehal, P.S. & Arkin, A.P. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Lowe, T.M. & Eddy, S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Laha, T. et al. The bandit, a new DNA transposon from a hookworm-possible horizontal genetic transfer between host and parasite. PLoS Negl. Trop. Dis. 1, e35 (2007).
Wheeler, T.J. et al. Dfam: a database of repetitive DNA based on profile hidden Markov models. Nucleic Acids Res. 41, D70–D82 (2013).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467 (2005).
Sayers, E.W. et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 40, D13–D25 (2012).
Schäffer, A.A. et al. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005 (2001).
Firczuk, M. & Bochtler, M. Folds and activities of peptidoglycan amidases. FEMS Microbiol. Rev. 31, 676–691 (2007).
UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198 (2014).
Acknowledgements
We thank C.T. Brown, P.K. Korhonen, S. Kumar and J.E. Stajich for advice on bioinformatics, T.A. Aydin, J. Liu and A.H.K. Roeder for comments on the manuscript, L. Schaeffer and V. Kumar for genome and RNA sequencing, and C.T. Brown for use of the Michigan State University High-Performance Computing Center (supported by grant 2010-65205-20361 from the US Department of Agriculture and grant IOS-0923812 from the National Institute of Food and Agriculture and the National Science Foundation). Sequencing was carried out at the Millard and Muriel Jacobs Genome Facility at the California Institute of Technology. This work was supported by US National Institutes of Health grants to P.W.S. (GM084389) and to R.V.A. (AI056189), by Cornell University salary and start-up funds to E.M.S. and by the Howard Hughes Medical Institute to P.W.S.
Author information
Authors and Affiliations
Contributions
E.M.S., P.W.S. and R.V.A. conceived of and managed the project. Y.H. isolated genomic DNA and RNA from all infection stages of A. ceylanicum and converted RNA to cDNA for sequencing. M.M.M. maintained A. ceylanicum and golden hamster cultures through full life cycles and performed the photography for Figure 1. I.A. constructed large-insert paired-end and jumping Illumina libraries and supervised both genomic and RNA-seq Illumina sequencing. E.M.S. conducted all bioinformatics and biological analyses. Writing was primarily carried out by E.M.S. but with input from all authors.
Corresponding author
Ethics declarations
Competing interests
E.M.S., Y.H., I.A., P.W.S. and R.V.A. have submitted preliminary patent applications based on this work via the Office of Technology Transfer at the California Institute of Technology. The purpose of these patent applications was to enable technological development. Otherwise, the authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Note, Supplementary Figures 1–7 and Supplementary Tables 1–3, 7, 11–19, 21, 23–26, 28–30, 32 and 33. (PDF 8281 kb)
Supplementary Table 4
Protein-coding gene predictions and annotations. (XLSX 19390 kb)
Supplementary Table 5
Identities of the amiD homologs shown in Supplementary Figure 1. (XLSX 164 kb)
Supplementary Table 6
Genomic and RNA-seq Illumina sequencing libraries for A. ceylanicum. (XLSX 48 kb)
Supplementary Table 8
Lists of GO terms associated with developmental transitions or drug- versus control-induced conditions in A. ceylanicum, H. contortus and C. elegans. (XLSX 173 kb)
Supplementary Table 9
More-extended, stage-by-stage tables of GO terms associated with developmental transitions or drug- versus control-induced conditions in A. ceylanicum. (XLSX 122 kb)
Supplementary Table 10
Protein motifs and orthology groups associated with developmental transitions or drug- versus control-induced conditions in A. ceylanicum. (XLSX 113 kb)
Supplementary Table 20
Protein domains associated with drug targets in A. ceylanicum. (XLSX 61 kb)
Supplementary Table 22
Data sources of C. elegans RNA-seq reads from modENCODE. (XLSX 70 kb)
Supplementary Table 27
Identities, species, types and references of the repetitive elements shown in Supplementary Figure 7. (XLSX 84 kb)
Supplementary Table 31
Sequences used in each lectin phylogeny. (XLSX 61 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article

Cite this article
Schwarz, E., Hu, Y., Antoshechkin, I. et al. The genome and transcriptome of the zoonotic hookworm Ancylostoma ceylanicum identify infection-specific gene families. Nat Genet 47, 416–422 (2015). https://doi.org/10.1038/ng.3237
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3237
- Springer Nature America, Inc.
This article is cited by
-
The parasitic nematode Strongyloides ratti exists predominantly as populations of long-lived asexual lineages
Nature Communications (2023)
-
The community-curated Pristionchus pacificus genome facilitates automated gene annotation improvement in related nematodes
BMC Genomics (2021)
-
Dafachronic acid and temperature regulate canonical dauer pathways during Nippostrongylus brasiliensis infectious larvae activation
Parasites & Vectors (2020)
-
The algal selenoproteomes
BMC Genomics (2020)
-
Speciation and adaptive evolution reshape antioxidant enzymatic system diversity across the phylum Nematoda
BMC Biology (2020)