
Abstract
Metastatic cancer cells acquire energy-intensive processes including increased invasiveness and chemoresistance. However, how the energy demand is met and the molecular drivers that coordinate an increase in cellular metabolic activity to drive epithelial–mesenchymal transition (EMT), the first step of metastasis, remain unclear. Using different in vitro and in vivo EMT models with clinical patient’s samples, we showed that EMT is an energy-demanding process fueled by glucose metabolism-derived adenosine triphosphate (ATP). We identified angiopoietin-like 4 (ANGPTL4) as a key player that coordinates an increase in cellular energy flux crucial for EMT via an ANGPTL4/14-3-3γ signaling axis. This augmented cellular metabolic activity enhanced metastasis. ANGPTL4 knockdown suppresses an adenylate energy charge elevation, delaying EMT. Using an in vivo dual-inducible EMT model, we found that ANGPTL4 deficiency reduces cancer metastasis to the lung and liver. Unbiased kinase inhibitor screens and Ingenuity Pathway Analysis revealed that ANGPTL4 regulates the expression of 14-3-3γ adaptor protein via the phosphatidylinositol-3-kinase/AKT and mitogen-activated protein kinase signaling pathways that culminate to activation of transcription factors, CREB, cFOS and STAT3. Using a different mode of action, as compared with protein kinases, the ANGPTL4/14-3-3γ signaling axis consolidated cellular bioenergetics and stabilized critical EMT proteins to coordinate energy demand and enhanced EMT competency and metastasis, through interaction with specific phosphorylation signals on target proteins.
Similar content being viewed by others


Introduction
The exponential growth of the primary tumor exposes cancer cells to various microenvironmental stresses, including hypoxic, acidic and inflammatory milieus.1, 2 Interestingly, though these constraints can kill cancer cells, they also exert selective pressures on cells to exploit adverse microenvironments by modifying their cellular behavior and selecting for a highly invasive phenotype that facilitates metastasis.3 Metastasis, the spread of cancer cells to distant tissues and/or organs, is a complex and multistep process, making it a challenging therapeutic target.4 Metastasis accounts for >90% of cancer mortality.5 Initiation of the metastatic process is considered a rate-limiting event and resembles the epithelial–mesenchymal transition (EMT) that occurs during embryogenesis and wound healing.1, 2 Microenviromental stimuli can initiate the EMT process by controlling the expression and function of specific transcription factors, such as Snai1, Slug, Twist and ZEB-1.6 During EMT in cancer, epithelial cells lose their cell polarity and cell–cell adhesions are weakened as they adopt a mesenchymal phenotype. This change is accompanied by energy-demanding processes such as cytoskeletal remodeling and increased chemoresistance, ultimately allowing the cells to gain motility and the ability to invade distant organs.7 However, the mechanism that secures the energy supply for EMT competency remains unknown.
Our current understanding of cancer cell metabolism is based primarily on a comparison of metabolic status between primary tumors and normal healthy cells. Cancer cells derive a substantial amount of adenosine triphosphate (ATP) from aerobic glycolysis (that is, the Warburg effect) instead of oxidative phosphorylation to support their anabolic growth and proliferation.8 Cancer cells also exploit glutaminolytic flux, amino acid and lipid metabolism, mitochondrial biogenesis, the pentose phosphate pathway and macromolecular biosynthesis to progress malignancy.9, 10 Recent studies have suggested that oncogenes and tumor suppressors function as critical modulators of metabolic reprogramming to support tumor progression.11, 12 Hypoxia and transforming growth factor-β (TGF-β), which are known initiators of EMT, can also modulate cancer cell metabolism during tumorigenesis.3, 13 Thus, these microenvironmental signals may also alter cancer cell bioenergetics to increase their motility and enhance their invasive capabilities. Despite the significance of EMT in metastasis, little is known about the changes in cellular bioenergetics that occur during this process and whether a demand for metabolic energy is a functional prerequisite. Furthermore, the molecular drivers of metabolic reprogramming in EMT remain unknown.
Using three different in vitro and in vivo EMT models to compliment human cancer biopsies, we identified angiopoietin-like 4 (ANGPTL4) as a key player that adenylates energy charge and coordinates the energy demands required for EMT. We further revealed an ANGPTL4/14-3-3 signaling influences biological processes by protein–protein interactions.
Results
ANGPTL4 increases cellular bioenergetics needed for EMT competency
ANGPTL4 is best known for its role as an adipocytokine involved in systemic glucose and lipid metabolism.14 ANGPTL4 has been identified as a prometastatic gene that is involved in tumor growth, anoikis resistance, angiogenesis and tumor invasion.15 However, its involvement in EMT, particularly its function in energy homeostasis in the cellular level, is unknown. Herein, to examine changes in energy demand during metastasis, we first measured the energy charge and examined the expression of ANGPTL4 in human tumor biopsies. Clinical samples showed significant correlations between the energy charge status and tumor grades, with the higher- grade metastatic tumors exhibiting between 26.7% and 106% increases in energy charge compared with cognate stage I tumors (Figure 1a; Supplementary Table S1). The energy charge status of the cells is indicative of the cellular metabolic activity.16, 17, 18 Importantly, our analysis also revealed that human metastatic cancers expressed higher ANGPTL4 expression compared with their lower-grade counterparts, suggesting a role for ANGPTL4 in regulating cellular bioenergetics for metastasis (Figure 1b).

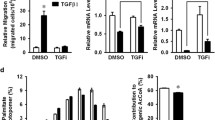
Increased cellular metabolic activity is essential for epithelial–mesenchymal transition. (a) Percentage increase in energy charge in breast, colorectal, gastric and head and neck tissues at various tumor stages. Comparison was made against respective stage I samples. (b) Immunoblot analysis of cANGPTL4 expression in patient biopsies at various tumor stages. (c) Immunofluorescence staining of E-cadherin (green) in hypoxia cells at the indicated time intervals. Cells were counterstained with DAPI (blue) for nuclei and phalloidin (red) for actin cytoskeleton. Scale bar=40 μm. (d) Relative mRNA expression of EMT markers (CDH1, DDR1, ERBB3, Snai1 and ZEB1) in hypoxia-exposed MKN74 cells at indicated time points. (e) Immunoblot analysis of E-cadherin and Snai2 expression in hypoxia-exposed MKN74 cells at indicated time points. (f) Fluorescence-activated cell sorting (FACS) analysis of the fluorescent glucose analog 2-NBDG uptake and (g) percentage increase in energy charge in MKN74 and MKN74Snai1ER cells after the indicated treatments. (h) Relative mRNA expression (left panel) and immunoblot analysis (right panel) of ANGPTL4 in MKN74 and MKN74Snai1ER cells after the indicated treatments. (i) Percentage increase in energy change in MCF7, HSC, II4 and HepG2 cells after indicated treatments. For immunoblot analyses, representative immunoblot pictures and densitometric quantification plots are shown. Loading controls for the immunoblot analyses were from the same sample. For qPCR, TBP was used as reference gene. Data are represented as mean±s.d. from at least three independent experiments. *P<0.05, **P<0.01, ***P<0.001; n.s., not significant.
Next, we carried out a series of molecular and biochemical analyses to examine whether change in cellular bioenergetics was observed in in vitro EMT of cancer cells. First, we established three in vitro EMT models using the polarized gastric carcinoma cell line MKN74. In two models, EMT was initiated by either hypoxia (1% O2, Figures 1c–e) or TGF-β (Supplementary Figures S1a–c) to mimic the microenvironment of stress-induced EMT. The third model utilized the transgenic MKN74 cell line harboring a Snai1-ER transgene (MKN74Snai1ER) that allows direct initiation of EMT by 4-hydroxytamoxifen (4-OHT) (Supplementary Figures S1d–f). Upon exposure to either microenvironmental stress or 4-OHT, the colonies of MKN74 underwent EMT, with diminished E-cadherin staining bordering the epithelial cells after 48–96 h (Figure 1c and Supplementary Figures S1c and f). Real-time PCR and immunoblot analysis showed the expression of critical epithelial genes (E-cadherin (CDH1); discoidin domain receptor 1 (DDR1); receptor-tyrosine kinase (ErBb3)) was downregulated, with a concomitant increase in mesenchymal gene expression (Snai1; zinc finger E-box-binding homeobox 1 (ZEB-1); Figures 1d and e and Supplementary Figures S1a, b, d and e). Regardless of the EMT stimuli, we detected an increase in glucose uptake and energy charge in cancer cells, indicative of increased cellular metabolic activity during EMT (Figures 1f and g). Importantly, ANGPTL4 mRNA and protein levels, specifically the C-terminal fibrinogen-like form (cANGPTL4), were elevated during EMT (Figure 1h). An increase in cANGPTL4 expression (Supplementary Figure S1g) and augmented cellular metabolic activity were also observed in hypoxia-induced EMT of MCF7, II4, HSC and HepG2 cancer cell lines (Figure 1i and Supplementary Figure S1h). Our observations indicate that an increase in cellular bioenergetics in cancer cells undergoing EMT is widespread.
To confirm a role for cANGPTL4 in metabolic activity of cancer cells during EMT, we investigated the effects of ANGPTL4 deficiency (through knockdown or immunoneutralization) and treatment of recombinant human cANGPTL4 (rh-cANGPTL4) on energy charge and EMT. The increases in glucose uptake and energy charge were suppressed by either neutralizing cANGPTL4 antibody (α-cANGPTL4) or small interfering RNA against ANGPTL4 (ΔANGPTL4) in MKN74 (Figures 2a and b and Supplementary Figures S2a and b), MCF7, II4, HSC and HepG2 cancer lines during hypoxia-induced EMT (Supplementary Figure S1h). Exogenous rh-cANGPTL4 alone elevated 2-deoxyglucose uptake associated with increased GLUT1 protein expression and augmented the energy charge status during EMT (Figures 2a–c and Supplementary Figure S2c). Importantly, an impaired ANGPTL4-mediated increase in energy charge delayed EMT. At 48 and 96 h after EMT induction, the ANGPTL4-deficient cells still retained their epithelial-like morphology characterized by clear E-cadherin staining (Figure 2d and Supplementary Figures S2d and f). Quantitative PCR (qPCR) and immunoblot analysis revealed a delay in the expression of critical EMT genes (Figure 2e and Supplementary Figures S2e and g). Conversely, MKN74 cells treated with rh-cANGPTL4 resulted in an incomplete disruption of E-cadherin staining (Figure 2f), repressed expression of epithelial markers and significant elevation of mesenchymal markers (Figure 2g). Taken together, these observations suggest that ANGPTL4 modulates cellular bioenergetics needed for EMT competency.

ANGPTL4 increases cellular bioenergetics for EMT competency. (a) Fluorescence-activated cell sorting (FACS) analysis of the fluorescent glucose analog 2-NBDG uptake and (b) percentage increase in energy charge in MKN74 and MKN74Snai1ER cells after the indicated treatments. (c) Immunoblot analysis of GLUT1 expression in MKN74 cells after the indicated treatments. (d, f) Immunofluorescence staining of E-cadherin in hypoxia-treated MKN74 cells in the presence of neutralizing human cANGPTL4 antibodies (α-cANGPTL4) (d) and recombinant human cANGPTL4 (rh-cANGPTL4) -treated MKN74 (f) at the indicated time intervals. Cells were counterstained with DAPI (blue) for nuclei and phalloidin (red) for actin cytoskeleton. Scale bar=40 μm. (e, g) Relative mRNA expression (left panel) and immunodetection (middle and right panels) of EMT markers in hypoxia-treated MKN74 cells exposed to α-cANGPTL4 (e) and rh-cANGPTL4 (g) at the indicated time intervals. For immunoblot analyses, representative immunoblot pictures and densitometric quantification plots are shown. Loading controls for the immunoblot analyses were from the same sample. For qPCR, TBP was used as reference gene. Data are represented as mean±s.d. from at least three independent experiments. *P<0.05, **P<0.01, ***P<0.001; n.s., not significant.
ANGPTL4 modulates energy charge in metastasis
EMT is an integral step of metastasis. An increase in cellular metabolic activity could fuel energy-demanding processes required for enhanced invasiveness to the cancer cells. To further strengthen the link between ANGPTL4 and energy-demanding EMT, we generated a dual-inducible EMT xenograft mouse model. The MKN74Snai1ER:shANGPTL4 cell line for tumor xenograft has a doxycycline (Dox)-inducible shANGPTL4 expression plasmid introduced into MKN74Snai1ER cells. This cell line undergoes EMT when treated with 4-OHT, whereas endogenous ANGPTL4 will be suppressed when treated with Dox (Supplementary Figure S3a). Mice on a normal diet and injected with 4-OHT harbored significantly more lung metastases than the vehicle control mice or mice treated with 4-OHT and fed a Dox diet (Figure 3a). Next, we used human-specific TBP primers to identify and quantify human MKN74Snai1ER cells that have metastasized to the recipient mouse lung. The 18S primers were used to detect both human and mouse cells for normalization. In line with the above observation, the qPCR result showed the presence of human cancer cells in mouse lung sections after 4-OHT induction (Figure 3a). The cANGPTL4 protein was elevated in the xenograft tumors of 4-OHT-treated mice, whereas mice fed simultaneously with a Dox diet exhibited in vivo suppression of cANGPTL4 in MKN74Snai1ER:shANGPTL4-derived tumors (Figure 3b). Histological analysis of tumor sections revealed that vehicle-treated control tumors showed strong defined E-cadherin and laminin 332 staining (Figure 3c). The 4-OHT-treated tumors lost E-cadherin staining at the cell–cell borders, with clear disruption of basement membrane laminin 332 compared with vehicle-treated tumors. After in vivo suppression of ANGPTL4 by the Dox diet, the punctuated E-cadherin staining was diminished (Figure 3c). Gelatin zymography revealed elevated activities for proinvasive matrix metalloproteinases 2 and 9 in 4-OHT-treated tumors, indicating an aggressive tumor phenotype (Supplementary Figure S3b). The in vivo 2-deoxyglucose imaging showed that 4-OHT-treated tumors had significantly elevated glucose uptake compared with vehicle-treated tumors (Figure 3d). The suppression of ANGPTL4 in 4-OHT-treated tumors reduced the 2-deoxyglucose uptake to levels comparable to vehicle-treated tumors (Figure 3d). Consistent with our findings, 4-OHT-treated tumors expressed higher levels of GLUT1 than vehicle- and Dox-treated tumors. ANGPTL4 suppression in 4-OHT-treated tumors resulted in decreased GLUT1 expression (Figure 3b). Importantly, in vivo xenograft tumors exhibited a similar elevation in the energy charge status during EMT, and this elevation was diminished by Dox diet-induced ANGPTL4 depletion (Figure 3e). Taken together, these observations indicate a role for cANGPTL4 in cellular metabolic changes during EMT in vivo.

ANGPTL4 modulates energy charge during metastasis. (a) Representative macroscopic images of lungs (upper panel) and the relative expression of human TBP from the indicated lungs (lower panel). Black arrows indicate micrometastases (n=8–10). The 18S was used as reference gene. (b) Immunodetection of cANGPTL4 and GLUT1 from tumor lysates exposed to the indicated treatments. (c) Representative hematoxylin and eosin (H&E) images and immunofluorescence images of E-cadherin and laminin 332 from MKN74Snai1ER:shANGPTL4-derived tumor sections exposed to the indicated treatments. Scale bar=40 μm (upper and middle panels) or 100 μm (lower panel). (d) The in vivo 2-deoxyglucose (2-DG) fluorescence imaging of mice bearing tumors and (e) the percentage increase in energy charge in xenograft tumors that received the indicated treatments. Data are represented as mean±s.d. from at least three independent experiments. *P<0.05, **P<0.01, ***P<0.001. Loading controls for the immunoblot analyses were from the same sample.
ANGPTL4 is a molecular driver of metabolic changes during EMT
The phosphatidylinositol-3-kinase (PI3K)/AKT and AMPK (adenosine monophosphate-activated protein kinase) pathways were identified as two major regulatory conduits for cellular bioenergetics in cancer cells.19 To gain further insights into how cellular bioenergetics affects EMT, dominant-negative (dn) and constitutive active forms (ca) of AKT and AMPK were introduced into MKN74 cells (Supplementary Figure S3c). As expected, MKN74CTRL cells underwent EMT after 48 h of hypoxia as evidenced by diminished immunofluorescence signal for E-cadherin at the cell–cell junction (Supplementary Figure S3d), reduced E-cadherin and concomitant increased Snai1 protein levels (Figure 4a). In normoxic condition, the colony morphology of MKN74 cells harboring dnAKT (MKN74dnAKT), caAMPK (MKN74caAMPK) and dnAMPK (MKN74dnAMPK) resembled that of MKN74CTRL (empty vector). The MKN74caAKT cells revealed a reduced E-cadherin signal compared with MKN74CTRL cells (Supplementary Figure S3d). Upon exposure to hypoxia-induced EMT, MKN74caAKT and MKN74caAMPK cells exhibited an earlier (24 h) and more pronounced EMT phenotype than corresponding MKN74CTRL cells (48 h) (Supplementary Figure S3d). Indeed, immunoblots for E-cadherin and Snai1 indicated that dnAKT or dnAMPK was attenuated (Figure 4a), whereas caAKT and caAMPK facilitated hypoxia-induced EMT (Figure 4a). Importantly, the enhanced EMT observed in MKN74caAKT and MKN74caAMPK cells was associated with an increase in energy charge compared with MKN74CTRL (Figure 4b). Conversely, the delayed EMT in MKN74dnAKT and MKN74dnAMPK was accompanied by the abolishment of the rise in energy charge for EMT of these cells (Figure 4b). Similar observations were also obtained using 4-OHT-induced EMT of MKNSnai1ER cells (Figure 4c).

ANGPTL4 is a molecular driver of metabolic changes during EMT. (a) Immunoblot analysis of E-cadherin and Snai1 and (b) percentage increase in energy charge in MKN74 cells (MKN74caAKT, MKN74dnAKT, MKN74caAMPK and MKN74dnAMPK) under normoxic or hypoxic conditions. (c) Percentage increase in energy charge in 4-OHT-induced EMT in MKN74Snai1ER cells under similar modifications in (b). (d–g) Immunoblot analysis of pAKT (S473), total AKT, pAMPK (T172) and AMPKα in MKN74ΔANGPTL4 and MKN74Ctrl (d), MKN74Snai1ER (e), MKN74Ctrl (f) cells and MKN74Snai1ER:shANGPTL4-derived xenograft tumors (g) after indicated treatments. (h) Immunoblot analysis of pGSK3α/β (S21/9) and total GSK3α/β in breast, colorectal, gastric and head and neck tissues at various tumor stages. For immunoblot analyses, representative immunoblot pictures and densitometric quantification plots are shown. Loading controls for the immunoblot analyses were from the same sample. Data are represented as mean±s.d. from at least three independent experiments. */#P<0.05, **/##P<0.01, ***/###P<0.001; n.s., not significant.
The above observations prompted us to examine whether ANGPTL4 could modulate these two pathways. MKN74CTRL cells exposed to hypoxia and 4-OHT-treated MKN74Snai1ER displayed increased phospho-activation of AKT and AMPK (Figures 4d and e). We examined the temporal phosphorylation profiles of AMPK and AKT during rh-cANGPTL4-induced EMT. The expression of phospho-AMPK peaked by 48 h and returned to basal level by 96 h. Notably, the expression of phospho-AKT peaked at 72 h and remained elevated even at 96 h (Figure 4f). This suggests that ANGPTL4-mediated activation of AMPK and AKT may play nonredundant roles to metabolically prime cancer cells for EMT. The analysis of MKN74Snai1ER:shANGPTL4-derived xenograft tumors and MCF7, II4, HSC and HepG2 cancer cell lines also revealed similar phospho-activation of AKT (Figure 4g and Supplementary Figures S3c and e). Higher AKT activity was also detected in patients’ metastatic tumors compared with lower-grade tumors (Figure 4h). Taken together, these observations suggest that ANGPTL4 modules the AKT and AMPK pathways and that the consolidation of cellular bioenergetics is important for EMT competency.
The ANGPTL4/14-3-3γ axis coordinates energy demand for EMT
The 14-3-3 protein family consists of adaptor proteins that play critical roles in numerous important cellular functions via binding to specific phosphorylation signals on target proteins.20 Previous studies showed that ANGPTL4 influenced the expression of 14-3-3 adaptor protein family during wound healing, reminiscent of EMT.21, 22 Thus, we hypothesize that ANGPTL4 consolidates cellular bioenergetics to enhance EMT by regulating several key regulatory networks.
In response to EMT-inducing signals, only the expression of 14-3-3γ was consistently elevated by ANGPTL4 in MKN74CTRL and MKN74Snai1ER (Figures 5a and b and Supplementary Figures S4a and b). The remaining 14-3-3 isotypes were unchanged. Consistently, the suppression of ANGPTL4 resulted in a significant decrease in the expression of 14-3-3γ, whereas the exposure of MKN74CTRL cells to rh-cANGPTL4 enhanced 14-3-3γ expression (Figure 5c and Supplementary Figure S4c). Similar changes in 14-3-3γ expression were also observed in four other cancer lines during EMT as well as in metastatic human tumor biopsies (Figure 5d and Supplementary Figures S4d–g). Notably, the knockdown of 14-3-3γ in MKN74CTRL attenuated the expression of mesenchymal markers (Supplementary Figures S4h and i). These observations suggest that ANGPTL4 may exert its effect on cancer cell bioenergetics and EMT competency by modulating the expression of 14-3-3γ.

ANGPTL4 regulates 14-3-3γ expression to facilitate EMT. (a–c) Relative fold change of the various 14-3-3 isoforms (protein level) in hypoxia-induced MKN74Ctrl and MKN74ΔANGPTL4 (a), 4-OHT-induced MKN74Snai1ER (b) and rh-cANGPTL4-induced MKN74Ctrl conditions (c). (d) Immunoblot analysis of 14-3-3γ in breast, colorectal, gastric and head and neck tissues at various tumor stages. (e) Analysis map of kinases (solid line) whose inhibition attenuates ANGPTL4-induced 14-3-3 expression and possible downstream transcriptional regulators (dotted line). (f) Relative fold changes of the various transcription factors (phospho- and total-proteins) in hypoxic-induced (left panel) and rh-cANGPTL4-treated (right panel) MKN74Ctrl cells. The plots were derived based on western blots shown on Supplementary Figure S4l. (g) Representative ChIP assay performed using preimmune IgG (p.i.) or antibodies against pCREB, pSTAT3 or p-cFOS in rh-cANGPTL4-treated MKN74 cells. Putative specific binding sites for respective transcription factors spanning the promoter region of 14-3-3γ are shown on the left panel, along with their cognate primers. A control region 2 kb upstream of the promoter served as a negative control. For the immunoblot analyses, densitometric quantification plots are shown. Loading controls were from the same sample. Data are represented as mean±s.d. from at least three independent experiments. *P<0.05, **P<0.01.
Many studies showed that 14-3-3 can associate with diverse proteins, modulating complex regulatory networks, but how 14-3-3 is transcriptionally regulated remains unclear. We observed that rh-cANGPTL4 increased the expression of 14-3-3γ mRNA in MKN74 cells in 6 h (Supplementary Figure S4j). To obtain further insight into the signaling pathways, we performed an unbiased kinase inhibitor screen to identify key signaling mediators whose inhibition would suppress ANGPTL4-mediated upregulation of 14-3-3γ mRNA (Supplementary Figure S4k). Coincidentally, qPCR followed by Ingenuity Pathway Analysis revealed a convergence to the PI3K/AKT and mitogen-activated protein kinase signaling cascades and implicated several transcription factors known to be involved in cancer progression (Figure 5e). Immunoblot analysis showed that three of these transcription factors, namely CREB, cFOS and Stat3, were phospho-activated during EMT (Figure 5f and Supplementary Figure S4l). Next, we performed chromatin immunoprecipitation (ChIP) analysis using antibodies against phosphorylated CREB (pCREB), pSTAT3 and p-cFOS on MKN74 cells treated with rh-cANGPTL4. Our result showed that these transcription factors were bound to their cognate binding sites along the promoter region of 14-3-3γ gene (Figure 5g). These observations indicate that ANGPTL4 can activate specific transcription factors via the PI3K/AKT pathway to differentially regulate specific 14-3-3γ isotype.
Next, we interrogated the ANnotation and Integrated Analysis of the 14-3-3 interactome (ANIA) database to find key signaling mediators of cancer cell metabolism and EMT.23 Interestingly, our analysis identified tuberin sclerosis complex (TSC2) and Snai1 as highly ranked interaction partners for 14-3-3. TSC2 is an important gatekeeper of cellular metabolism that integrates extracellular signals to direct anabolic or catabolic activities.24, 25 During EMT, TSC2 phosphorylation at Ser939 (TSC2S939) was increased, indicating TSC2 inhibition (Supplementary Figures S5a–c). Co-immunoprecipitation and proximity ligation assay (PLA) revealed increased 14-3-3γ/TSCS939 complex formation in in vitro EMT culture (Figures 6a and b and Supplementary Figure S5d), MKN74Snai1ER:shANGPTL4-derived xenografts (Figure 6c and Supplementary Figure S5e) and patients’ tumor biopsies (Figure 6d and Supplementary Figures S5f–h), further confirming that TSC2 was suppressed during EMT. Conversely, ANGPTL4 deficiency diminished 14-3-3γ/TSCS939 complex formation (Figures 6a–c and Supplementary Figures S5d and e). To further clarify the role for ANGPTL4 in the elevation of adenylate charge, we performed transient small interfering RNA knockdown of TSC2 in MKN74 cells (MKN74ΔTSC2) that successfully suppressed the endogenous TSC2 level by ~70% (Supplementary Figure S5i left panel). The MKN74ΔTSC2 cells were subjected to hypoxic condition in the presence or absence of rh-cANGPTL4 protein for 48 h before energy charge was measured. Consistent with our finding, the ANGPTL4-dependent elevation in adenylate charge was abolished by ~45% because of TSC2 knockdown, resulting in an abrogation of EMT under hypoxia. The rh-cANGPTL4 treatment failed to rescue the increase in energy charge and therefore failed to induce EMT, indicating that TSC2 is one downstream effector of the ANGPTL4/14-3-3γ axis that regulates energy flux during EMT (Supplementary Figure S5i, middle and right panels). Taken together, all these results evidenced that ANGPTL4 coordinates energy demand for EMT through a TSC2-dependent pathway.

The ANGPTL4/14-3-3γ axis coordinates energy demand during EMT. (a, e) Immunoblot analysis of 14-3-3γ expression after TSC2S939 (a) or Snai1 (e) immunoprecipitation in MKN74Ctrl and MKN74ΔANGPTL4 cells. (b–d and f, h–i) Proximity ligation assay (PLA) quantification of 14-3-3γ/TSC2S939 and 14-3-3γ/Snai1 interactions in MKN74CTRL and MKN74ΔANGPTL4 cells (b, f), xenograft tumors (c, h), and tumor biopsies from patients (d, i) after the indicated treatments or with indicated disease stages. (g) Immunodetection of the indicated proteins in hypoxic-induced MKN74CTRL and MKN74ΔANGPTL4 cells at 0 and 48 h. (j) Representative ChIP results using anti-Snai1 (upper panel) followed by re-ChIP with anti-14-3-3γ (lower panel) in MKN74CTRL and MKN74ΔANGPTL4 cells under normoxic or hypoxic conditions. Preimmune serum (IgG) served as control. (k) Schematic diagram depicting the coordination of metabolic reprogramming by the ANGPTL4/14-3-3 axis during EMT. Data are represented as mean±s.d. from at least three independent experiments. *P<0.05, **P<0.01. Loading controls for the immunoblot analyses were from the same sample.
The in vitro co-immunoprecipitation analysis and PLA of Snai1 and 14-3-3 revealed a significant increase in 14-3-3γ/Snai1 complex formation during hypoxia-induced EMT. This interaction was diminished in the absence of ANGPTL4 (Figures 6e and f and Supplementary Figure S6a). No change in complex formation between Snai1 and 14-3-3η or τ was observed. The 14-3-3β, which does not interact with Snai1, served as a negative control (Figure 6g). Similarly, PLA analysis revealed significantly more 14-3-3γ/Snai1 complexes in 4-OHT-treated tumors than in vehicle-treated tumors from mice fed a normal diet (Figure 6h and Supplementary Figure S6b). PLA analysis of tumor biopsies from patients further confirmed that higher-grade, more aggressive tumors harbored significantly more 14-3-3γ/Snai1 complexes than their benign counterparts, highlighting the importance of 14-3-3γ/Snai1 complexes during malignancy (Figure 6I and Supplementary Figures S6c–e). These observations underscore the ANGPTL4/14-3-3 signaling axis as an important driver of the observed changes in bioenergetics during EMT.
The interaction of 14-3-3 with Snai1 was proposed to stabilize Snai1 binding on the E-cadherin promoter to facilitate transcriptional repression.26 We showed that reduced 14-3-3γ/Snai1 complex in MKN74ΔANGPTL4 decreased Snai1 occupancy of the E-cadherin promoter during hypoxia-induced EMT, indicating that the repression of E-cadherin gene expression was impaired (Figure 6j). Furthermore, EMT was delayed in the absence of ANGPTL4, highlighting the importance of 14-3-3γ/Snai1 complexes during malignancy. Altogether, our data revealed a previously unknown mechanism where the ANGPTL4/14-3-3γ signaling axis coordinates energy demand and stability of critical EMT proteins necessary to drive EMT (Figure 6k).
Discussion
Metastatic cancer cells acquire energy-intensive processes including increased invasiveness and chemoresistance.2 However, how the energy demand is met and the mechanisms that coordinate cellular metabolic activity with EMT remain unclear. Using experimental and clinical metastatic tumors, we showed that an increase in adenylate energy charge during cancer cell EMT is widespread and is required for metastasis competency. We identified ANGPTL4, a matricellular protein with no intrinsic enzymatic activity, as a novel extracellular driver of cellular metabolic activity during EMT. ANGPTL4 exploited the PI3K/AKT and mitogen-activated protein kinase signaling cascades to regulate the expression of 14-3-3γ adaptor protein. This ANGPTL4/14-3-3γ signaling axis, through interaction with specific phosphorylation signals on target proteins, consolidated cellular bioenergetics and stabilized critical EMT proteins to coordinate energy demand and enhanced EMT.
Many studies have linked ANGPTL4 to metastasis, from cell motility, angiogenesis to vascular dysfunction to facilitate distal dissemination.27, 28, 29 Indeed, ANGPTL4 has been recently implicated in EMT of non-small-cell lung cancer;30 however, a role for ANGPTL4 in cellular metabolic changes is still unknown. We identified ANGPTL4 as a molecular driver of EMT-enriched metabolic changes as supported by several lines of experimental evidence. First, in vitro and in vivo EMT models showed that cancer cells deficient in ANGPTL4 were unable to increase the cellular energy charge during EMT, resulting in reduced metastasis. Molecular and biochemical analyses confirmed the down- and up-regulation of epithelial and mesenchymal markers, respectively. Histological analysis of xenograft tumors also showed increased matrix metalloproteinase activities and a disrupted laminin 332 phenotype. Second, ANGPTL4 activated the proglycolysis AKT and AMPK pathways to enhance the glycolytic phenotype and increase the cellular energy charge necessary to fuel biological processes of EMT. The active remodeling of the actin cytoskeleton requires ATP binding and hydrolysis to confer invasive abilities to the cancer cells.31, 32 AMPK has been proposed to play both ‘conditional’ tumour suppressor and ‘contextual’ oncogene roles.33, 34 In cancer cells, AMPK functions as an energy sensor that inhibits the main anabolic processes that cancer cells needed for proliferation and growth, similarly observed in cells at 0 h. During EMT, AMPK was rapidly phospho-activated to carry out its ‘contextual’ oncogene role to increase glucose uptake and ATP production. Upon completion of EMT, AMPK returned to basal level. Independently, phospho-activation of AKT modulates the mTOR machinery regulating energy flux for EMT progression. After EMT, phospho-activated AKT remained elevated for subsequent metastatic events, such as resistance to anoikis.29 Finally, ANGPTL4 regulates the expression of specific 14-3-3 proteins that coordinates the energy supply during EMT. Notably, these changes were also detected in clinical tumor biopsies and strongly correlated with the stage and aggressiveness of the tumors, further underscoring the clinical relevance of our findings. TGF-β and hypoxia are well-established stimuli of EMT. The human metastasis-related ANGPTL4 gene is transcriptional upregulated by TGF-β28 and hypoxia14 Hypoxia stabilizes the hypoxia-inducible factor-1α that upregulates several genes to promote metastasis.35, 36 The hypoxia-inducible factor-1α has been shown to directly upregulate ANGPTL4 expression that facilitates transendothelial migration and increases angiogenesis.37, 38, 39, 40 We showed that the expression of ANGPTL4 was increased during hypoxia-induced EMT. The ANGPTL4-mediated upregulation of 14-3-3γ expression stabilizes Snai1-mediated repression of E-cadherin and elevated adenylate charge to fuel the various biological activities required for EMT.
Metastasis necessitates the cooperative effects of multiple signaling events and biological progresses.1, 13, 41 A key mechanistic insight is that ANGPTL4 regulates 14-3-3γ expression and contributes to an important new signaling axis that coordinates multiple biological processes for metastasis. Unlike protein kinases, 14-3-3γ adaptor protein associates with many mediators of key signaling hubs to either augment or attenuate the activities of specific pathways. We showed that ANGPTL4/14-3-3γ coordinated cellular metabolic activity during EMT via interaction with TCS and Snai1, respectively. The TSC2 pathway coordinates catabolic processes, such as glycolysis, to provide the necessary substrates for the PI3K/AKT signaling pathways to signal anabolic metabolism. The phospho-inhibition of TSC2 by ANGPTL4-mediated AKT activation altered cellular energy charge and primed cancer cells for EMT. ANGPTL4 also modulated Snai1 complex stabilization through the 14-3-3 γ/Snai1 interaction to stably suppress E-cadherin gene expression and augment the EMT progression. Interestingly, the loss of E-cadherin has been reported to trigger a glycolytic switch that favors EMT.42, 43 Previous studies showed that ANGPTL4 triggers the 14-3-3 adaptor protein to sequester the pro-apoptotic Bad protein from mitochondria, conferring resistance to anoikis and favoring tumor survival and growth that has been suggested to be a prerequisite for EMT.29 In addition, ANGPTL4/14-3-3 is involved in rapid integrin recycling during cell migration, an important biological process for metastasis.21
Our study shows that an increase in adenylate energy charge by angiopoietin-like 4 protein enhances EMT by inducing 14-3-3γ expression. Our findings of the ANGPTL4/14-3-3γ signaling axis revealed the integration of protein–protein interactome with kinome network to synchronize cancer cell metabolic activities with EMT.
Materials and methods
Antibodies
Otherwise indicated, all antibodies were from Cell Signaling (Boston, MA, USA); N-cadherin and Snai2 from Millipore (Billerica, MA, USA); β-tubulin, Snai1, hypoxia-inducible factor-1α and 14-3-3σ from Santa Cruz (Dallas, TX, USA); Vimentin, laminin 332 and mouse monoclonal anti-human cANGPTL4 mAb11F6C4 from Abcam (Cambridge, MA, USA); pan-cytokeratin and IRdye 680-conjugated secondary antibodies from Thermo Scientific (Waltham, MA, USA); and Alexa Fluor 488-conjugated secondary antibodies and Alexa Fluor 594-conjugated phalloidin from Molecular Probes (Waltham, MA, USA).
Human tumor biopsies
Fresh frozen breast, colon, gastric and head and neck tumor biopsies (Proteogenex, Culver City, CA, USA) were used for protein, energy charge and immunofluorescence analysis (Supplementary Table S1).
Expression and purification of recombinant cANGPTL4 proteins
The expression and purification of recombinant cANGPTL4 were performed as previously described.21, 22
Cell culture
MKN74 human gastric carcinoma (JCRB0255, Osaka, Japan) was cultured in RPMI-1640 supplemented with 10% fetal bovine serum in a humidified atmosphere of 5% CO2 at 37 °C. All other cell lines were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. HSC and II4 were from German Cancer Research Center (DKFZ, Heidelberg, Germany), and MCF7 (HTB-22) and HepG2 (HB-8065) were from American Type Culture Collection (ATCC, Manassas, VA, USA). Smartpool small interfering RNAs (Thermo Scientific) against human ANGPTL4 (L-007807-00-0050) and TSC2 (L-003029-00-0005) were used to knockdown respective genes in cells of interest. AKT activity was measured using the AKT activity kit (Cell Signaling).
Retrovirus transduction
MKN74Snai1ER cells were generated using retroviral transduction as previously described.44 Briefly, HEK 293T cells were transfected with pCL-10A1 retrovirus packaging vector (Imgenex, Littleton, CO, USA) and pWZL-Snai1-ER (Addgene, Cambridge, MA, USA). Harvested pseudoviruses were used to infect MKN74 cells. Following transduction, cells were selected with 5 μg/ml of blasticidin hydrochloride (Sigma Aldrich, St Louis, CO, USA).
Generation of dual-inducible MKN74Snai1ER/shANGPTL4 cell line
Dox-inducible pSingle-tTS-shRNA vector carrying either shANGPTL4 or scrambled shRNA was introduced into MKN74Snai1ER cells using Fugene HD reagent as per the manufacturer protocol (Promega, Madison, WI, USA). MKN74Snai1ER:shANGPTL4 cells were selected with 1 mg/ml of G418 (Sigma Aldrich). The efficiency of knockdown was verified by qPCR and immunoblotting after treatment with various concentrations of Dox (up to 10 μg/ml).
Glucose uptake assay
Briefly, cells were incubated with 10 μM of 2-NBDG for 12 h. The cells were then harvested and subjected to fluorescence-activated cell sorting analysis using BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA). Data were analyzed using FlowJo (Ashland, OR, USA) and plotted based on mean intensity.
In vitro induction of EMT
Cells were seeded at a density of 5.25 × 102 cell per cm2. Hypoxia treatment (1% O2 concentration) was conducted in hypoxic chamber (Stem Cell Technology, Cambridge, MA, USA). For TGF-β-induced and Snai1-mediated EMT, cells were treated with 10 ng/ml of TGF-β for 2 days and with 20 ng/ml of 4-OHT for 4 days, respectively.
In vivo induction of EMT
Six-week-old male NSG mice (20–22 g; NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were injected subcutaneously with 1 × 106 cancer cells resuspended in matrigel. A week after the xenograft injection, each xenograft group was randomly divided into four subgroups. Two subgroups received intraperitoneal injection of 4-OHT (4 mg/kg) and were fed on either normal chow diet or Dox diet (625 mg/kg, Harlan Laboratories, Indianapolis, IN, USA). The other two subgroups received vehicle (sunflower seed oil) and similarly fed on the two different diets. After 8 weeks, tumors and lungs were harvested for analysis. Power analysis was used to determine the required number of animal necessary to ensure adequate power to detect a prespecified effect size. All the experimental groups were allocated with double-blinded randomization. Animal experiments were carried out in accordance to the guidelines of the institutional animal care and use committee (ARF-SBS/NIE-A0250AZ, -A0324 and -A0321) of Nanyang Technological University, Singapore.
ChIP and Re-ChIP
ChIP experiments were carried out as previously described.45 Sonicated chromatin complexes were immunoprecipitated using antibodies against Snai1, pCREB, pSTAT3 or p-cFOS and Re-ChIP using anti-14-3-3γ. Immunocomplexes were affinity precipitated by Protein A/G Sepharose (Santa Cruz). The ChIP primer sequences are listed in Supplementary Table S2.
Energy charge determination
Cellular levels of AMP, ADP and ATP were determined by high-performance liquid chromatography. A Shimadzu LC-20AD (Kyoto, Japan) series Quaternary Gradient HPLC system with PDA detector using a Hypersil ODS 150 mm × 4.6 mm I.D. C18 reverse-phase column with 3 μm particle size and 5 μm Hypersil ODS C18 guard column (Thermo Scientific) was used. Nucleotides were extracted as previously described.46 Nucleotides standards were from Sigma Aldrich. Separation of the nucleotides was carried out with a mobile phase containing 60 mmol/l of KH2PO4, 0.45 mmol/l of tetrabutylammonium bromide and 1.26 mol/l of acetonitrile. The pH of the mobile phase was adjusted to 3.20 using concentrated phosphoric acid. Retention times of the individual nucleotides were matched to that of the respective nucleotide standards. Areas under the peaks for the respective nucleotides were tabulated and energy charge was calculated using the formula: Energy Charge=[ATP]+½[ADP]/[ATP]+[ADP]+[AMP].
Kinase inhibitor array
MKN74 cells were treated with 95 different kinase inhibitors (SYN-2103; Synkinase, Victoria, Australia) in the absence and presence of rh-cANGPTL4 (10 μg/ml) for 6 h. RNA was isolated and reverse transcribed using the iScript Reverse Transcription Supermix (Bio-Rad, Hercules, CA, USA). qPCR was done using the KAPA SYBR qPCR Universal Master Mix (KAPABiosystems). The qPCR primer sequences are listed in Supplementary Table S3.
Immunofluorescence staining
EMT was identified by immunofluorescence staining for E-cadherin and Snai2. MKN74 cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min and blocked with 3% normal goat serum (NGS) containing 0.05% Triton X-100 in a humidified chamber for 1 h at room temperature. Following three washes in phosphate-buffered saline, cells were incubated overnight at 4 °C with either anti-human-E-cadherin (1:200) or Snai1 (1:100) antibodies in 3% NGS. After three washes, cells were incubated for 1 h at room temperature with Alexa488-secondary antibodies (1:250) and counterstained with Alexa594-phalloidin for F-actin and DAPI (4',6-diamidino-2-phenylindole) for nuclei. Immunostainings performed without primary antibodies served as negative controls. Images were acquired using Zeiss (Munich, Germany) LSM710 confocal microscope with a Plan-APOCHROMAT 40 × /1.4 oil objective and ZEN2009 software.
PLA and analysis
MKN74CTRL or MKN74ΔANGPTL4 (5 × 103 cells) were subjected to either normoxia or hypoxia for 2 days at 37 °C. At indicated time, MKN74 cells were fixed, permeabilized and blocked like immunofluorescence staining. Next, MKN74 cells were incubated with monoclonal mouse-anti human 14-3-3γ antibody (1:200) and monoclonal rabbit anti-human Snai1 antibody (1:200) in 3% NGS overnight at 4 °C. PLA was carried out as per the manufacturer’s protocol (Olink Bioscience, Watertown, MA, USA). Images were taken using Carl Zeiss confocal microscope LSM710 using a Plan-APOCHROMAT 63 × /1.4 oil DIC objective, and ZEN2012 LE software with constant exposure and gain. Number of protein interaction was quantified using the BlobFinder software.47
Immunoprecipitation and immunoblot
For immunoprecipitation, at the indicated time points after treatments, cells were lysed using IP lysis buffer (25 mM Tris-HCl pH7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA). The lysate was then incubated with indicated antibodies overnight at 4 °C with constant rotation. Following that, the antibodies were affinity precipitated using protein A/G beads (Santa Cruz). Proteins were released by boiling for 10 min in Lamelli’s buffer. Protein extracts were resolved using 12% SDS–polyacrylamide gel electrophoresis and electrotransferred (25 mM Tris, 192 mM glycine, 20% methanol) onto a low fluorescence polyvinylidene difluoride membrane for immunoblot analysis. Membranes were stripped and reprobed for other proteins as previously described.48 Protein bands were detected using Odyssey CLx Infrared Imaging system (LI-COR, Lincoln, NE, USA).
Statistical analysis
Statistical analyses were performed using two-tailed Mann–Whitney or one-way analysis of variance test with SPSS software (New York, NY, USA)where appropriate. A P-value of <0.05 is considered significant.
References
Chiang AC, Massagué J . Molecular basis of metastasis. N Engl J Med 2008; 359: 2814–2823.
Klymkowsky MW, Savagner P . Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol 2009; 174: 1588–1593.
Finger EC, Giaccia AJ . Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev 2010; 29: 285–293.
Steeg PS, Theodorescu D . Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol 2008; 5: 206–219.
Siegel R, Ma J, Zou Z, Jemal A . Cancer statistics, 2014. CA Cancer J Clin 2014; 64: 9–29.
Thiery JP, Sleeman JP . Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7: 131–142.
Savagner P . Leaving the neighborhood: molecular mechanisms involved during epithelial-mesenchymal transition. Bioessays 2001; 23: 912–923.
Vander Heiden MG, Cantley LC, Thompson CB . Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 2009; 324: 1029–1033.
Dang CV, Semenza GL . Oncogenic alterations of metabolism. Trends Biochem Sci 1999; 24: 68–72.
Ward PS, Thompson CB . Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21: 297–308.
Dang CV, Lewis BC, Dolde C, Dang G, Shim H . Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. J Bioenerg Biomembr 1997; 29: 345–354.
Jones RG, Thompson CB . Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009; 23: 537–548.
Lunt SJ, Chaudary N, Hill RP . The tumor microenvironment and metastatic disease. Clin Exp Metastasis 2009; 26: 19–34.
Zhu P, Goh YY, Chin HF, Kersten S, Tan NS . Angiopoietin-like 4: a decade of research. Biosci Rep 2012; 32: 211–219.
Tan MJ, Teo Z, Sng MK, Zhu P, Tan NS . Emerging roles of angiopoietin-like 4 in human cancer. Mol Cancer Res 2012; 10: 677–688.
Atkinson DE, Walton GM . Adenosine triphosphate conservation in metabolic regulation. Rat liver citrate cleavage enzyme. J Biol Chem 1967; 242: 3239–3241.
Live TR, Kaminskas E . Changes in adenylate energy charge in Ehrlich ascites tumor cells deprived of serum, glucose, or amino acids. J Biol Chem 1975; 250: 1786–1789.
Ridge JW . Hypoxia and the energy charge of the cerebral adenylate pool. Biochem J 1972; 127: 351–355.
Dang CV . Links between metabolism and cancer. Genes Dev 2012; 26: 877–890.
Bridges D, Moorhead GB . 14-3-3 proteins: a number of functions for a numbered protein. Sci STKE 2005; 2005: re10.
Goh YY, Pal M, Chong HC, Zhu P, Tan MJ, Punugu L et al. Angiopoietin-like 4 interacts with integrins beta1 and beta5 to modulate keratinocyte migration. Am J Pathol 2010; 177: 2791–2803.
Goh YY, Pal M, Chong HC, Zhu P, Tan MJ, Punugu L et al. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J Biol Chem 2010; 285: 32999–33009.
Tinti M, Madeira F, Murugesan G, Hoxhaj G, Toth R, Mackintosh C . ANIA: ANnotation and Integrated Analysis of the 14-3-3 interactome. Database (Oxford) 2014; 2014: bat085.
Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J et al. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol 2006; 173: 279–289.
Mak BC, Yeung RS . The tuberous sclerosis complex genes in tumor development. Cancer Invest 2004; 22: 588–603.
Hou Z, Peng H, White DE, Wang P, Lieberman PM, Halazonetis T et al. 14-3-3 binding sites in the snail protein are essential for snail-mediated transcriptional repression and epithelial-mesenchymal differentiation. Cancer Res 2010; 70: 4385–4393.
Huang R-L, Teo Z, Chong HC, Zhu P, Tan MJ, Tan CK et al. ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood 2011; 118: 3990–4002.
Padua D, Zhang XHF, Wang Q, Nadal C, Gerald WL, Gomis RR et al. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008; 133: 66–77.
Zhu P, Tan MJ, Huang R-L, Tan CK, Chong HC, Pal M et al. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2(-):H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell 2011; 19: 401–415.
Zhu X, Guo X, Wu S, Wei L . ANGPTL4 Correlates with NSCLC Progression and Regulates Epithelial-Mesenchymal Transition via ERK Pathway. Lung 2016; 194: 637–646.
Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D et al. Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 2016; 164: 433–446.
Shiraishi T, Verdone JE, Huang J, Kahlert UD, Hernandez JR, Torga G et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2015; 6: 130–143.
Liang J, Mills GB . AMPK: a contextual oncogene or tumor suppressor? Cancer Res 2013; 73: 2929–2935.
Zadra G, Batista JL, Loda M . Dissecting the dual role of AMPK in cancer: from experimental to human studies. Mol Cancer Res 2015; 13: 1059–1072.
Schito L, Rey S . Hypoxic pathobiology of breast cancer metastasis. Biochim Biophys Acta 2017; 1868: 239–245.
Tsai YP, Wu KJ . Hypoxia-regulated target genes implicated in tumor metastasis. J Biomed Sci 2012; 19: 102.
Hu K, Babapoor-Farrokhran S, Rodrigues M, Deshpande M, Puchner B, Kashiwabuchi F et al. Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget 2016; 7: 7816–7828.
Khong TL, Thairu N, Larsen H, Dawson PM, Kiriakidis S, Paleolog EM . Identification of the angiogenic gene signature induced by EGF and hypoxia in colorectal cancer. BMC Cancer 2013; 13: 518.
Li H, Ge C, Zhao F, Yan M, Hu C, Jia D et al. Hypoxia-inducible factor 1 alpha-activated angiopoietin-like protein 4 contributes to tumor metastasis via vascular cell adhesion molecule-1/integrin beta1 signaling in human hepatocellular carcinoma. Hepatology 2011; 54: 910–919.
Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 2012; 31: 1757–1770.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Chu K, Boley KM, Moraes R, Barsky SH, Robertson FM . The paradox of E-cadherin: role in response to hypoxia in the tumor microenvironment and regulation of energy metabolism. Oncotarget 2013; 4: 446–462.
Sousa B, Ribeiro AS, Nobre AR, Lopes N, Martins D, Pinheiro C et al. The basal epithelial marker P-cadherin associates with breast cancer cell populations harboring a glycolytic and acid-resistant phenotype. BMC cancer 2014; 14: 734.
Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–715.
Montagner A, Delgado MB, Tallichet-Blanc C, Chan JSK, Sng MK, Mottaz H et al. Src is activated by the nuclear receptor peroxisome proliferator-activated receptor β/δ in ultraviolet radiation-induced skin cancer. EMBO Mol Med 2014; 6: 80–98.
Yang N-C, Ho W-M, Chen Y-H, Hu M-L . A convenient one-step extraction of cellular ATP using boiling water for the luciferin-luciferase assay of ATP. Anal Biochem 2002; 306: 323–327.
Allalou A, Wählby C . BlobFinder, a tool for fluorescence microscopy image cytometry. Comput Methods And Programs Biomed 2009; 94: 58–65.
Yeung Y-G, Stanley ER . A solution for stripping antibodies from polyvinylidene fluoride immunoblots for multiple reprobing. Anal Biochem 2009; 389: 89–91.
Acknowledgements
This work was supported by grants from Ministry of Education, Singapore (MOE2014-T2-1-012 and 2012-T1-001-036).
Author contributions
ZT performed experiments, analyzed the results and wrote the article; MKS, JSKC, YL, MMKL, TP, JYHL, ZWT and PZ performed experiments and contributed to discussion. JSKC, YL, LL and PZ performed and analyzed the results from the animal experiments. NST analyzed the results, contributed to discussion, reviewed and edited the article.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Teo, Z., Sng, M., Chan, J. et al. Elevation of adenylate energy charge by angiopoietin-like 4 enhances epithelial–mesenchymal transition by inducing 14-3-3γ expression. Oncogene 36, 6408–6419 (2017). https://doi.org/10.1038/onc.2017.244
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2017.244
- Springer Nature Limited
This article is cited by
-
YWHAG promotes colorectal cancer progression by regulating the CTTN-Wnt/β-catenin signaling axis
Medical Oncology (2024)
-
Age-related epithelial defects limit thymic function and regeneration
Nature Immunology (2024)
-
Transcriptomic and functional analysis of ANGPTL4 overexpression in pancreatic cancer nominates targets that reverse chemoresistance
BMC Cancer (2023)
-
Spatial probabilistic mapping of metabolite ensembles in mass spectrometry imaging
Nature Communications (2023)
-
STAT3 induces breast cancer growth via ANGPTL4, MMP13 and STC1 secretion by cancer associated fibroblasts
Oncogene (2022)