
Abstract
Multiple system atrophy (MSA) is a neurodegenerative disorder characterized by various combinations of autonomic failure, parkinsonism, and cerebellar ataxia. To elucidate variants associated with MSA, we have been conducting short-read-based whole-genome sequence analysis. In the process of the association studies, we initially focused on GBA1, a previously proposed susceptibility gene for MSA, to evaluate whether GBA1 variants can be efficiently identified despite its extraordinarily high homology with its pseudogene, GBA1LP. To accomplish this, we conducted a short-read whole-genome sequence analysis with alignment to GRCh38 as well as Sanger sequence analysis and compared the results. We identified five variants with inconsistencies between the two pipelines, of which three variants (p.L483P, p.A495P–p.V499V, p.L483_M489delinsW) were the results of misalignment due to minor alleles in GBA1P1 registered in GRCh38. The miscalling events in these variants were resolved by alignment to GRCh37 as the reference genome, where the major alleles are registered. In addition, a structural variant was not properly identified either by short-read or by Sanger sequence analyses. Having accomplished correct variant calling, we identified three variants pathogenic for Gaucher disease (p.S310G, p.L483P, and p.L483_M489delinsW). Of these variants, the allele frequency of p.L483P (0.003) in the MSA cases was higher than that (0.0011) in controls. The meta-analysis incorporating a previous report demonstrated a significant association of p.L483P with MSA with an odds ratio of 2.85 (95% CI; 1.05 – 7.76, p = 0.0400).
Similar content being viewed by others
Introduction
GBA1 (HGNC:4177) is the gene encoding a lysosomal enzyme, glucocerebrosidase, located on chromosome 1q21. GBA1 consists of 11 exons spanning 7.6 kb, and a highly homologous pseudogene (GBA1LP, HGNC:4178) is located approximately 16 kb apart from the functional GBA1 gene [1,2,3]. Unequal pairing with rearrangement between GBA1 and GBA1LP is a frequent cause leading to the generation of gene–pseudogene rearrangements, many of which are causative variants for Gaucher disease. Of these rearrangements, nonreciprocal recombination (gene conversion) events are the most frequent, in which a portion of the functional gene sequence is replaced by the corresponding part of the pseudogene [1, 2, 4].
Biallelic pathogenic variants of GBA1 cause deficient glucocerebrosidase activities leading to the development of Gaucher disease [4]. Starting from the observation that family members of patients with Gaucher disease have a considerably high prevalence of Parkinson disease (PD) [5], numerous studies have consistently shown that deleterious variants of GBA1 are strong risk factors for the development of PD [6,7,8,9]. Furthermore, we have reported that GBA1 variants pathogenic for Gaucher disease are also significantly, albeit with a small odds ratio, associated with an increased risk of developing multiple system atrophy (MSA), which is also a neurodegenerative disorder characterized by various combinations of autonomic failure, parkinsonism, and cerebellar ataxia along with the accumulation of α-synuclein within oligodendroglia [10]. Other studies, however, have not demonstrated the associations of GBA1 variants with MSA, possibly owing to their small sample sizes [11,12,13].
Given the remarkable progress of short-read sequencing technologies employing next-generation sequencers, comprehensive mutational analysis has become a standard procedure for large-scale studies. Owing to the extraordinarily high homology between GBA1 and GBA1LP, however, short-read sequencing techniques without specific amplification of GBA1 pose a challenge to the accurate sequence analysis of GBA1. Therefore, PCR designed to specifically amplify GBA1 followed by Sanger sequencing is usually needed for validation of the genotypes in this locus [14].
To investigate the molecular basis of MSA, we have been conducting short-read whole-genome sequence (WGS) analysis of large-scale MSA cases to explore genes associated with MSA. As the initial step for the association studies, we focused on GBA1, which we previously reported to be associated with MSA [10]. In this study, we extracted all the variants mapped to GBA1 as well as those mapped to GBA1LP from the WGS data obtained employing short-read sequencers. During the analysis, we noticed a critical pitfall in GBA1–GBA1LP genotyping that leads to the miscalling of variants based on the short-read sequence data depending on the reference genome. We herein report the details underlying the miscalling events. We also briefly present the results of a potential association of the GBA1 variants with MSA.
Materials and methods
Subjects
Genomic DNA was extracted from peripheral blood leukocytes with written informed consent from 500 patients with MSA registered in the Japan Multiple System Atrophy Registry (Japan MSA registry) (https://msajp.org/) [15] from August 2016 through September 2022. The Japan MSA registry is a multicenter-based prospective cohort study participated by 13 institutions in Japan that enrolls patients with possible or probable MSA on the basis of the revised Gilman criteria [16]. There was no overlap of the MSA cases with those described in our previous study [10]. As the control dataset, variant information data obtained from 9474 healthy individuals or patients with some common diseases were provided from the National Center Biobank Network (NCBN) [17]. Of the control samples, 358 with the registered diagnosis classified into the category of neurodegenerative diseases were not included in the association study. For the association study of GBA1 variants with MSA, relatedness between samples was checked with KING [18] using the data of the whole genome sequence data described below, and duplicated samples or those with 2nd degree or higher relationships were removed. Ancestry estimation was conducted for all the samples with Somalier using the whole genome sequence data [19] and those with ancestry estimation other than East Asian origin were removed. As the result, 499 MSA cases and 8777 controls were used for the following association study. The research protocol was approved by the institutional review board of each participating institution.
Sanger sequence analysis of polymerase chain reaction (PCR) products
PCR was conducted employing three primer pairs designed to selectively amplify exons 1–5, 5–7, and 8–11 of GBA1 but not those of GBA1LP, as previously described [6, 10, 20]. Direct nucleotide sequence analysis of the 11 coding regions and the splice sites of GBA1 was conducted employing a 3730xl Genetic Analyzer (Life Technologies, Carlsbad, CA). The sequences were analyzed using Sequence Scanner (Version 1.0, Applied Biosystems, 2005) and compared with the human GBA1 sequence using the GRCh38/hg38 Assembly. GBA1 variants were annotated based on RefSeq NM_00157.4 (NP_000148.2).
Short-read WGS analysis
WGS analysis was conducted employing the NovaSeq 6000 (Illumina, San Diego, CA) platform with 150-bp paired-end reads with a target depth of 30× for all 500 samples. WGS analysis of the 168 MSA samples was conducted at the International University of Health and Welfare (IUHW). WGS analysis of the 332 MSA samples and the 9116 control samples of the NCBN was conducted at the National Center for Global Health and Medicine (NCGM) [17]. Alignment to GRCh38 and variant calling of all the short reads were conducted employing the Parabricks v3.1.0 (Nvidia, Santa Clara, CA, US), which provides the capability to perform the analysis recommended by GATK at high speed using a GPU [21]. Generated gVCF files were then joint-called using the gVCFtyper program of the Sentieon package [22]. Alignment of the short reads was conducted for both GRCh37 and GRCh38 to compare the results of the variant calling because there are differences in the registered alleles in the GBA1–GBA1LP regions between the two reference genomes, as described below.
Long-read WGS analysis
We additionally conducted a long-read WGS analysis employing PacBio Sequel II (Pacific Biosciences, Menlo Park, CA) to further confirm the accurate variant calling of the GBA1–GBA1LP region, for which it was difficult to conclusively explain the inconsistencies between the results obtained by the Sanger and those by the short-read sequence analyses from a case with a structural variant. The HiFi reads were aligned to the reference genome of GRCh38 using Minimap2 [23] with the default parameter settings. The reads aligned to the GBA1–GBA1LP locus were visualized with IGV and reviewed manually to confirm the structural variation [24].
Extraction of variants pathogenic for Gaucher disease in patients with MSA
The pathogenicity of GBA1 variants for Gaucher disease was determined based on whether they were previously reported as pathogenic for Gaucher disease. We referred to the Human Gene Mutation Database (HGMD®) Professional 2023.2 to identify previously published reports [25]. The allele frequency of each variant in the MSA samples was compared with that in the control samples obtained from mapping to GRCh37 and our previous report [10], and Fisher’s exact test was conducted using R version 2.8.0.
Results
In the 500 MSA cases, we identified a total of 13 variants including eight missense (p.I20V, p.R202Q, p.R301H, p.S310G, p.V334I, p.L483P, p.A495P, and p.I528V) variants, two synonymous (p.V499V and p.K505K) variants, and three variants including a splicing variant (c.115+1G>A) and two indels (p.H313del and c.del1447_1466insTG) by either short-read or Sanger sequence analysis (Supplementary Table 1). Notably, inconsistencies in variant calling between the Sanger and the short-read sequence analyses were observed in variant calls for the five variants (p.A495P–p.V499V and c.115+1G>A were called only by short-read sequence analysis, whereas p.H313del, p.L483P and c.del1447_1466insTG were called only by Sanger sequence analysis) (Table 1).
Misalignment attributable to rare variants registered in GBA1LP of the reference genome (GRCh38)
Case 1: p.L483P was detected by Sanger sequence analysis, but undetected by short-read sequence analysis with alignment to GRCh38
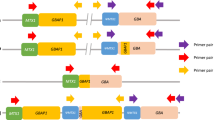
Chr1:155,233,639 – chr1:155,235,252 containing exons 10–11 of GBA1 and adjacent MTX1LP have an extraordinarily high sequence homology with the corresponding GBA1LP and MTX1 regions located in the vicinity of GBA1 on chromosome 1. In GBA1LP, GRCh38 registers a minor allele C with an allele frequency (AF) of 0.0017 instead of a major allele G at chr1:155,214,576, a minor allele C with an AF of 0.0107 instead of a major allele G at chr1:155,214,590, a minor allele C with an AF of 0.0108 instead of a major allele T at chr1:155,214,276, and a minor allele A with an AF of 0.213 instead of a major allele G at chr1:155,214,266 (Fig. 1). In contrast, GRCh37 registers major alleles at these four positions in GBA1LP. When the short reads were aligned to GRCh38, the reads containing C–C–G completely matched with the reference at chr1:155,214,576, chr1:155,214,590, and chr1:155,214,625 in GBA1LP, while the reads showed a mismatch at chr1:155,235,252 in GBA1. Consequently, p.L483P was not called a variant in GBA1 by the short-read sequence analysis (Fig. 2A). When the reads were aligned to GRCh37, the reads containing C–C–G showed two mismatches at chr1:155,184,367 and chr1:155,184,381 in GBA1LP, whereas the reads showed only one mismatch at chr1:155,205,043 in GBA1, resulting in correct alignment of all the short reads containing A at chr1: 155,205,043 (GRCh37) to the GBA1 locus. Consequently, p.L483P was called a variant in GBA1.

Physical map of GBA1, GBA1LP, MTX1, and MTX1LP. The positions of the major alleles different between GBA1 and GBA1LP are shown. Allele frequencies from the Japanese population (8.3KJPN) are displayed in red at the positions on GBA1LP (GRCh38) where the minor alleles, identical to the major alleles in GBA1, are registered. In GRCh37, the major alleles are registered at the corresponding positions

Misalignment of short reads attributable to rare variants registered in GBA1LP of the reference genome (GRCh38) in cases 1 and 2. Short-read alignments to GBA1 and GBA1LP in each case are displayed using IGV (with the mapping quality threshold = 20). The tables below the images of IGV show the allele depths retrieved from VCF files at the base positions on chromosome 1 of the reference genomes used for alignment. Note that the number of read bars shown in the IGV based on BAM files does not necessarily completely match with the read depths retrieved from the VCF. A Case 1 No variants were called by short-read sequence analysis, whereas p.L483P was detected by Sanger sequence analysis. When the short reads were aligned to GRCh38, the reads with C–C–G showed a complete match at chr1:155,214,576, chr1:155,214,590, and chr1:155,214,625 in GBA1LP, whereas the reads showed a mismatch at chr1:155,235,252 in GBA1. Consequently, p.L483P was not called a variant in GBA1. When the reads were aligned to GRCh37, the reads containing C–C–G showed two mismatches at chr1:155,184,367 and chr1:155,184,381 in GBA1LP. Consequently, p.L483P is called a variant in GBA1. B Case 2 p.A495P and p.V499V were called by short-read sequence analysis, whereas no variants were detected by Sanger sequence analysis. When the short reads were aligned to GRCh38, the reads with G–G–A showed two matches at chr1:155,235,203 and chr1:155,235,217 in GBA1, and the reads showed three mismatches at chr1:155,214,576, chr1:155,214,590, and chr1:155,214,625 in GBA1LP, resulting in alignment of the reads containing G–G–A to both GBA1 and GBA1LP. Consequently, p.A495P and p.V499V were called variants in GBA1. When the reads were aligned to GRCh37, the reads containing G–G–A had two mismatches to the reference genome of GBA1 at chr1:155,235,203 and chr1:155,235,217, whereas they had only one mismatch to the reference genome of GBA1LP at chr1: 155,184,416, and all the reads were correctly aligned to the GBA1LP sequences. As a result, neither p.A495P nor p.V499V was called at GBA1
Given the above results, we reanalyzed the variant calling of the other two cases with p.L483P in detail (Cases 5 and 6). In Cases 5 and 6, p.L483P was identified by Sanger sequence analysis as well as by short-read sequence analysis even with alignment to GRCh38. However, the allele depths in the VCF files obtained from alignment to GRCh38 were 13 and 4 (ref/alt) in Case 5, and 15 and 3 (ref/alt) in Case 6, indicating markedly biased allele balances, and reduced reliability in the variant calling. When a filtering option was set based on allele balance or read depth, p.L483P may well be missed in variant calling. In contrast, mapping to GRCh37 resulted in more reliable variant calling, with allele depths of 13 and 15 (ref/alt) and 16 and 7 (ref/alt) in Case 5 and Case 6, respectively.
Case 2: p.A495P and p.V499V were called by short-read sequence analysis with alignment to GRCh38, whereas the variants were not detected by Sanger sequence analysis
Similar to Case 1, due to the minor alleles registered in the GBA1LP in the reference genome (GRCh38), a misalignment of reads was observed. When the reads were aligned to GRCh38, the reads containing G–G–A showed two mismatches at chr1:155,235,203 and chr1:155,235,217 in GBA1, and the reads showed three mismatches at chr1:155,214,576, chr1:155,214,590, and chr1:155,214,625 in GBA1LP (Fig. 2B). Thus, the short-read sequence analysis employing GRCh38 as the reference genome could not unequivocally determine whether the reads containing G–G–A are derived from GBA1 or GBA1LP. Indeed, while the majority of the short reads containing G–G–A were aligned to GBA1LP, a limited number of short reads containing G–G–A were aligned to GBA1. Consequently, p.A495P and p.V499V were called by short-read sequence analysis with alignment to GRCh38. When the reads were aligned to GRCh37 (major alleles are registered in GBA1LP at the four positions), the reads containing G–G–A showed two mismatches to the reference genome of GBA1 at chr1:155,235,203 and chr1:155,235,217, whereas they showed only one mismatch to the reference genome of GBA1LP. As a result, reads containing G–G–A were fully aligned to the GBA1LP locus, and the p.A495P and p.V499V variants were not called in GBA1.
Case 3: c.del1447_1466insTG was detected by Sanger sequence analysis, but not called by short-read sequence analysis with alignment to GRCh38
Similar to Cases 1 and 2, the registration of four minor alleles in GBA1LP in GRCh38 increases the homology between GBA1 and GBA1LP, leading to fewer reads being specifically aligned to GBA1. Consequently, c.del1447_1466insTG was not called by short-read sequence analysis with alignment to GRCh38. When a read does not contain a GBA1-specific variant, it could be aligned to either GBA1 or GBA1LP, leading to a MAPQ value of 0. As a result, there are only three reads with a MAPQ > 20 (Supplementary Fig. 1), and the variant was missed through the variant calling process. Conversely, when the reads were aligned to GRCh37, which registers major alleles in GBA1LP, it became easier to align the reads specifically to GBA1. Consequently, there are twelve reads with a MAPQ > 20, and the variants were accurately called employing GRCh37 as the reference genome through the variant calling process.
Misalignment attributable to a structural variant involving GBA1/GBA1LP
Case 4: c.115+1G>A was called by short-read sequence analysis, while p.H313del was called by Sanger sequence analysis
When the read depth data of the short-read sequence analysis were analyzed, we noticed that the read depths in the part of the GBA1 region were increased to about 1.5, whereas those in the GBA1LP region were decreased to about 0.5, raising the possibility that the copy numbers of GBA1 and GBA1LP regions are three and one, respectively (Fig. 3A). Of note, long-read sequence analysis revealed that the long-reads 1 and 2 indeed contained a chimeric structure containing the GBA1LP–MTX1 and the GBA1 regions. The long-reads 3 and 4 contained a chimeric structure containing the GBA1LP–MTX1 and the GBA1–MTX1LP regions (Fig. 3B). Based on these results, we concluded that a gene conversion event occurred in the GBA1LP–MTX1 region due to the extraordinarily high homology between the GBA1–MTX1LP and GBA1LP–MTX1 regions. The region spanning the intron 2 of GBA1LP and the intron 5 of MTX1 (NC_00001.11:g.155213012_155218391 indicated by a box in Fig. 3B) was replaced by the region spanning the intron 2 of GBA1 and the intron 5 of MTX1LP (NC_00001.11:g.155233640_155240628). Of note, there is a deletion of CAC in this region (NC_00001.11:g.155237401_155237403del). Taken together with these observations, the complex structural variant is described as NC_00001.11:g.155213012_155218391delins[NC_00001.11:g.155233640_155237400; NC_00001.11:g.155237404_155240628] according to the guidelines of the Human Genome Variation Society (https://www.hgvs.org/content/guidelines). Consistent with the above interpretation, one of the paired-end reads spanning the breakpoint between GBA1 (upstream) and GBA1LP (downstream) contained GBA1-specific sequences derived from GBA1. On the other hand, the other reads contained sequences derived from GBA1LP, whose sequences are identical to that of GBA1 except for a mismatched base corresponding to c.115+1G>A in GBA1. Consequently, the reads were aligned to GBA1 resulting in the miscalling of c.115+1G>A in GBA1 (Fig. 3C). Regarding the p.H313del, which was called only by Sanger sequence analysis as the variant in GBA1, it was found to be a variant located in the structural variant involving GBA1LP as determined by the long-read sequence analysis (Fig. 3B). Thus, both Sanger sequence and short-read sequence analyses failed to correctly call the variants.

Structural variant involving the region spanning intron 2 of GBA1LP and intron 5 of MTX1. A The read depth ratios of the short reads aligned to GBA1, GBA1LP, MTX1. and MTX1LP of Case 4 were calculated as the ratio to the average read depth of the five control subjects, and plotted with chromosomal positions. The depth ratio in the GBA1 region was increased to about 1.5, while that in the GBA1LP region was decreased to about 0.5, raising the possibility that the copy numbers of GBA1 and GBA1LP are three and one, respectively. B Physical maps of the GBA1–GBA1LP locus in the reference genome and the structural variant derived from gene conversion involving the GBA1LP–MTX1 locus in Case 4 are shown. The region spanning intron 2 of GBA1LP (ENST00000689630.1) and intron 5 of MTX1 was replaced by the region spanning intron 2 of GBA1 and intron 5 of MTX1LP with a deletion of CAC (c.937_939del) in this region (NC_00001.11:g.155237400_155237404), which was clearly demonstrated by the four long-reads 1–4. C Across the breakpoint of GBA1 and GBA1LP of the complex structural variant, there are several sets of paired-end reads that were preferentially aligned to GBA1 because one of the paired-end reads contains an increased number of bases specific to the GBA1 reference sequence than those specific to GBA1LP. Consequently, c.115+1G>A was miscalled at GBA1 by the short-read sequence analysis. The regions with nucleotide sequences identical between GBA1 and GBA1LP are shown in gray. The regions with nucleotide sequences specific to GBA1 are shown in blue, whereas those specific to GBA1LP are shown in red
Variants pathogenic for Gaucher disease identified in MSA cases
Pathogenic variants for Gaucher disease identified in the MSA cases include p.S310G, p.L483P, and c.del1447_1466insTG (Table 2). Of the 499 MSA cases, three were heterozygous for p.L483P (AF: 0.003), one was heterozygous for c.del1447_1466insTG (AF: 0.001), and one was heterozygous for p.S310G (AF: 0.001) (Supplementary Table 1). In summary, five of the 499 (AF: 0.005) MSA cases were carriers of GBA1 variants pathogenic for Gaucher disease. The demographic features of the five MSA patients with the GBA1 variants are described in Supplementary Table 2. The ages at onset of the five patients ranged from 43 to 73 with the mean age at onset (standard deviation) of 58.6 (9.6), which was not significantly different from those of non-carriers. The clinical phenotypes of these five patients included three MSA-C and two MSA-P patients. Four patients were classified as probable MSA, while one was classified as possible MSA. The combined allele frequency of all the pathogenic variants in the MSA cases was higher than those described in the previous report [10], which, however, did not reach statistical significance. Of note, the allele frequency of p.L483P (0.003) in the MSA cases in this study was comparable to that (0.0035) described in the previous report and higher than that (0.0011) in control samples, which, however, did not reach a statistical significance, either (Table 2). We then conducted a meta-analysis of p.L483P combining the results of the current study (3/998 alleles for the disease group vs 20/17,554 alleles derived from the control group) and those of our previous study (4/1148 alleles for the disease group vs 2/1800 alleles for the control group) [10]. Since between-study variability was not observed (I2 = 0%, τ2 = 0, p = 0.87), the common effects model was employed for the meta-analysis, which demonstrated the odds ratio of 2.85 (95% CI; 1.05 – 7.76, p = 0.0400), indicating that the allele frequency of p.L483P is significantly higher in MSA than in control samples in the Japanese population (Fig. 4)

Forest plot showing the results of the meta-analysis of the association of p.L483P with MSA. The forest plot shows the results of the meta-analysis of the association of p.L483P with MSA combining the current study and the previous report [10]. Squares and horizontal lines represent estimated ORs and 95% CIs for individual series. The size of each square represents the statistical weight, the mean of the effect sizes using the inverse variance of the individual studies. Diamonds show the overall effects with 95% CIs. Since between-study variability was not observed (I2 = 0%, τ2 = 0, p =0.87), the common effects model was employed for the meta-analysis, which demonstrated the odds ratio of 2.85 (95% CI; 1.05 – 7.76, p = 0.0400), indicating that the allele frequency of p.L483P is significantly higher in MSA than in control samples in the Japanese population
Discussion
In this study, we analyzed GBA1 in a large number of Japanese MSA cases using both Sanger and short-read whole-genome sequence analyses to investigate the potential association of variants pathogenic for Gaucher disease with MSA. In the course of the sequence analysis, we noticed that miscalling could occur in variant calling based on short-read sequence analysis, which is attributable to the minor variants registered in GBA1LP in the GRCh38 reference genome but not in the GRCh37 reference genome. In addition, we noticed both Sanger and short-read sequence analyses failed to correctly call a large structural variant involving the GBA1LP locus.
As mentioned above, the four minor alleles at chr1:155,214,576, chr1:155,214,590, chr1:155,214,276, and chr1:155,214,266 of GRCh38 reference genome in GBA1LP, which indeed correspond to the major alleles of GBA1. The presence of these minor alleles in the GRCh37 causes misalignment, wherein short reads derived from GBA1 are aligned to GBA1LP or those derived from GBA1LP are aligned to GBA1. Specifically, this misalignment caused the disappearance of p.L483P due to the reads derived from GBA1 being mapped to GBA1LP, and the incorrect calling of p.A495P–p.V499V in GBA1, which actually originated from reads derived from GBA1LP. Although p.L483P was called by the short-read sequence analysis with alignment to GRCh38 in two of the three samples with MSA, these two cases also exhibited variant calling results with reduced confidences (Cases 5 and 6). When a filtering option was set based on allele balance or read depth, p.L483P may well be missed in variant calling using GRCh38 as the reference genome. Since p.L483P is one of the most prevalent pathogenic variants for Gaucher disease worldwide [1, 26] and one with the biggest impact of increasing the risk of PD across different ancestries [27], this issue is critical in clinical sequencing for suspected Gaucher disease and familial as well as sporadic PD.
Similar miscalling of variants is presumably present in the allele frequency databases based on GRCh38. For instance, gnomAD v2.1.1, which is based on alignment to GRCh37, registers p.L483P with an allele frequency of 0.0006703, whereas gnomAD v3.1.2, which is based on alignment to GRCh38, registers p.L483P with an allele frequency of 0.0002369. The lower frequency of p.L483P in v3.1.2 than that in v2.1.1, despite targeting similar ancestry groups, is presumably due to the underestimation of the p.L483P frequency depending on alignment to GRCh38. Additionally, the Japanese allele frequency database, jMorp (8.3KJPN) (https://jmorp.megabank.tohoku.ac.jp/), was based on data obtained with alignment to GRCh37, whereas databases thereafter (up to the latest 54KJPN) are based on data obtained with alignment to GRCh38, suggesting that similar errors in the allele frequency may be present. Therefore, it is necessary to recognize the reference genome used for mapping and carefully interpret the allele frequency database of the GBA1 p.L483P variant.
In Case 3, it is likely that c.1447_1466delinsTG was not called during the joint calling process due to a low number of reads specifically mapped to exon 9 of GBA1. This issue is also attributed to the presence of the four minor alleles registered in the GBA1LP. The existence of these minor alleles in GBAP1 in GRCh38 results in completely identical sequences at chr1:155,233,639 – chr1:155,235,252 (GRCh38) of GBA1–MTX1LP and the corresponding regions of GBA1LP–MTX1. This leads to a significant decrease in MAPQ values and exclusion of reads from variant calling (Supplementary Fig. 1). Therefore, special attention is required in the variant calling of this region containing exons 10–11 of GBA1. As described above, the presence of extraordinarily high homologous regions between GBA1 and GBA1LP is a frequent cause of gene–pseudogene rearrangements, many of which are causative variants for Gaucher disease. These variants may easily be missed based on the short-read sequence analysis. To overcome these problematic variant callings of GBA1, mapping to GRCh37 or T2T-CHM13/hs1 can prevent misalignment events because major alleles are registered as the reference sequences at these four positions in GBA1LP. Furthermore, a software tool, Gauchian, has recently been developed to detect recombinant alleles and single nucleotide variants in the GBA1 locus [28]. Gauchian is reported to have a more accurate genotyping performance than the BWA-GATK pipeline [28]. However, particularly in exons 9 to 11 of GBA1, false positive and false negative calls are not infrequently observed, and mapping to GRCh37 results in fewer errors than GRCh38, indicating that the presence of the four minor alleles registered in GRCh38 may contribute to some errors even with Gauchian [29]. Furthermore, recombinant alleles of GBA1 are mistakenly called a copy number change in the GBA1–GBA1LP locus in some cases, hence results obtained employing Gauchian should be interpreted with caution [29]. When comprehensive variant calling of GBA1 is pursued, alternative sequencing pipelines including Sanger or long-read sequence analyses would be required for accurate variant calling of these regions.
We also identified that a large structural variant involving the GBA1–GBA1LP locus can be misinterpreted as a single nucleotide variant within the GBA1 locus based on the short-read sequence analysis as well as on the Sanger sequence analysis. The splice donor site variant of c.115+1G>A, miscalled using short-read sequence analysis in Case 4, is known to be a pathogenic variant for Gaucher disease and associated with an increased risk of developing PD in the Ashkenazi Jewish population via exon 2 skipping of glucocerebrosidase mRNA [8, 9, 30]. Furthermore, a previous study showed that the frequency of structural variations in the GBA1–GBA1LP locus is considerably high [28]. As such, long-read sequence analysis is preferred for accurate variant calling and should be considered if a large structural variant is suspected when a biased distribution of read depths is observed with short-read sequence data.
Our present study showed that the allele frequency of p.L483P is higher than that in the control samples, which, however, did not reach statistical significance (Table 2). This may be due to a type II error, that is because allele counts of 9948 may be required in each group to reach a statistical significance for a variant with an allele frequency of 0.0030 vs 0.0011 with 80% power and alpha of 0.05. Thus, sufficiently large sample sizes would be required to demonstrate associations of rare variants with MSA. For this reason, we conducted a meta-analysis of p.L483P combining the independent datasets of the current study and those of our previous study [10], which indeed showed a significant association of p.L483P with MSA (Fig. 4). It has been proposed that GBA1 variants might contribute to the accumulation of α-synuclein in patients with PD [31, 32], while the role of GBA variants in the pathogenesis of MSA, which is also an α-synucleinopathy, remains unclear. Since we have recently reported siblings with MSA-C and PD sharing a GBA1 variant pathogenic for Gaucher disease, the observation may suggest a role of GBA1 variants as a common genetic basis underlying PD and MSA [33].
In conclusion, given the extraordinarily high homology between GBA1 and GBA1LP, variant calling should be interpreted with caution because different results may be obtained depending on the analysis pipelines. We emphasize the importance of alignment to the appropriate reference genome and utilizing long-read sequencing technology, particularly for this gene locus.
Change history
20 September 2024
A Correction to this paper has been published: https://doi.org/10.1038/s10038-024-01293-y
References
Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, Barranger JA, et al. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med. 1987;316:570–5. https://doi.org/10.1056/NEJM198703053161002
Zampieri S, Cattarossi S, Bembi B, Dardis A. GBA Analysis in Next-Generation Era. J Mol Diagn. 2017;19:733–41. https://doi.org/10.1016/j.jmoldx.2017.05.005
Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. 1989;4:87–96. https://doi.org/10.1016/0888-7543(89)90319-4
Tayebi N, Stubblefield BK, Park JK, Orvisky E, Walker JM, LaMarca ME, et al. Reciprocal and nonreciprocal recombination at the glucocerebrosidase gene region: implications for complexity in Gaucher disease. Am J Hum Genet. 2003;72:519–34. https://doi.org/10.1086/367850
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41:937–40. https://doi.org/10.1136/jmg.2004.024455
Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol. 2009;66:571–6. https://doi.org/10.1001/archneurol.2009.72
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–61. https://doi.org/10.1056/NEJMoa0901281
Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. 2015;84:880–7. https://doi.org/10.1212/WNL.0000000000001315
Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain. 2015;138:2648–58. https://doi.org/10.1093/brain/awv179
Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Dürr A, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. 2015;2:417–26. https://doi.org/10.1002/ACN3.185
Srulijes K, Hauser A-K, Guella I, Asselta R, Brockmann K, Schulte C, et al. No association of GBA mutations and multiple system atrophy. Eur J Neurol. 2013;20:e61–2. https://doi.org/10.1111/ene.12086
Wernick AI, Walton RL, Koga S, Soto-Beasley AI, Heckman MG, Gan-Or Z, et al. GBA variation and susceptibility to multiple system atrophy. Parkinsonism Relat Disord. 2020;77:64–9. https://doi.org/10.1016/j.parkreldis.2020.06.007
Segarane B, Li A, Paudel R, Scholz S, Neumann J, Lees A, et al. Glucocerebrosidase mutations in 108 neuropathologically confirmed cases of multiple system atrophy. Neurology. 2009;72:1185–6. https://doi.org/10.1212/01.wnl.0000345356.40399.eb
Woo EG, Tayebi N, Sidransky E. Next-generation sequencing analysis of GBA1: the challenge of detecting complex recombinant alleles. Front Genet. 2021;12:684067. https://doi.org/10.3389/fgene.2021.684067
Chikada A, Orimo K, Mitsui J, Matsukawa T, Ishiura H, Toda T, et al. The Japan MSA registry: a multicenter cohort study of multiple system atrophy. Neurol Clin Neurosci. 2024. https://doi.org/10.1111/ncn3.12809
Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. https://doi.org/10.1212/01.wnl.0000324625.00404.15
Kawai Y, Watanabe Y, Omae Y, Miyahara R, Khor SS, Noiri E, et al. Exploring the genetic diversity of the Japanese population: insights from a large-scale whole genome sequencing analysis. PLoS Genet. 2023;19. https://doi.org/10.1371/journal.pgen.1010625
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73. https://doi.org/10.1093/bioinformatics/btq559
Pedersen BS, Bhetariya PJ, Brown J, Kravitz SN, Marth G, Jensen RL, et al. Somalier: Rapid relatedness estimation for cancer and germline studies using efficient genome sketches. Genome Med 2020;12. https://doi.org/10.1186/s13073-020-00761-2
Koprivica V, Stone DL, Park JK, Callahan M, Frisch A, Cohen IJ, et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am J Hum Genet. 2000;66:1777–86. https://doi.org/10.1086/302925
Franke KR, Crowgey EL. Accelerating next generation sequencing data analysis: an evaluation of optimized best practices for Genome Analysis Toolkit algorithms. Genom Inf. 2020;18:e10. https://doi.org/10.5808/GI.2020.18.1.e10
Freed D, Aldana R, Weber JA, Edwards JS. The Sentieon Genomics Tools - a fast and accurate solution to variant calling from next-generation sequence data. bioRxiv [Preprint]. 2017. Available from: https://doi.org/10.1101/115717
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–100. https://doi.org/10.1093/bioinformatics/bty191
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. https://doi.org/10.1038/nbt.1754
Stenson PD, Mort M, Ball EV, Shaw K, Phillips AD, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. https://doi.org/10.1007/s00439-013-1358-4
Horowitz M, Zimran A. Mutations causing gaucher disease. Hum Mutat. 1994;3:1–11. https://doi.org/10.1002/humu.1380030102
Zhang Y, Shu L, Sun Q, Zhou X, Pan H, Guo J, et al. Integrated genetic analysis of racial differences of common GBA variants in Parkinson’s disease: a meta-analysis. Front Mol Neurosci. 2018;11:43. https://doi.org/10.3389/fnmol.2018.00043
Toffoli M, Chen X, Sedlazeck FJ, Lee C-Y, Mullin S, Higgins A, et al. Comprehensive short and long read sequencing analysis for the Gaucher and Parkinson’s disease-associated GBA gene. Commun Biol. 2022;5:670. https://doi.org/10.1038/s42003-022-03610-7
Tayebi N, Lichtenberg J, Hertz E, Sidransky E. Is Gauchian genotyping of GBA1 variants reliable? medRxiv [Preprint]. 2023. Available from: https://doi.org/10.1101/2023.10.26.23297627
He GS, Grabowski GA. Gaucher disease: a G+1––A+1 IVS2 splice donor site mutation causing exon 2 skipping in the acid beta-glucosidase mRNA. Am J Hum Genet. 1992;51:810–20.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. https://doi.org/10.1016/j.cell.2011.06.001
Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, et al. Acid β-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol. 2011;69:940–53. https://doi.org/10.1002/ana.22400
Matsukawa T, Porto KJL, Mitsui J, Chikada A, Ishiura H, Takahashi Y, et al. Clinical and genetic features of multiplex families with multiple system atrophy and Parkinson’s disease. Cerebellum. 2024;23:22–30. https://doi.org/10.1007/s12311-022-01426-z
Acknowledgements
We thank Mio Takeyama, Keiko Hirayama, and Zhenghong Wu for their support in laboratory experiments. This work was supported in part by KAKENHI (19H03425 and 22H02823) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and grants (16kk0205001h0001, 17kk0205001h0002, 18kk0205001h0003, 20ek0109491h0001, 21ek0109491h0002, 22ek0109491h0003, and 23ek0109673h0001) from the Japan Agency for Medical Research and Development (AMED) to ST. This work was supported in part by Grants-in-Aid from the Research Committee of CNS Degenerative Diseases, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health, Labour and Welfare Sciences Research Grants, the Ministry of Health, Labour and Welfare, Japan to ST and grants (21ek0109573h0001, 22ek0109573h0002, and 23ek0109573h0003) from the AMED to JM.
Funding
Open Access funding provided by The University of Tokyo.
Author information
Authors and Affiliations
Consortia
Contributions
KO and ST designed and conceptualized the study, analyzed the data; wrote the draft of the manuscript. JM designed and conceptualized the study; wrote the draft of the manuscript. MT, JN, YO, YK, and KT contributed to the whole-genome sequence analysis. TM, HI and TT wrote the draft of the manuscript. NCBN Controls WGS Consortium contributed to the collection of the control samples.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Institutional Review Board of the University of Tokyo.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Corrections have been made to the article: In the abstract, “The meta-analysis incorporating a previous report demonstrated a significant association of p.L483P with MSA with an odds ratio of 2.92 (95% CI; 1.08 – 7.90, p = 0.0353).” should read “The meta-analysis incorporating a previous report demonstrated a significant association of p.L483P with MSA with an odds ratio of 2.85 (95% CI; 1.05 – 7.76, p = 0.0400).” Figure 4 has been updated. In the results section, “Since between-study variability was not observed (I2= 0%, τ2 =0, p= 0.83), the common effects model was employed for the meta-analysis, which demonstrated the odds ratio of 2.92 (95% CI; 1.08 – 7.90, p = 0.0353)…” should read “Since between-study variability was not observed (I2= 0%, τ2 =0, p= 0.87), the common effects model was employed for the meta-analysis, which demonstrated the odds ratio of 2.85 (95% CI; 1.05 – 7.76, p = 0.0400).
Supplementary information
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Orimo, K., Mitsui, J., Matsukawa, T. et al. Association study of GBA1 variants with MSA based on comprehensive sequence analysis -Pitfalls in short-read sequence analysis depending on the human reference genome-. J Hum Genet (2024). https://doi.org/10.1038/s10038-024-01266-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s10038-024-01266-1
- Springer Nature Singapore Pte Ltd.