
Abstract
Within the tumor microenvironment (TME), regulatory T cells (Tregs) play a key role in suppressing anticancer immune responses; therefore, various strategies targeting Tregs are becoming important for tumor therapy. To prevent the side effects of nonspecific Treg depletion, such as immunotherapy-related adverse events (irAEs), therapeutic strategies that specifically target Tregs in the TME are being investigated. Tumor-targeting drug conjugates are efficient drugs in which a cytotoxic payload is assembled into a carrier that binds Tregs via a linker. By allowing the drug to act selectively on target cells, this approach has the advantage of increasing the therapeutic effect and minimizing the side effects of immunotherapy. Antibody–drug conjugates, immunotoxins, peptide–drug conjugates, and small interfering RNA conjugates are being developed as Treg-targeting drug conjugates. In this review, we discuss key themes and recent advances in drug conjugates targeting Tregs in the TME, as well as future design strategies for successful use of drug conjugates for Treg targeting in immunotherapy.
Similar content being viewed by others

Introduction
Over the past few years, advancements in anticancer immunotherapies have increasingly aided treatment of solid and hematological malignancies. Immune checkpoint inhibition (ICI) and adoptive transplantation of genetically engineered T cells (CAR-T cells) have been successfully used to treat malignancies. However, there are some obstacles to the current strategy of immunotherapy: (1) lack of T-cell access due to disorganized neovasculature and stromal barriers, tumor antigens and the mutational burden, and tumor heterogeneity and (2) T-cell exhaustion due to suppressive tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and hypoxia in the suppressive tumor microenvironment (TME) (Fig. 1). Therefore, new approaches have been adopted to overcome these challenges. For example, TAMs, Tregs, immunosuppressive cytokines, and hypoxia have been targeted to enhance T-cell access and overcome TME-mediated immune suppression1.

Regulatory T cells (Tregs), M2 macrophages (MΦ), and myeloid-derived suppressor cells (MDSCs) are predominant in the immunosuppressive TME during tumor development.
The TME is similar to the play Othello; it is similar to a zero-sum game. On a game board, antitumor immune cells and immunosuppressive actions strongly oppose each other. White discs, such as effector T cells, M1 macrophages, and natural killer cells, compete with black discs, such as Tregs, M2 macrophages, and tumor cells. All the cells play strategically. When an effector T-cell kills a tumor cell, similar to turning a black disc into a white disc, the Tregs hidden in the diagonal corners turn the white disc all at once and cause numerous effector T cells to disappear instantly (Fig. 1).
Tregs accumulate at high rates in the immunosuppressive TME during tumor development. They activate effector T-cell suppression by the following mechanisms: (1) regulation of antigen-presenting cell (APC) function through competitive blockade of CD80 and CD28 binding costimulation by CTLA-4, (2) secretion of immunosuppressive molecules such as IL-10 and TGF-β, (3) suppression of effector T-cell growth by inducing depletion of the local IL-2 pool due to high CD25 (IL-2Ra) expression, and (4) secretion of perforins and granzymes to cause apoptosis of effector T cells. Therefore, the effectiveness of immunotherapy can be enhanced by targeting Tregs, enabling antitumor treatment1,2.
A drug conjugate combines a cytotoxic drug with a carrier capable of binding to a specific target using chemical linkers. Drug conjugates are being actively developed for targeted delivery to specific regions and effectively kill target cells with remarkable potency, even at small dosages. Recently, drug conjugates targeting Tregs have been developed and shown to be successful3. They represent innovative immunotherapeutic agents and act by altering the immunosuppressive environment in a manner different from the existing method of targeting cancer cells4.
In this review, we focus on the immunosuppressive TME and discuss current approaches for targeting/depleting Tregs to overcome TME-mediated immune suppression. In addition, we review drug conjugates for targeting/depleting Tregs in novel trials.
Immunosuppressive cells in TME
The TME is a highly complex and heterogeneous ecosystem that includes malignant cells and host-interacting cells such as endothelial cells, stromal fibroblasts, and various immune cells. In this ‘nest’, tumor cells undergo differentiation, epigenetic changes, dissemination, and immune evasion5.
Transformation of normal cells into tumor cells relies on irreversible genetic alterations that trigger oncogenic signaling pathways. According to Swann and Smyth, “Transformed cells that escape intrinsic control are subjected to extrinsic tumor suppressor mechanisms that detect and eliminate developing tumors before they become clinically apparent.” This is the elimination phase of a broader process known as cancer immunoediting6.
Immunoediting consists of immune surveillance and tumor progression in three stages: elimination, equilibration, and escape. The immune elimination phase includes both innate and adaptive immune responses against tumor cells. Most tumor cells are destroyed at this stage; however, some can survive and reach equilibrium with the immune system. During the equilibrium phase, tumor cells with a nonimmunogenic phenotype are selected and grow. Tumor cell variants that acquire clearance resistance enter the escape phase. During the escape phase, tumor cells continue to grow and expand in an uncontrolled manner, eventually leading to malignancy7.
Tumor immunogenicity is modified throughout these phases, and immunosuppressive mechanisms that enable disease progression are acquired8. The immunosuppressive TME is enriched in immunosuppressive cells such as Tregs, TAMs, myeloid-derived suppressor cells, tumor-associated neutrophils, and tumor-associated dendritic cells (Fig. 1). Cytokines and related molecules secreted by these cells and tumor cells promote tumor progression and mediate immune escape9.
Tregs in the TME and specific markers
Tregs are one of the representative immunosuppressive cells and functionally ambivalent. They have several positive roles, including a role in immune tolerance, inhibition of autoimmune diseases, prevention of tissue damage, and regulation of inflammation after infection; negative roles include interference with cancer immunity. Tregs are chemoattracted to the TME by a chemokine gradient secreted by tumor cells and are then activated to suppress the antitumor immune response and release immunosuppressive cytokines such as IL-10, TGF-β, and IL-35. In addition, Tregs suppress other immune cells, such as basophils, eosinophils, and mast cells, and release perforins and granzymes upon binding to T-cell receptors that target cytolysis of effector T cells and APCs5.
The Sakaguchi group noted in a recent review paper that “Tregs are one of the important roadblocks in tumor treatment, so it is necessary to remove them before tumor defeat”10. Targeting Tregs is an efficient strategy for cancer treatment; however, nonspecific systemic Treg depletion causes problems such as immunotherapy-related side effects. Hence, there are several criteria for selecting Tregs in tumors through their specific markers for targeting.
Sakaguchi et al. classified CD4 + FOXP3 + T cells in humans into the following three subsets based on expression of CD45RA, a cell surface marker of naïve T cells, and the transcription factor FOXP3: fraction 1 (Fr. 1) naïve Tregs, as defined as FOXP3low (CD25low) CD45RA+ cells; fraction 2 (Fr. 2) effector Tregs (eTregs), as defined as FOXP3high (CD25high) CD45RA− cells; and fraction 3 (Fr. 3), and non-Tregs, as defined as FOXP3low (CD25low) CD45RA cells11.
Naive Tregs (Fr. 1) are Tregs that leave the thymus and have weak immunosuppressive activity. Upon receiving T-cell receptor stimulation, they differentiate into eTregs (Fr. 2), which have strong immunosuppressive activity. In most cancers, the frequency of eTregs is 2–5% in peripheral blood but 10–50% in tumor tissue; thus, they are mostly distributed in tumor tissue. Non-Tregs (Fr. 3) do not have immunosuppressive properties and produce inflammatory cytokines such as interferon (IFN)-γ and IL-1712.
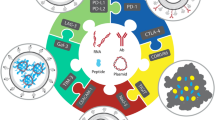
Tregs in the TME have several membrane targets, such as CD25, CTLA-4, PD-1, ICOS, GITR, OX40, CCR4, and CCR8, that can be exploited to deplete these cells. Treg-targeting drug conjugates target several of these markers (Fig. 2)13.

ADC antibody–drug conjugate, DT diphtheria toxin, PE Pseudomonas exotoxin A, siRNA small-interfering RNA.
CD25 is an IL-2 receptor, and IL-2 is an important factor for T-cell maintenance. As eTregs are CD25high cells, CD25 can be targeted to deplete eTregs. CD25 has been used as a target for the antibody–drug conjugate (ADC) ADCT-301 as well as the immunotoxins 2E4-PE38, Denileukin Diftitox, and LMB-23,14,15,16.
CTLA-4 is an important molecule for Tregs and functions as an immune checkpoint protein that negatively regulates T-cell activation and contributes peripherally. Tregs express CTLA-4 to suppress APCs. CTLA-4 binds to CD80 and CD86 with higher affinity than CD28 and acts as a competitive inhibitor of CD28 on APCs. It has been used as a target for small interfering RNA (siRNA) conjugates (CTLA4apt–STAT3 siRNA, NPsiCTLA-4, cSNPs, and hybrid SNPs)17,18,19.
Because Tregs express costimulatory receptors such as GITR, OX40, and ICOS, targeting them can lead to Treg depletion and functional regulation. GITR, which plays an essential role in Treg expansion, is expressed at high levels by Tregs but at low levels by resting CD4+ and CD8 + T cells. OX40 is constitutively expressed by a subset of Tregs but is also found on effector T cells. ICOS, which is essential for Treg function and homeostasis, is highly expressed by activated Tregs among the tumor-infiltrating lymphocytes (TILs) of patients with gastric cancer13.
Chemokine receptors that allow Tregs to migrate to the TME are candidate molecules for Treg depletion. CCR4 is highly expressed by eTregs but not by most effector T cells, except for naive Tregs and some Th2 and Th17 cells in peripheral blood13. CCR8 is a chemokine receptor expressed at high levels on the surface of tumor-infiltrating Tregs but not on peripheral Tregs or effector T cells20,21,22. Reanalysis of T-cell scRNA-seq datasets of non-small cell lung cancer23 and colorectal cancer24 has shown that CCR4 and CCR8 are more selectively expressed in tissue-associated Tregs than in Tregs in peripheral or healthy tissues25.
FOXP3 is considered a master regulator of the immunosuppressive phenotype of Tregs26. Naive and memory CD4 + T cells differentiate into Tregs by inducing FOXP3 expression27. FOXP3 is a promising target for Treg suppression; however, because of its nuclear localization, targeting requires strategies or efficient delivery of suppressive molecules into cells28. Cell-penetrating peptides or short RNA strands can be used as efficient FOXP3 target molecules. P60, CM1315, and FOXP3 393–403 are peptides developed to target FOXP329,30. siRNAs targeting FOXP3 are predesigned and marketed for use in gene silencing31, or they can be designed and produced directly based on target sequences32. Manrique-Rincón et al. synthesized a 4-1BB aptamer conjugated with a small antisense RNA (sasRNA) for Foxp3 silencing in melanoma-bearing mice28.
Antibody–drug conjugates
ADCs are drug conjugates in which monoclonal antibodies (mAbs) are coupled to cytotoxic payloads via a linker (Fig. 2). Since Paul Ehrlich coined the term “magic bullet” 100 years ago, postulating that some compounds can directly access a desired target in cells to treat diseases, ADC use has become an important option in cancer treatment33.
The ADC gemtuzumab ozogamicin (Mylotarg®) was approved for the first time by the US Food and Drug Administration (FDA) in 2000. To date, 14 ADCs have been approved, and more than 100 ADC candidates are currently in the clinical stage34.
Monoclonal antibodies are specific for cell surface targets, provide specificity and efficacy, and can be used to deplete or disrupt Treg function35. Treg-targeting mAbs such as daclizumab (CD25), ipilimumab (CTLA-4), nivolumab (Pd-1), and mogamlizumab (CCR4) are used to induce antitumor immunity, including antibody-dependent cytotoxicity, complement-dependent cytotoxicity, and antibody-dependent cellular phagocytosis effects36.
Linkers play a pivotal role in the stability and homogeneity of ADCs by maintaining their stability in systemic circulation and releasing the payload after internalization at the target site. Unstable linkers cause systemic toxicity by releasing small cytotoxic molecules into the bloodstream. Linkers are subdivided into two categories based on their mechanism of payload release: cleavable and noncleavable37.
Payloads are largely divided into DNA-damaging agents (e.g., pyrrolobenzodiazepines [PBDs], calicheamicins, duocarmycins, and SN-38) and microtubule-disrupting agents (e.g., auristatins and maytansines)38. Calicheamicins, duocarmycins, and PBDs are natural antibiotics that bind to DNA minor grooves and cause strand breaks39; SN-38 inhibits TOP1 (topoisomerase I), causing S-phase-specific cytotoxicity and mediating DNA single-strand breaks40. Auristatin monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF) induce G2/M phase cell cycle arrest by interfering with microtubule polymerization when binding to the β-subunit of the tubulin dimer41., DM1 (a derivative of maytansine 1) is a maytansinoid used in T-DM142.
A good ADC has a high internalization rate, low immunogenicity, high binding specificity and affinity, a strong payload, and a stable linker. It provides improved efficacy compared to traditional antibody drugs and enables targeted delivery of cytotoxic drugs. Although ADCs have been developed to limit toxicity, with progression of clinical trials, the limitations of ADCs, such as hepatic, neurological, and ocular toxicity as side effects, have been noted, as has drug resistance due to payload internalization and retention37.
ADCs targeting Tregs in the TME
The existing ADCs were mainly developed to target cancer cells directly. However, the study of Zammarchi et al. relied on ADCs that directly target immune cells rather than tumor cells, providing a proof-of-concept for an entirely new application of ADCs as immunotherapeutics. Using camidanlumab tesirine (ADCT-301), which targets human CD25 based on a pyrrolobenzodiazepine (PBD) dimer, the authors demonstrated that Treg depletion and antitumor immunity can eradicate tumors (Fig. 2).
Camidanlumab tesirine (ADCT-301) is being evaluated in several clinical trials (Table 1). These studies include the following: a phase 2 clinical trial in patients with relapsed or refractory Hodgkin’s lymphoma (NCT04052997)43; a phase 1 with relapsed or refractory Hodgkin’s lymphoma and non-Hodgkin’s lymphoma (NCT02432235)44; a phase 1 in acute myeloid leukemia or acute lymphoblastic leukemia (NCT02588092)45; and a phase 1 in solid tumors (NCT03621982)4.
Immunotoxins
An immunotoxin is an immunoconjugate that induces death of a target cell by combining an antibody with target-specific high-affinity binding activity with other molecules, such as radioisotopes, chemicals, siRNA, and cytotoxic proteins (Fig. 2)46. To date, three cytotoxins and immunotoxins, namely, Denileukin diftitox (Ontak®)15, Tagraxofusp (Elzonris®)47, and Moxetumomab pasudotox (Lumoxiti®)48, have been FDA-approved for treatment of several forms of hematological cancer, and more than 20 treatments are being clinically tested49.
Immunotoxins have a mechanism of action similar to that of ADCs, in which fragments of immunotoxin mAbs bind to the target cell surface, and the protein toxin enters the cell and interferes with cellular processes; however, in contrast to ADCs, immunotoxins have potent protein toxins as payloads and can effectively kill quiescent, nondividing cells50. The payloads used for immunotoxins are cytotoxic proteins derived from bacteria, plants, or humans. Representative examples include Pseudomonas aeruginosa exotoxin A (PE), diphtheria toxin (DT), ricin, saporin, gelonin, proteases, and ribonucleases (RNases)46. ADCs can induce off-target toxicity due to improper payload separation from chemical linkers; however, modern recombinant immunotoxins do not have such problems because specific intracellular proteases to separate the recombinant peptide linkers are required51.
Immunotoxins can induce vascular leak syndrome through enzymatic activity against the endothelium. As they are immunogenic molecules, anti-immunotoxin antibodies are produced when administered to patients. Various strategies have been studied to overcome this problem51. Because immunotoxins are relatively large molecules, they cannot readily penetrate solid tumors. For improved solid tumor penetration and antitumor efficacy, the target moiety of an immunotoxin is usually a single-chain variable fragment (scFv) or a novel scaffold52.
Immunotoxins for targeting Tregs in the TME
Anti-Tac (Fv)‐PE38 (LMB-2) is a recombinant immunotoxin composed of a variable domain (Fv) of antibody binding to Tac (CD25) and Pseudomonas exotoxin A (PE38). LMB-2 is an effective treatment for hairy cell leukemia53. LMB-2 can eliminate Tregs expressing CD4, CD25, and FOXP3 in humans but not in mice54. Powell et al. studied the improvement in the clinical effect of immunotherapy in patients with melanoma by inducing elimination of Tregs by administering LMB-2 and demonstrated the capacity of a CD25-directed immunotoxin to selectively mediate a transient partial reduction in circulating and tumor-infiltrating Tregs in vivo16. A phase 2 trial of LMB-2—Fludarabine and Cyclophosphamide for Adult T-Cell Leukemia— (NCT00924170)55 and a phase 2 trial —LMB-2 to Treat Hairy cell Leukemia— (NCT00321555)56 are ongoing (Table 1).
Denileukin diftitox (DAB389-IL-2, Ontak®) is an engineered immunotoxin combining IL-2 and diphtheria toxin46. It was FDA-approved in 2008 for treatment of persistent or recurrent cutaneous T-cell lymphoma and has been used to eliminate CD25+ lymphoma cells and Tregs57. However, its development was stopped in 2011 due to severe toxicity, and it was voluntarily withdrawn from the market in 2014 due to manufacturing difficulties. Second-generation denileukin diptitox (s-DAB-IL-2 (V6A)) with reduced vascular leakage was developed by Cheung et al. 58, and E7777 (I/Ontak), a purified version of denileukin diptitox, was developed. E7777 is undergoing clinical research59. The phase 3 trial “Persistent or Recurrent Cutaneous T-Cell Lymphoma” (NCT01871727) and phase 2 trial “Peripheral/Cutaneous T-cell Lymphoma” (NCT02676778) have been completed60. “T-regulatory Cell Depletion With E7777 Combined With Pembrolizumab in Recurrent or Metastatic Solid Tumors” is a phase 2 trial in the recruiting stage (NCT05200559).
2E4-PE38 is a single-chain recombinant anti-CD25 immunotoxin in which the Fv portion of rat mAb 2E4 is genetically fused to a 38-kDa fragment of PE and has strong cytotoxic activity. Onda et al. treated mice with breast cancer, mesothelioma, and colon cancer by intratumoral injection of 2E4-PE38 to kill Tregs and found that the immunotoxin caused complete regression of the treated tumors, as well as most distal noninjected tumors, inducing systemic antitumor immunity3.
DT390‐BiscFv(1567)‐6xHis is a diphtheria toxin-based recombinant anti-human CCR4 immunotoxin. Wang et al. developed it using the scFv of an anti-CCR4 monoclonal antibody (mAb1567)61 and diphtheria toxin (DT390)62. They demonstrated the binding and in vivo efficacy of DT390‐BiscFv(1567)‐6xHis on nonhuman primate (NHP) CCR4+ FOXP3+ monkey Tregs. Its administration in monkeys led to depletion of 78-89% of CCR4+ FOXP3+ monkey Tregs for 10 days, while not affecting other cells, including CD8+ T cells, B cells, and natural killer cells63. Wang et al. subsequently synthesized an IL2‐CCR4/CCR4‐IL2 bispecific immunotoxin by recombining Ontak®‐like human IL2 fusion toxin and ccr4 immunotoxin. They demonstrated that the efficacy of bispecific human IL2‐CCR4 immunotoxin in mouse cutaneous T-cell lymphoma (CTCL) was more effective than when used alone64.
Peptide–drug conjugates
Peptide–drug conjugates (PDCs) are next-generation targeted therapies that were developed after ADCs. Currently, there are two drugs approved for marketing by the FDA65.
Because most mAbs have a molecular weight of ~150 kDa, they are too large to infiltrate tumor tissues. They may be immunogenic and aggregate in excretory organs, such as the kidney and liver66,67,68. Additionally, mAb generation is expensive and time-consuming69.
Peptides well penetrate solid tissues due to their low molecular weight and are considered safe due to their low immunogenicity and nontoxic metabolite production70. They are also inexpensive, easily synthesized, and transformable71.
PDCs consist of a homing peptide, payload, and linker. The peptides in the PDC are target cell specific and should induce receptor-mediated endocytosis of the conjugate. Payloads include highly toxic maytansine, camptothecin derivatives, auristatin, or doxorubicin. Linkers are chosen to allow sufficient circulation time for the conjugate to reach the target cell72.
The peptides used in PDC can be broadly divided into two categories: cell-penetrating peptides (CPPs) and cell-targeting peptides (CTPs). CPPs use specific amino acid sequences to effectively transport cell-impermeable compounds or drugs across cell membranes to reach their intracellular targets. CTP is an ideal homing molecule because, similar to mAbs, it binds with high affinity to receptors overexpressed on the target cell surface. Most homing peptides are linear and bind well but have several drawbacks, including enzymatic degradation at the termini, chemical instability, and fast renal clearance. To compensate for this, cyclization or peptide stapling of linear peptides has been used, as have conjugation to gold nanoparticles and antibody Fc or albumin73,74.
Peptides can be selected by screening peptide libraries according to their binding affinity to the target. Peptide libraries are systematic combinations of diverse peptides widely used to identify gene- or cell-targeted therapies for cancer treatment. Depending on the type and method of displaying the target, such libraries include ribosome display, phage display, mRNA display, and protein fragment complementation analyses30.
Peptides for targeting Tregs in the TME
Until now, development of PDCs as a Treg target has not been reported, but studies on peptides that inhibit Tregs are being actively conducted. CPPs can be used as a payload in a drug conjugate, and P60 conjugated with a CD28 aptamer is a good example75. CTPs with high binding affinity to the Treg membrane target have the potential to be used as a homing molecule for PDCs. Studies on peptides for targeting Tregs are mostly in the preclinical stage (Table 2).
P60 is a peptide that binds to FOXP3 and prevents its nuclear translocation. Lozano et al. used phage display screening and synthesized P60 (RDFQSFRKMWPFFAM), a 15-amino acid sequence that binds well to FOXP3. P60 penetrates cell membranes, binds to FOXP3, prevents nuclear translocation, reduces FOXP3’s ability to inhibit transcription factors NF-κB and NFAT, and impairs Treg activity76. CM-1315 is a high-tech product of P60 with improved FOXP3-binding ability and metabolic stability via modification of several base sequences of P60 to form a ring77. As peptides can penetrate all cells, they require a high dose to exert an antitumor effect. Lozano et al. conjugated a CD28 aptamer78 to P60 to address this drawback. CD28 is a T-cell-specific receptor that is abundant in Tregs. In an in vivo mouse experiment, when CD28Apt-P60 was coadministered with the AH1 vaccine, an antitumor effect on CT26 was shown at a lower dose than with free P6079.
FOXP3 393–403 is derived from FOXP3 and responsible for DNA binding. FOXP3 393–403 disrupts interaction between FOXP3 and NFAT1, enhances expression of proinflammatory cytokines and downregulates immunosuppressive molecules80.
SAH-BCL9B is an inhibitory peptide that blocks binding of β-catenin/B-cell lymphoma 9 (BCL9). Mutations and overexpression of beta-catenin are associated with many cancers. Kawamoto et al. analyzed the crystal structure of the BCL9 complex and developed this BCL9-derived peptide81. Tumor-specific Wnt/β-catenin signaling promotes immune evasion of tumor cells, enhances Treg production, inhibits CD8 + T-cell activation and invasion, converts DCs to an immunoregulatory phenotype, and differentiates CD8 + T cells into effector cells82. When SAH-BCL9B was administered in vivo, the Treg population in tumors decreased, whereas the populations of CD8+ effector T cells and CD103+ DCs increased81. Feng et al. synthesized hsBCL9CT-24 to improve the activity of SAH-BCL9B based on the BCL9 homology domain 2. Because hsBCL9CT-24 has a stronger binding affinity for β-catenin than BCL9-HD2A, it effectively reduces Tregs and increases DCs, promotes cytotoxic T-cell infiltration into a tumor, and shows a strong synergistic effect with anti-PD-183.
P144 and P17 are peptide inhibitors that act on the TGF-β signaling pathway. P144 (TSLDASIIWAMMQN) is hydrophobic and forms the TGF-β1 binding region of human type III TGF-β1 receptor (TGFβRIII)84. P17 (KRIWFIPRSSWYERA) is a TGF-β-binding peptide identified by phage display screening85. TGF-β is produced at high levels by Tregs and suppresses the immune response by downregulating activity of T cells and natural killer cells. Llopiz et al. found that administration of P144 and P17 in combination with administration of the adjuvant molecule poly (I: C) and an agonist anti-CD40 antibody to mice bearing T-cell lymphoma increases antitumor immune responses86.
Fc-TPP-11 is synthesized by fusion of the NRP-1 binding peptide and the immunoglobulin Fc region of an antiangiogenic agent87. NRP1 is a membrane-bound coreceptor to a tyrosine kinase receptor for vascular endothelial growth factor and semaphorin family member88. Jung et al. demonstrated that Fc-TPP-11 inhibits Treg proliferation and function by antagonizing NRP-1. Fc-TPP-11 selectively suppresses intratumoral Treg function without compromising peripheral Treg function89.
Peptide-R29 is a novel CXCR4 antagonist derived from peptide R with higher avidity and stability. Santagata et al.90 demonstrated that CXCR4 is highly expressed in Tregs and that CXCR4 antagonism induced by a novel peptide antagonist, peptide-R29, efficiently reverses Treg suppression of effector T-cell proliferation.
Small interfering RNA
siRNA is a double-stranded RNA (dsRNA) molecule 21–23 nucleotides in length that specifically causes RNA interference (RNAi), a posttranscriptional method of silencing gene expression. siRNA-based therapies have been developed for 20 years, and to date, four siRNA formulations have been FDA-approved for management of rare metabolic diseases, namely, patisiran, givosiran, lumasiran, and inclisiran91.
siRNAs exert their effects at the posttranscriptional level. The dsRNA is unwound into a single-stranded siRNA (guide strand) by an RNase III-like enzyme called Dicer, and the siRNA guide strand is loaded into a multiprotein component complex called the RNA-induced silencing complex (RISC). Once the RISC complex and the target mRNA are aligned, the mRNA is cleaved through the action of the catalytic RISC protein, a member of the Argonaute family (Ago2)92.
siRNA development has promoted new therapeutic approaches for a variety of diseases, as it has enabled selective inhibition of expression of specific genes and downregulation of specific proteins in the body through the design of custom oligonucleotide molecules using target mRNA transcript sequences. However, clinical application of siRNA-based therapeutics has been limited because of their off-target effects of rapid degradation and renal clearance, low cellular release, and evasion of intracellular uptake. In addition, negatively charged RNAi molecules have poor cellular uptake because cell membranes are composed of negatively charged bilayers of phospholipids and functional proteins93.
Therefore, an efficient delivery ‘carrier’ that can prevent siRNA degradation in vivo and deliver siRNA specifically to the cytoplasm of target cells is needed. Considering the characteristics of siRNAs, effective delivery systems for target cells have been investigated. Viruses, cationic lipids, polymers, and nanoparticles have been proposed as various approaches for in vivo siRNA delivery (Fig. 2)94.
siRNA conjugates
siRNA conjugates are one of the most promising approaches involving covalent association of siRNAs with biomolecules (lipophilic molecules, antibodies, aptamers, ligands, peptides, or polymers). GalNAc siRNA conjugates have been approved by the FDA for treatment of acute hepatic porphyria95,96. siRNA conjugates are classified as aptamer-siRNA, peptide-siRNA, carbohydrate-siRNA, nanostructured material-siRNA conjugates, lipid-siRNA and polymer-siRNA96.
Aptamers are single-stranded oligonucleotides that recognize their targets through their unique three-dimensional complementarity. Because they are similar to mAbs due to their high affinity and specificity for the target molecule, they are called nucleic acid antibodies. However, they have several advantages over mAbs, including little to no immunogenicity or toxicity, a longer shelf life, lower production costs, and lower batch-to-batch variability. They have been utilized for the targeted delivery of siRNA to various cells (Fig. 2)97,98.
Peptides are suitable for siRNA delivery owing to their easy-to-define structures and various biological functions. High-yield peptide-siRNAs can be produced via various linkages, such as disulfide bonds, amide bonds, thioether bonds, thiol-maleimide bonds, the carboxy terminus of peptides, or additional cysteine residues. Peptide-siRNA conjugates can include CPP, CTP, soluble peptides, and carbohydrate peptides96.
Nanostructures are engineered structures with features at a nanoscale of fewer than 100 nanometers and include nanotextured surfaces, nanoparticles, nanotubes, and more complex nanoscale structures. They are readily taken up by target cells, act as specific ligands for receptors explicitly overexposed on the cell surface, avoid endosomes, and are nontoxic to healthy cells. Therefore, they can be useful carriers for siRNA conjugates for cancer treatment99. Nanostructure materials include gold nanoparticles, carbon nanotubes, magnetic nanoparticles, quantum dots, and mesoporous silica nanoparticles (Fig. 2)100.
siRNA conjugates for targeting Tregs in the TME
CTLA4apt-STAT3 siRNA is a siRNA conjugate in which CTLA4 binding RNA aptamer and mouse STAT3 siRNA are linked. FOXP3+ Tregs in the TME are a significant cause of tumor-induced immunosuppression and highly express CTLA4101, and the transcription factor STAT3 is necessary and sufficient for Treg development. Inhibition of STAT3 signaling has been shown to inhibit tumor growth and improve survival; therefore, it is a molecular target for cancer treatment102. Herrmann et al. synthesized CTLA4apt-STAT3 siRNA for gene silencing and demonstrated blocking of tumor Treg accumulation and inhibition of tumor growth in various mouse tumor models17.
NPsiCTLA-4 is a nanostructure material-siRNA conjugate in which siRNA-targeting CTLA-4 mRNA is surrounded by nanoparticles composed of PEG5k–PLA11k and BHEM-Chol103. Li et al. constructed NPsiCTLA-4 to promote T-cell activation and proliferation. NPsiCTLA-4 delivers CTLA-4-siRNA to CD4+ and CD8 + T-cell subsets in the TME while reducing the ratio of Tregs among TILs. In addition, NPsiCTLA-4 effectively inhibits tumor growth and extends survival time in mouse models of melanoma18.
Hybrid SNPs are spherical nucleotide nanoparticles (SNPs) loaded with a CTLA-4-siRNA aptamer (cSNP) and PD-1 siRNA (pSNP) in a nanoparticle comprising an amphiphilic polymer of PLGA-S-S-PEG as the core and the cationic lipid DOTAP. Zhang et al. designed a hybrid SNP that combined the blocking strategies of CTLA-4 and PD-1. Hybrid SNPs show synergistic immunostimulatory effects by blocking CTLA-4 and PD-1, partially regulating the immunosuppressive function of Tregs and allowing effector T-cell expansion. Antitumor efficacy was demonstrated by inhibiting the growth of melanoma tumors and colorectal adenocarcinomas in mice19.
Manrique-Rincón et al. created an aptamer chimera by linking an aptamer that binds to 4-1BB, which is abundant in the Treg cell membrane, to a small antisense RNA (sasRNA) targeting FOXP3. The authors exploited a transcriptional gene silencing mechanism to suppress Foxp3 expression using sasRNA. Unlike posttranscriptional gene silencing, which represses mRNA expression, transcriptional gene silencing is driven by promoter or enhancer regions and is associated with DNA methylation. They showed that aptamer-sasRNA-mediated transcriptional gene silencing of Foxp3 promotes an antitumor response in combination treatment using a melanoma-bearing mouse model28.
Conclusion
Existing drug conjugates have been developed to directly remove cancer cells. However, as current cancer treatments target immune-suppressive cells in the tumor microenvironment, drug conjugates are developing accordingly. Although the function of each moiety is known, when they are combined, the result may cause a synergistic effect beyond expectation or may have unexpected side effects.
Due to advanced synthesis technology, delivery efficiency and drug efficacy have been improved and can lower toxicity and side effects. Diverse conjugates with new drug-targeting molecules are increasing, and clinical trials with drug conjugates targeting immune-suppressive cells appropriately are driving innovative therapeutic design. This approach would be a promising strategy for overcoming the limitations of current anticancer drugs and specifically targeting the immunosuppressive TME.
References
Waibl Polania, J., Lerner, E. C., Wilkinson, D. S., Hoyt-Miggelbrink, A. & Fecci, P. E. Pushing past the blockade: advancements in T cell-based cancer immunotherapies. Front. Immunol. https://doi.org/10.3389/fimmu.2021.777073 (2021).
Li, C., Jiang, P., Wei, S., Xu, X. & Wang, J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 19, 116 (2020).
Onda, M., Kobayashi, K. & Pastan, I. Depletion of regulatory T cells in tumors with an anti-CD25 immunotoxin induces CD8 T cell-mediated systemic antitumor immunity. Proc. Natl Acad. Sci. USA 116, 4575–4582 (2019).
Zammarchi, F. et al. CD25-targeted antibody–drug conjugate depletes regulatory T cells and eliminates established syngeneic tumors via antitumor immunity. J. Immunother. Cancer 8, e000860 (2020).
Labani-Motlagh, A., Ashja-Mahdavi, M. & Loskog, A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front. Immunol. https://doi.org/10.3389/fimmu.2020.00940 (2020).
Swann, J. B. & Smyth, M. J. Immune surveillance of tumors. J. Clin. Investig. 117, 1137–1146 (2007).
Kim, R., Emi, M. & Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 121, 1–14 (2007).
O’Donnell, J. S., Teng, M. W. L. & Smyth, M. J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167 (2019).
Tie, Y., Tang, F., Wei, Y.-Q. & Wei, X.-W. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J. Hematol. Oncol. 15, 61–61 (2022).
Tay, C., Tanaka, A. & Sakaguchi, S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell 41, 450–465 (2023).
Nishikawa, H. & Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J. Immunother. Cancer 9, e002591 (2021).
Miyara, M. et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 30, 899–911 (2009).
Ohue, Y. & Nishikawa, H. Regulatory T (Treg) cells in cancer: can Treg cells be a new therapeutic target? Cancer Sci. 110, 2080–2089 (2019).
Flynn, M. J. et al. ADCT-301, a pyrrolobenzodiazepine (PBD) dimer-containing antibody-drug conjugate (ADC) targeting CD25-expressing hematological malignancies. Mol. Cancer Ther. 15, 2709–2721 (2016).
Lansigan, F., Stearns, D. M. & Foss, F. Role of denileukin diftitox in the treatment of persistent or recurrent cutaneous T-cell lymphoma. Cancer Manag. Res. 2, 53–59 (2010).
Powell, D. J. Jr. et al. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J. Immunol. (Baltim., Md. 1950) 179, 4919–4928 (2007).
Herrmann, A. et al. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J. Clin. Investig. 124, 2977–2987 (2014).
Li, S.-Y. et al. Restoring anti-tumor functions of T cells via nanoparticle-mediated immune checkpoint modulation. J. Control. Release 231, 17–28 (2016).
Zhang, J. et al. Hybrid spherical nucleotide nanoparticles can enhance the synergistic anti-tumor effect of CTLA-4 and PD-1 blockades. Biomater. Sci. 8, 4757–4766 (2020).
Campbell, J. R. et al. Fc-optimized anti-CCR8 antibody depletes regulatory T cells in human tumor models. Cancer Res. 81, 2983–2994 (2021).
De Simone, M. et al. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of tumor-infiltrating T regulatory cells. Immunity 45, 1135–1147 (2016).
Plitas, G. et al. Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45, 1122–1134 (2016).
Guo, X. et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 24, 978–985 (2018).
Zhang, Y. et al. Deep single-cell RNA sequencing data of individual T cells from treatment-naïve colorectal cancer patients. Sci. Data 6, 131 (2019).
Cinier, J. et al. Recruitment and expansion of tregs cells in the tumor environment—how to target them? Cancers 13, 1850 (2021).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Allan, S. E. et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol. Ther. 16, 194–202 (2008).
Manrique-Rincón, A. J. et al. Aptamer-mediated transcriptional gene silencing of Fox p 3 inhibits regulatory T cells and potentiates antitumor response. Mol. Ther. Nucleic Acids 25, 143–151 (2021).
Lozano, T. et al. Searching for peptide inhibitors of T regulatory cell activity by targeting specific domains of FOXP3 transcription factor. Biomedicines https://doi.org/10.3390/biomedicines9020197 (2021).
Xu, P. et al. Screening of specific binding peptides using phage-display techniques and their biosensing applications. TrAC Trends Anal. Chem. 137, 116229 (2021).
Yongsheng, Y., Xiaoliang, L., Zhenghao, T. & Guoqing, Z. siRNA-mediated knockdown of FoxP3 promotes the ratio of T-helper 1 (Th1) to Th2 in chronic hepatitis B patients. Turk. J. Gastroenterol. 22, 587–593 (2011).
Amarzguioui, M. et al. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat. Protoc. 1, 508–517 (2006).
Strebhardt, K. & Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 8, 473–480 (2008).
Fu, Z., Li, S., Han, S., Shi, C. & Zhang, Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 7, 93 (2022).
Kim, J.-H., Kim, B. S. & Lee, S.-K. Regulatory T cells in tumor microenvironment and approach for anticancer immunotherapy. Immune Netw. 20, e4 (2020).
Coleman, N., Yap, T. A., Heymach, J. V., Meric-Bernstam, F. & Le, X. Antibody-drug conjugates in lung cancer: dawn of a new era? npj Precis. Oncol. 7, 5 (2023).
McKertish, C. M. & Kayser, V. Advances and limitations of antibody drug conjugates for cancer. Biomedicines https://doi.org/10.3390/biomedicines9080872 (2021).
Lambert, J. M. & Morris, C. Q. Antibody–drug conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv. Ther. 34, 1015–1035 (2017).
Smith, A. L. & Nicolaou, K. C. The enediyne antibiotics. J. Med. Chem. 39, 2103–2117 (1996).
Pommier, Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer 6, 789–802 (2006).
Francisco, J. A. et al. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 102, 1458–1465 (2003).
Oroudjev, E. et al. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther. 9, 2700–2713 (2010).
Herrera, A. F. et al. Preliminary results of a phase 2 study of Camidanlumab Tesirine (Cami), a novel pyrrolobenzodiazepine-based antibody-drug conjugate, in patients with relapsed or refractory hodgkin lymphoma. Blood 136, 21–23 (2020).
Hamadani, M. et al. Camidanlumab tesirine in patients with relapsed or refractory lymphoma: a phase 1, open-label, multicentre, dose-escalation, dose-expansion study. Lancet Haematol. 8, e433–e445 (2021).
Goldberg, A. D. et al. Camidanlumab tesirine, an antibody-drug conjugate, in relapsed/refractory CD25-positive acute myeloid leukemia or acute lymphoblastic leukemia: a phase I study. Leuk. Res. 95, 106385 (2020).
Kim, J.-S., Jun, S.-Y. & Kim, Y.-S. Critical issues in the development of immunotoxins for anticancer therapy. J. Pharm. Sci. 109, 104–115 (2020).
Jen, E. Y. et al. FDA approval summary: Tagraxofusp-erzs for treatment of blastic plasmacytoid dendritic cell neoplasm. Clin. Cancer Res. 26, 532–536 (2020).
Lin, A. Y. & Dinner, S. N. Moxetumomab pasudotox for hairy cell leukemia: preclinical development to FDA approval. Blood Adv. 3, 2905–2910 (2019).
Hamamichi, S., Fukuhara, T. & Hattori, N. Immunotoxin screening system: a rapid and direct approach to obtain functional antibodies with internalization capacities. Toxins (Basel) https://doi.org/10.3390/toxins12100658 (2020).
Bruins, W. S. C., Zweegman, S., Mutis, T. & van de Donk, N. Targeted therapy with immunoconjugates for multiple myeloma. Front Immunol. 11, 1155 (2020).
Alewine, C., Hassan, R. & Pastan, I. Advances in anticancer immunotoxin therapy. Oncologist 20, 176–185 (2015).
Li, M., Mei, S., Yang, Y., Shen, Y. & Chen, L. Strategies to mitigate the on- and off-target toxicities of recombinant immunotoxins: an antibody engineering perspective. Antib. Ther. 5, 164–176 (2022).
Kumar, P., Kumar, A., Parveen, S., Murphy, J. R. & Bishai, W. Recent advances with Treg depleting fusion protein toxins for cancer immunotherapy. Immunotherapy 11, 1117–1128 (2019).
Attia, P. et al. Selective elimination of human regulatory T lymphocytes in vitro with the recombinant immunotoxin LMB-2. J. Immunother. 29, 208–214 (2006).
Kreitman, R. J. et al. Complete remissions of adult T-cell Leukemia with Anti-CD25 recombinant Immunotoxin LMB-2 and chemotherapy to block immunogenicity. Clin. Cancer Res. 22, 310–318 (2016).
Basheer, F., Bloxham, D. M., Scott, M. A. & Follows, G. A. Hairy cell leukemia - immunotargets and therapies. Immunotargets Ther. 3, 107–120 (2014).
Kaminetzky, D. & Hymes, K. B. Denileukin diftitox for the treatment of cutaneous T-cell lymphoma. Biologics 2, 717–724 (2008).
Cheung, L. S. et al. Second-generation IL-2 receptor-targeted diphtheria fusion toxin exhibits antitumor activity and synergy with anti-PD-1 in melanoma. Proc. Natl Acad. Sci. USA 116, 3100–3105 (2019).
Foss, F. M. et al. Efficacy and safety of E7777 (improved purity Denileukin diftitox [ONTAK]) in patients with relapsed or refractory cutaneous T-cell lymphoma: results from pivotal study 302. Blood 140, 1491–1492 (2022).
Kawai, H. et al. Phase II study of E7777 in Japanese patients with relapsed/refractory peripheral and cutaneous T-cell lymphoma. Cancer Sci. 112, 2426–2435 (2021).
Chang, D.-K. et al. Humanization of an Anti-CCR4 antibody that kills cutaneous T-cell lymphoma cells and abrogates suppression by T-regulatory cells. Mol. Cancer Ther. 11, 2451–2461 (2012).
Wang, Z. et al. Diphtheria-toxin based anti-human CCR4 immunotoxin for targeting human CCR4+ cells in vivo. Mol. Oncol. 9, 1458–1470 (2015).
Wang, Z. et al. Treg depletion in non-human primates using a novel diphtheria toxin-based anti-human CCR4 immunotoxin. Mol. Oncol. 10, 553–565 (2016).
Wang, H. et al. Bispecific human IL2-CCR4 immunotoxin targets human cutaneous T-cell lymphoma. Mol. Oncol. 14, 991–1000 (2020).
Fu, C. et al. Peptide–drug conjugates (PDCs): a novel trend of research and development on targeted therapy, hype or hope? Acta Pharmaceut. Sin. B 13, 498–516 (2023).
Borsi, L. et al. Selective targeting of tumoral vasculature: comparison of different formats of an antibody (L19) to the ED-B domain of fibronectin. Int. J. Cancer 102, 75–85 (2002).
van Schouwenburg, P. A. et al. A novel method for the detection of antibodies to adalimumab in the presence of drug reveals “hidden” immunogenicity in rheumatoid arthritis patients. J. Immunol. Methods 362, 82–88 (2010).
Carrasco-Triguero, M. et al. Immunogenicity assays for antibody-drug conjugates: case study with ado-trastuzumab emtansine. Bioanalysis 5, 1007–1023 (2013).
Nejadmoghaddam, M. R. et al. Antibody-drug conjugates: possibilities and challenges. Avicenna J. Med. Biotechnol. 11, 3–23 (2019).
Ahrens, V. M., Bellmann-Sickert, K. & Beck-Sickinger, A. G. Peptides and peptide conjugates: therapeutics on the upward path. Future Med. Chem. 4, 1567–1586 (2012).
Firer, M. A. & Gellerman, G. Targeted drug delivery for cancer therapy: the other side of antibodies. J. Hematol. Oncol. 5, 70 (2012).
Alas, M., Saghaeidehkordi, A. & Kaur, K. Peptide–drug conjugates with different linkers for cancer therapy. J. Med. Chem. 64, 216–232 (2021).
Chavda, V. P., Solanki, H. K., Davidson, M., Apostolopoulos, V. & Bojarska, J. Peptide-drug conjugates: a new hope for cancer management. Molecules 27, 7232 (2022).
Kalimuthu, K. et al. Gold nanoparticles stabilize peptide-drug-conjugates for sustained targeted drug delivery to cancer cells. J. Nanobiotechnol. 16, 34 (2018).
Lozano, T. et al. Targeting inhibition of Foxp3 by a CD28 2′-Fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials 91, 73–80 (2016).
Casares, N. et al. A peptide inhibitor of FOXP3 impairs regulatory T cell activity and improves vaccine efficacy in mice. J. Immunol. 185, 5150–5159 (2010).
Lozano, T. et al. Blockage of FOXP3 transcription factor dimerization and FOXP3/AML1 interaction inhibits T regulatory cell activity: sequence optimization of a peptide inhibitor. Oncotarget 8, 71709–71724 (2017).
Pastor, F. et al. CD28 aptamers as powerful immune response modulators. Mol. Ther. Nucleic Acids 2, e98 (2013).
Lozano, T. et al. Targeting inhibition of Foxp3 by a CD28 2’-Fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials 91, 73–80 (2016).
Lozano, T. et al. Inhibition of FOXP3/NFAT interaction enhances T cell function after TCR stimulation. J. Immunol. 195, 3180–3189 (2015).
Kawamoto, S. A. et al. Analysis of the interaction of BCL9 with beta-catenin and development of fluorescence polarization and surface plasmon resonance binding assays for this interaction. Biochemistry 48, 9534–9541 (2009).
Li, X. et al. WNT/β-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front. Immunol. 10, 2293–2293 (2019).
Feng, M. et al. Pharmacological inhibition of β-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating T(reg) cells. Sci. Adv. 5, eaau5240 (2019).
Ezquerro, I. J. et al. A synthetic peptide from transforming growth factor beta type III receptor inhibits liver fibrogenesis in rats with carbon tetrachloride liver injury. Cytokine 22, 12–20 (2003).
Dotor, J. et al. Identification of peptide inhibitors of transforming growth factor beta 1 using a phage-displayed peptide library. Cytokine 39, 106–115 (2007).
Llopiz, D. et al. Peptide inhibitors of transforming growth factor-β enhance the efficacy of antitumor immunotherapy. Int. J. Cancer 125, 2614–2623 (2009).
Kim, Y.-J. et al. Immunoglobulin Fc-fused, neuropilin-1-specific peptide shows efficient tumor tissue penetration and inhibits tumor growth via anti-angiogenesis. J. Control. Release 216, 56–68 (2015).
Niland, S. & Eble, J. A. Neuropilins in the context of tumor vasculature. Int. J. Mol. Sci. 20, 639 (2019).
Jung, K. et al. A neuropilin-1 antagonist exerts antitumor immunity by inhibiting the suppressive function of intratumoral regulatory T cells. Cancer Immunol. Res. 8, 46–56 (2020).
Santagata, S. et al. Targeting CXCR4 reverts the suppressive activity of T-regulatory cells in renal cancer. Oncotarget 8, 77110–77120 (2017).
Padda, I. S., Mahtani, A. U. & Parmar, M. in StatPearls (StatPearls PublishingCopyright © 2023, StatPearls Publishing LLC., 2023).
Alshaer, W. et al. siRNA: mechanism of action, challenges, and therapeutic approaches. Eur. J. Pharmacol. 905, 174178 (2021).
Bholakant, R. et al. Recent advances of polycationic siRNA vectors for cancer therapy. Biomacromolecules 21, 2966–2982 (2020).
Dana, H. et al. Molecular mechanisms and biological functions of siRNA. Int. J. Biomed. Sci. 13, 48–57 (2017).
Zhang, L. et al. The therapeutic prospects of N-acetylgalactosamine-siRNA conjugates. Front. Pharmacol. https://doi.org/10.3389/fphar.2022.1090237 (2022).
Chernikov, I. V., Vlassov, V. V. & Chernolovskaya, E. L. Current development of siRNA bioconjugates: from research to the clinic. Front. Pharmacol. https://doi.org/10.3389/fphar.2019.00444 (2019).
Fu, Z. & Xiang, J. Aptamers, the nucleic acid antibodies, in cancer therapy. Int J. Mol. Sci. 21, 2793 (2020).
Kruspe, S. & Giangrande, P. H. Aptamer-siRNA chimeras: discovery, progress, and future prospects. Biomedicines 5, 45 (2017).
Ashique, S. et al. siRNA-based nanocarriers for targeted drug delivery to control breast cancer. Adv. Cancer Biol. Metastasis 4, 100047 (2022).
Lee, S. H., Kang, Y. Y., Jang, H.-E. & Mok, H. Current preclinical small interfering RNA (siRNA)-based conjugate systems for RNA therapeutics. Adv. Drug Deliv. Rev. 104, 78–92 (2016).
Shevach, E. M. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2, 389–400 (2002).
Jia, H. et al. Antitumor effects of Stat3-siRNA and endostatin combined therapies, delivered by attenuated Salmonella, on orthotopically implanted hepatocarcinoma. Cancer Immunol. Immunother. 61, 1977–1987 (2012).
Liu, Y. et al. Triple negative breast cancer therapy with CDK1 siRNA delivered by cationic lipid assisted PEG-PLA nanoparticles. J. Control. Release 192, 114–121 (2014).
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. RS-2023-00208347).
Author information
Authors and Affiliations
Contributions
J.Y. and H.B. contributed to the conception and design of the article. J.Y. also contributed to investigating and interpreting the relevant literature. H.B. supervised this review. All authors wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, J., Bae, H. Drug conjugates for targeting regulatory T cells in the tumor microenvironment: guided missiles for cancer treatment. Exp Mol Med 55, 1996–2004 (2023). https://doi.org/10.1038/s12276-023-01080-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01080-3
- Springer Nature Limited