
Abstract
Adipose tissues, composed of various cell types, including adipocytes, endothelial cells, neurons, and immune cells, are organs that are exposed to dynamic environmental challenges. During diet-induced obesity, white adipose tissues experience hypoxia due to adipocyte hypertrophy and dysfunctional vasculature. Under these conditions, cells in white adipose tissues activate hypoxia-inducible factor (HIF), a transcription factor that activates signaling pathways involved in metabolism, angiogenesis, and survival/apoptosis to adapt to such an environment. Exposure to cold or activation of the β-adrenergic receptor (through catecholamines or chemicals) leads to heat generation, mainly in brown adipose tissues through activating uncoupling protein 1 (UCP1), a proton uncoupler in the inner membrane of the mitochondria. White adipose tissues can undergo a similar process under this condition, a phenomenon known as ‘browning’ of white adipose tissues or ‘beige adipocytes’. While UCP1 expression has largely been confined to adipocytes, HIF can be expressed in many types of cells. To dissect the role of HIF in specific types of cells during diet-induced obesity, researchers have generated tissue-specific knockout (KO) mice targeting HIF pathways, and many studies have commonly revealed that intact HIF-1 signaling in adipocytes and adipose tissue macrophages exacerbates tissue inflammation and insulin resistance. In this review, we highlight some of the key findings obtained from these transgenic mice, including Ucp1 KO mice and other models targeting the HIF pathway in adipocytes, macrophages, or endothelial cells, to decipher their roles in diet-induced obesity.
Similar content being viewed by others


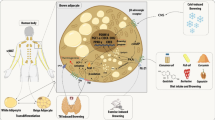
Oxygen sensing in obesity – hypoxia and hypoxia-inducible factor (HIF)
The World Health Organization defines obesity as excessive fat accumulation that might impair health1. Obesity substantially increases the risk of metabolic diseases, cardiovascular diseases, musculoskeletal diseases, Alzheimer’s disease, depression, and some types of cancers1. Recently, the World Obesity Federation has declared obesity a chronic progressive disease that requires intervention2.
In the process of diet-induced obesity, adipocytes undergo major structural and functional changes, including hypertrophy, which leads to adipose tissue expansion, resulting in inefficient blood flow to the tissue3. This may then create tissue hypoxia, in which the affected cells in adipose tissues, such as adipocytes, immune cells, and endothelial cells, activate the transcription factor hypoxia-inducible factor (HIF) to adapt to such hypoxic conditions4. Indeed, visceral adipose tissues obtained from obese human individuals have demonstrated a significantly lower level of oxygen tension, accompanied by high expression of HIF1A and other genes involved in inflammation and fibrosis5. Interestingly, severe adipose tissue hypoxia detected by pimonidazole, a nitroimidazole compound that is reduced, thereby binding to sulfhydryl groups of various molecules forming pimonidazole adducts6, has been detected as early as 3 days in mice fed a high-fat diet7.
HIF is a heterodimeric transcription factor composed of an O2-sensitive α subunit and an O2-insensitive β subunit8. To date, three α forms (HIF-1α, HIF-2α, and HIF-3α) are known to exist. These α forms (at least the human form) contain a basic helix-loop-helix (bHLH) domain, two Per-Arnt-Sim (PAS) domains, a PAS-associated COOH-terminal (PAC) domain, and an oxygen-dependent degradation (ODD) domain containing an NH2-terminal transactivation domain (N-TAD)9. Only HIF-1α and HIF-2α have a COOH-terminal transactivation domain (C-TAD)9. HIF-3α has been reported to exist as multiple variants, and some of the variants may lack one or more of the domains described above9. HIF-1α, HIF-2α, and HIF-3α protein stability is regulated by prolyl hydroxylases (PHD; Fig. 1), which require O2 and 2-oxoglutarate as substrates and ascorbate as a cofactor10. Hydroxylated Pro402 and Pro564 of the HIF-α form then allow von Hippel‒Lindau (VHL) tumor suppressor protein to bind and mediate the ubiquitination of HIF-α protein10 (Fig. 1). The transactivation capacity of HIF-1α and HIF-2α (but not HIF-3α because it lacks a C-TAD domain) can be further controlled by an asparagine hydroxylase (factor inhibiting HIF (FIH); Fig. 1), which also requires O2 and oxoglutarate as substrates, hydroxylating Asn803 in the C-TAD domain10. This hydroxylation at Asn803 of HIF-1α or HIF-2α prevents HIF from recruiting the transcriptional coactivators CBP and p30010.

HIF-1 can be activated selectively under hypoxic conditions (yellow color; left side of the diagram), regulating many enzymes involved in the glycolysis pathway, including GLUT1, HK, and PDK. PDK in turn blocks pyruvate conversion to acetyl-CoA in the mitochondria, thereby decreasing oxidative phosphorylation (OXPHOS)-mediated ROS production. Under normoxic conditions (sky blue color; right side of the diagram), UCP1 can generate heat in the process of electron transport chain-mediated OXPHOS by uncoupling the proton gradient. The net result of UCP1 is also to reduce ROS production in the mitochondria.
Once the HIF-α form is stabilized in the cytoplasm, it binds to HIF-1β, which translocates the HIF heterodimer into the nucleus, where it binds to the hypoxia-responsive element (HRE) of the downstream target genes10 (Fig. 1). Although HIF-1 and HIF-2 share a number of common downstream target genes, including vascular endothelial growth factor (VEGF) and adrenomedullin (ADM)11, there are genes that are exclusively regulated either by HIF-1 or HIF-2. HIF-1 has been reported to regulate many genes involved in glycolysis metabolism, including glucose transporter-1 (GLUT-1), phosphoglycerate kinase-1 (PGK-1), and lactate dehydrogenase A (LDHA) (Fig. 1), while HIF-2-specific downstream target genes include erythropoietin (EPO)12 and adipose differentiation-related protein (ADRP)11. HIF-3 is quite different from HIF-1 and HIF-213 such that HIF-3 may compete with HIF-1 and HIF-2, thereby negatively regulating target genes of HIF-1 and HIF-2 under hypoxic conditions13. However, a recent study reported contradictory findings that HIF-3 can share some target genes with HIF-1 and HIF-2, such as GLUT1, EPO and angiopoietin like-4 (ANGPTL4)14. HIF-3 does not seem to turn on HRE-driven reporter expression, indicating that HIF-3 regulates its target genes by binding non-canonical sites of cis-elements15.
HIF and adipocytes
Given the importance of tissue hypoxia in adipose tissues during diet-induced obesity, a number of researchers have investigated the role of HIF in adipocytes by generating tissue-specific knockout (KO) mice targeting HIF pathways (Table 1). A number of studies have consistently demonstrated that HIF-1α expression in white adipocytes exacerbates insulin resistance and tissue inflammation (Table 1). For example, high-fat diet-fed adipocyte-specific Hif-1α KO mice with the aP2 promoter have demonstrated improved insulin sensitivity resistance7,16,17,18 (Table 1), increased oxidative metabolism mediated by PPAR-gamma coactivator 1 alpha (PGC-1α)19, and decreased tissue inflammation7,20. However, there are also some studies reporting contradictory findings where mice expressing the dominant negative form of human HIF-1A in adipose tissues exhibit increased body weight gain, insulin resistance, and adipose tissue fibrosis20,21.
While there are fewer studies on the role of HIF-2α in adipocytes, the results seem consistent: intact HIF-2 signaling in adipocytes has a protective role against diet-induced obesity (Table 1). For example, adipocyte-specific Hif-2α KO mice with the fatty acid binding protein 4 (Fabp4) promoter exhibit increased body weight gain, insulin resistance, and tissue inflammation under high-fat diet conditions22 (Table 1). Another study that used knock-in of a single amino acid substitution (S305M) within the HIF-2α PAS-B domain in all tissues in mice reported that these mice gain more body weight23 (Table 1) and exhibited increased expression of genes involved in adipogenesis, including adiponectin, leptin, lipase, Fabp4, peroxisome proliferator-activated receptor gamma (Pparγ), CCAAT/enhancer-binding protein alpha (Cebpα), CCAAT/enhancer-binding protein beta (Cebpβ), and ATP citrate lyase (Acly), in visceral adipose tissues23.
Mice with deletion of Phd2, thereby constitutively upregulating HIF-1α, HIF-2α, and HIF-3α in adipocytes, exhibited decreased body weight gain, improved insulin sensitivity, and increased oxygen consumption (but not carbon dioxide production) upon high-fat diet feeding24 (Table 1). Visceral adipose tissues isolated from these mice exhibited increased vascularity, decreased adipose tissue macrophage (ATM) infiltration, and increased expression of many glycolytic genes, including Glut-1, Pdk-1, Gapdh, and Ldha24.
Adipocyte-specific Vhl KO mice using aP2 and Fabp4 dual promoters, whereby HIF-1α, HIF-2α, and HIF-3α are constitutively upregulated in the affected adipocytes, demonstrate no reduction in body weight gain, although visceral fat mass is significantly decreased25 (Table 1). These mice have a profound cardiac hypertrophy phenotype25, which may have compromised the investigators’ attempt to clearly dissect the role of HIF in adipocytes.
HIF and stromal cells in adipose tissues
Adipose tissue macrophages (ATM)
ATM are known to be the most abundant immune cells in the adipose tissues of obese individuals26. While they comprise fewer than 10% of the total immune cells in lean adipose tissues, this fraction rises to more than 50% in obese adipose tissues26. Furthermore, the number of ATM has been shown to correlate well with total adiposity and adipose cell size27. Macrophages are recruited to adipose tissues by various cytokines and chemokines, including C-X-C motif chemokine 12 (CXCL12)28 and semaphorin 3E29 secreted from adipocytes, which may be activated by the HIF transcriptional program. Generally, macrophages are known to be polarized either to the proinflammatory M1 or anti-inflammatory M2 phenotype30. In diet-induced obesity, extensive evidence suggests that M1-polarized macrophages dominate the adipose tissues of obese mice27 and humans31. A recent study with single-cell RNA sequencing technology, however, demonstrated that several classical M1/M2 signature genes are rarely expressed in ATM, although inflammatory signaling pathways, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), Rho GTPases, mammalian target of rapamycin (mTOR), and p38 mitogen activated protein kinase (MAPK) signaling pathways, do seem to be ‘activated in M1-like’ and ‘transitional M1-like’ ATM in obese mice but not in lean mice32. It has also been suggested that a high-fat diet leads to an endoplasmic reticulum stress response in adipocytes, which may in turn affect macrophage polarization33. ATM often exhibit a crown-like structure, which has been shown to be a result of scavenging lipid droplets released from dead adipocytes, forming multinucleated giant cells34.
As with adipocytes, a number of researchers have generated macrophage-specific Hif-KO mice using the lysozyme M (LysM) promoter to study the role of HIF in ATM in obesity (Table 2). Takikawa and colleagues35 demonstrated that HIF-1 deficiency in ATM leads to improved whole-body glucose clearance and tissue insulin sensitivity, reduced tissue inflammatory gene expression, enhanced angiogenesis and reduced hypoxia in adipose tissues (Table 2). Sharma and colleagues36 observed less ATM infiltration and Ki67-positive proliferating cells in the white adipose tissues (WAT) of macrophage-specific Hif-1α KO mice (Table 2). In contrast to these studies, Kihira and colleagues17 have reported that glucose clearance and ATM infiltration signatures are not different between the mutant (macrophage-specific Hif-1α KO mice) and their wild-type (WT) counterpart mice (Table 2). Nonetheless, all studies have commonly found that there is no difference in body weight gain between mutant and WT mice upon high-fat diet feeding17,35. The role of HIF-2α in macrophages in obesity is somewhat unclear. High-fat diet-fed macrophage-specific Hif-2α KO mice with the LysM promoter demonstrated no difference in insulin sensitivity between the mutant and WT mice22, whereas Hif-2α+/− heterozygous mice treated with clodronate liposomes, a macrophage-depleting agent, demonstrated improved glucose clearance37 (Table 2). Macrophage-specific Phd2 KO mice fed a high-fat diet have been reported to exhibit increased insulin resistance, adipose tissue fibrosis, and tissue inflammation38 (Table 2).
T cells
T cells are the second largest population of immune cells in obese adipose tissues26. Obesity has been shown to dramatically increase the frequency of T helper (Th) 1, Th17, and CD8 + T cells in WAT, whereas the frequency of Th2 and regulatory T (Treg) cells is decreased in WAT39. As T-cell activation requires aerobic glycolysis and glutamine catabolism triggered by downstream signaling cascades of the T-cell receptor, costimulatory molecules and cytokines40, HIF-1 has been shown to be essential in promoting glucose uptake and glycolysis to mediate the cytotoxic T-cell response41. Furthermore, HIF-1α is strongly induced in T cells undergoing Th17 differentiation, while the lowest levels are detected in cells undergoing Treg differentiation42. Despite the importance of HIF in the regulation of T-cell activation and inflammation43, there are no reports to date, as far as the authors know, investigating how HIF in T cells impacts diet-induced obesity.
Endothelial cells
Evidence suggests that obesity is accompanied by endothelial cell dysfunction and decreased vascular density44 and that modulation of endothelial cell function through some of the key molecules, including transcription factors (PGC-1α, PPARγ, and NF-κB), angiogenic signaling (VEGF/vascular endothelial growth factor receptor 2 (VEGFR2) and angiopoietin 2 (ANGPT2)/TEK tyrosine kinase (TIE2)), insulin signaling (insulin receptor), and mediators of fatty acid transporters (CD36 and PPARγ), is sufficient to regulate the progression of obesity45.
Endothelial-specific Hif-1α KO mice with the Tie2 promoter have demonstrated elevated basal glucose levels, decreased glucose clearance, delayed insulin release in the blood upon glucose load, and decreased glucose uptake into tissues, including the brain and heart46 (Table 2). Endothelial-specific Hif-2a KO mice with a stem cell leukemia promoter, on the other hand, exhibited no differences in body weight gain, fat mass, insulin sensitivity, or tissue vascularity compared to their WT controls when fed a high-fat diet22 (Table 2).
Factors other than molecular oxygen that activate HIF in obesity
HIF is well known to be regulated by the level of molecular oxygen. However, there are several factors other than molecular oxygen that can activate HIF. Some of these factors include reactive oxygen species (ROS) reducing Fe2+ to Fe3+, thereby inhibiting PHD activity47, increased concentrations of succinate and fumarate inhibiting the PHD reaction22, lipopolysaccharide48 activating the protein kinase C and phosphatidylinositol 3-kinase pathway, thereby increasing HIF-1α transcription and translation49, sphingosine-1-phosphate from apoptotic cell debris activating nuclear factor of activated T cells and hence HIF-1α translation50, and proinflammatory cytokines such as tumor necrosis factor-α and interleukin-1β increasing DNA binding of HIF-151,52.
From the obesity perspective, He and colleagues53 have demonstrated that insulin treatment alone is sufficient to increase HIF-1α mRNA and protein levels in differentiated 3T3-L1 adipocytes53. Interestingly, high glucose levels in normal cultured cells have been reported to reduce HIF-1α protein levels and increase mitochondrial ROS production54, perhaps through elevated fatty acid metabolism, lowering succinate availability for HIF-1α protein stabilization55. In addition, an increase in 2-methylglyoxal that accompanies high glucose conditions such as obesity and diabetes stimulates HIF-α degradation by inhibiting HIF-α-HIF-1β dimer formation and recruitment of the p300/CBP regulatory complex56.
HIF inhibitors as a potential anti-obesity drug
Given the importance of HIF in obesity, some HIF inhibitors have been shown to be effective in attenuating the obese phenotype in mice fed a high-fat diet. PX-478, a selective inhibitor of HIF-1α, effectively reduced weight gain accompanied by decreased fibrosis and inflammation57. YC1, a suppressor of HIF-1α accumulation58, has also been shown to induce lipolysis in Raw264.7 foam cells58 and rat visceral fat cells59. PT2399, an HIF-2α-specific antagonist, has been shown to protect mice from diet-induced obesity by decreasing body weight, adipogenesis and lipogenesis when administered for 8 weeks23. Digoxin, a cardiac glycoside that increases heart contraction forces by reversibly inhibiting the activity of the sodium-potassium ATPase pump, also identified through compound library screening as a small molecule HIF-1 inhibitor60, has been reported to prevent diet-induced obesity in mice, although the suggested mechanism is through inhibition of the IL-17A phosphorylation of PPARγ in adipocytes61. Berberine, an ingredient in the Chinese herbal medicine Coptis chinensis62, and anti-sense oligonucleotides to HIF-1α63 have been shown to attenuate body weight gain in mice fed a high-fat diet through inhibition of HIF-1α expression.
Temperature sensing in obesity – uncoupling protein (UCP) in adipose tissues
Uncoupling proteins (UCP) are members of the mitochondrial anion carrier family64, and five UCP homologs have been identified to date. UCP1, the first UCP identified and the most extensively studied member, is predominantly expressed in brown adipose tissue (BAT) and allows an alternative route for protons to enter other than ATP synthase, generating heat as a result of the dissipation of the energy from the electrochemical gradient65 (Fig. 1). In doing so, free fatty acids have been shown to act as secondary messengers to facilitate proton conductance mediated by UCP1, whereas the binding of purine nucleotides such as ADP and GDP has been shown to inhibit UCP1 activity65,66. UCP1 is regulated at the transcriptional level, and many physiological signals, including the cold-induced release of catecholamines (epinephrine and norepinephrine), act on β3-adrenergic receptors in BAT66. Recent evidence suggests that UCP1 can also be expressed in WAT under certain circumstances, such as cold-induced catecholamine activation of β3-adrenergic receptors, exercise-induced release of fibroblast growth factor (FGF)−21 acting on FGF receptors, and thyroid hormone synergizing with the activation of β3-adrenergic receptors67. Under these conditions, the phenomenon is also known as ‘browning’ of WAT or ‘beige adipocytes’; white adipocytes undergo lipolysis and FFA release through induction of UCP1 expression67. Ucp1 KO mice have been shown to be non-obese upon high-fat diet challenge and extremely cold sensitive68. UCP2 expression has been observed in many tissues, including adipose tissue, muscle, heart, kidney, digestive tract, brain, spleen, and thymus69, while UCP3 is primarily expressed in skeletal muscle in humans and rodents and in the heart and BAT in rodents70. Although UCP2 and UCP3 have 59% and 57% amino acid identity, respectively, to UCP171 – hence, they were originally thought to be thermogenic genes – it has been revealed that they are unlikely to be involved in adaptive thermogenesis based on the following evidence: the mitochondrial uncoupling by UCP2 has been shown to occur at much higher expression levels than the endogenous one66 and Ucp272 or Ucp373 KO mice are not cold sensitive.
Relationship between oxygen and temperature sensing in adipocytes – a possible link between HIF and UCP
Can adipocytes sense oxygen levels and temperature at the same time, and if so, what would be the consequences? It seems that molecular oxygen, glucose, and fatty acids are commonly shared signaling molecules by both HIF and UCP. Thermogenically activated BAT has been shown to take up large amounts of glucose, although there is no significant release of lactate74, indicating that glucose is utilized mainly in oxidative but not glycolytic metabolism. Then, is UCP required for glucose uptake by adipocytes? Inokuma and colleagues75 demonstrated that Ucp1 KO mice lack the stimulatory effect of norepinephrine on glucose uptake into BAT, suggesting that UCP1 is required for glucose uptake into adipocytes. Glucose taken up by adipocytes may then be utilized to synthesize tricarboxylic acid intermediates or converted to free fatty acids76. Since HIF-1 regulates the expression of GLUT-1, a transporter essential for glucose uptake11, and pyruvate dehydrogenase (PDH), an enzyme that converts pyruvate to acetyl-CoA77, HIF-1 may promote UCP1-mediated glucose uptake and oxidative phosphorylation (Fig. 1). However, given that HIF-1 activation occurs with either low oxygen tension or other factors, such as ROS, it is unlikely that HIF-1 and UCP1 cooperate in adipocytes at the same time. In line with this, the fact that free fatty acids, which are required for UCP1-mediated thermogenesis, prevent hypoxia-induced HIF-1α accumulation55 indicates a mutually exclusive relationship between HIF-1 and UCP1. Interestingly, however, there are a couple of studies demonstrating a possible interlinked regulation between UCP and HIF. For example, Han and colleagues78 have reported that cold exposure or CL-316,243, a β3-adrenoceptor agonist, treatment in mice results in an increased protein expression of HIF-1α and HIF-2α in thermogenic adipose tissues (inguinal and brown adipose tissues, but not in visceral adipose tissues), which closely coincided with the increased expression of UCP1 in these tissues. Similarly, Basse and colleagues79 demonstrated that isoproterenol, a β-adrenergic stimulator that normally increases UCP1 expression in adipocytes80, not only increases HIF-1α mRNA and protein levels but also increases the expression of glycolytic genes, including Glut-1, hexokinase (Hk), Pgk-1, and Ldha. In another study, leptin-deficient ob/ob mice treated with deferoxamine, a well-known iron chelator and PHD inhibitor, which thereby stabilizes HIF-α81, demonstrated increased UCP1 protein expression and mitochondrial biogenesis and improved insulin sensitivity markers in adipose tissues82.
Nonetheless, the end result of the signaling by both HIF-1 and UCP seems to be a reduction in mitochondrial ROS production (Fig. 1). UCP has been generally reported to decrease mitochondrial ROS by inducing proton leakage (Fig. 1), which allows mitochondria to avoid the oversupply of electrons/reducing equivalents into respiratory complexes and minimizes the likelihood of electron interaction with oxygen83. UCP2 and UCP3 have also been reported to catalyze proton leakage, thereby decreasing mitochondrial ROS84. HIF-1 can decrease mitochondrial ROS production by inhibiting the uptake of pyruvate into the mitochondria through the expression of pyruvate dehydrogenase kinase-1 (PDK1), which inactivates PDH, thereby inhibiting the conversion of pyruvate to acetyl-CoA in the tricarboxylic acid cycle77 (Fig. 1).
Conclusion
Based on the critical role played by HIF and UCP in physiological as well as pathological settings, there are numerous studies on how each of these molecules is regulated under the setting of diet-induced obesity, for example, by treatment with free fatty acids. However, one should bear in mind that the role of these molecules in obesity can be significantly different from those in, for example, cancer. For instance, while fatty acid metabolism decreases HIF-1α protein stabilization in the setting of obesity55, treatment of hepatocellular carcinoma cells with free fatty acids, such as oleic acid, can increase HIF-1α protein expression under both normoxia and hypoxia85. One solution to this complex situation may be to use an appropriate reporter system. With the latest technology, mice expressing ubiquitous ROSA26 ODD-driven luciferase+/− have been created, and these mice emit bioluminescence signals in mice breathing 8% oxygen or injected with hypoxia mimetics86. ThermoMouse87 and a dual Ucp1 reporter mouse88 expressing Ucp1-driven luciferase 2 and Ucp1-driven firefly luciferase and near-infrared red fluorescent protein, respectively, have been generated; these mice demonstrate the highest luciferase activity and thereby UCP1 expression in BAT of mice exposed to cold temperature, CL-316,243, or rosiglitazone (a PPARγ agonist). Using these transgenic mice, the development of HIF inhibitors or UCP1 activators as anti-obesity drugs may be facilitated, although low sensitivity due to the penetration depth in these mice remains a major obstacle to overcome. There may be a growing interest in developing UCP1 activators as a therapeutic strategy to combat diabetes and obesity. In fact, dinitrophenol (DNP), a benzene-based chemical, was widely used as a weight-loss drug in the 1930s89. It turned out that it acts as a proton permeabilizer, thereby uncoupling oxidative phosphorylation and causing fat calories to be dissipated as heat89. However, DNP is associated with some serious side effects, including rare idiosyncrasies and loss of vision90. With the known mechanism of UCP and thermogenesis, Cavalieri and colleagues91 utilized protein thermostability shift analysis to identify novel activating ligands of UCP, including tetradecylthioacetic acid (a β-oxidation-resistant synthetic fatty acid that activates PPAR), TUG-891 (a G protein-coupled receptor 120 agonist), and ibuprofen (a widely used non-steroidal anti-inflammatory drug). Retinoic acid92 and the cJun kinase inhibitor SP60012593 have also been reported as UCP1 activators. Although further studies of these compounds are warranted, it would be exciting if these compounds reveal paths to treat and prevent diet-induced obesity.
References
Blüher, M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 15, 288–298 (2019).
Bray, G., Kim, K., Wilding, J. & Federation, W. O. Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 18, 715–723 (2017).
Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 93, 1–21 (2013).
Goossens, G. H. & Blaak, E. E. Adipose tissue dysfunction and impaired metabolic health in human obesity: a matter of oxygen? Front. Endocrinol. 6, 55 (2015).
Cifarelli, V. et al. Decreased adipose tissue oxygenation associates with insulin resistance in individuals with obesity. J. Clin. Investig. 130, 6688–6699 (2020).
Ljungkvist, A. S. et al. Changes in tumor hypoxia measured with a double hypoxic marker technique. Int. J. Radiat. Oncol. Biol. Phys. 48, 1529–1538 (2000).
Lee, Y. S. et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352 (2014).
Hewitson, K. S. & Schofield, C. J. The HIF pathway as a therapeutic target. Drug Discov. Today 9, 704–711 (2004).
Duan, C. Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am. J. Physiol. Cell Physiol. 310, C260–C269 (2016).
Semenza, G. L. O2-regulated gene expression: transcriptional control of cardiorespiratory physiology by HIF-1. J. Appl. Physiol. 96, 1173–1177 (2004).
Hu, C.-J., Wang, L.-Y., Chodosh, L. A., Keith, B. & Simon, M. C. Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol. Cell. Biol. 23, 9361–9374 (2003).
Lee, J.-W., Bae, S.-H., Jeong, J.-W., Kim, S.-H. & Kim, K.-W. Hypoxia-inducible factor (HIF-1)α: its protein stability and biological functions. Exp. Mol. Med. 36, 1–12 (2004).
Yang, S. L., Wu, C., Xiong, Z. F. & Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function. Mol. Med. Rep. 12, 2411–2416 (2015).
Tolonen, J.-P. et al. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Cell. Mol. Life Sci. 77, 3627–3642 (2020).
Heikkilä, M., Pasanen, A., Kivirikko, K. I. & Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3α variants in the hypoxia response. Cell. Mol. Life Sci. 68, 3885–3901 (2011).
Jiang, C. et al. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet–fed mice. Diabetes 60, 2484–2495 (2011).
Kihira, Y. et al. Deletion of hypoxia-inducible factor-1α in adipocytes enhances glucagon-like peptide-1 secretion and reduces adipose tissue inflammation. PLoS ONE 9, e93856 (2014).
Lee, K. Y., Gesta, S., Boucher, J., Wang, X. L. & Kahn, C. R. The differential role of Hif1β/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 14, 491–503 (2011).
Krishnan, J. et al. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 26, 259–270 (2012).
Halberg, N. et al. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 29, 4467–4483 (2009).
Zhang, X. et al. Adipose tissue-specific inhibition of hypoxia-inducible factor 1α induces obesity and glucose intolerance by impeding energy expenditure in mice. J. Biol. Chem. 285, 32869–32877 (2010).
García-Martín, R. et al. Adipocyte-specific hypoxia-inducible factor 2α deficiency exacerbates obesity-induced brown adipose tissue dysfunction and metabolic dysregulation. Mol. Cell. Biol. 36, 376–393 (2016).
Feng, Z. et al. Modulation of HIF-2α PAS-B domain contributes to physiological responses. Proc. Natl Acad. Sci. USA 115, E5990–E5999 (2018).
Matsuura, H. et al. Prolyl hydroxylase domain protein 2 plays a critical role in diet-induced obesity and glucose intolerance. Circulation 127, 2078–2087 (2013).
Lin, Q. et al. Activation of hypoxia‐inducible factor‐2 in adipocytes results in pathological cardiac hypertrophy. J. Am. Heart Assoc. 2, e000548 (2013).
Park, C.-S. & Shastri, N. The role of T cells in obesity-associated inflammation and metabolic disease. Immune Netw. 22, e13 (2022).
Lu, J., Zhao, J., Meng, H. & Zhang, X. Adipose tissue-resident immune cells in obesity and type 2 diabetes. Front. Immunol. 10, 1173 (2019).
Kim, D. et al. CXCL12 secreted from adipose tissue recruits macrophages and induces insulin resistance in mice. Diabetologia 57, 1456–1465 (2014).
Schmidt, A. M. & Moore, K. J. The semaphorin 3E/PlexinD1 axis regulates macrophage inflammation in obesity. Cell Metab. 18, 461–462 (2013).
Yunna, C., Mengru, H., Lei, W. & Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 877, 173090 (2020).
Morris, D. L., Singer, K. & Lumeng, C. N. Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr. Opin. Clin. Nutr. Metab. Care 14, 341 (2011).
Li, C. et al. Single-cell transcriptomics–based MacSpectrum reveals macrophage activation signatures in diseases. JCI Insight 4, e126453 (2019).
Suzuki, T. et al. ER stress protein CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity. Cell Rep. 18, 2045–2057 (2017).
Cinti, S. et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 46, 2347–2355 (2005).
Takikawa, A. et al. HIF-1α in myeloid cells promotes adipose tissue remodeling toward insulin resistance. Diabetes 65, 3649–3659 (2016).
Sharma, M. et al. Enhanced glycolysis and HIF-1α activation in adipose tissue macrophages sustains local and systemic interleukin-1β production in obesity. Sci. Rep. 10, 1–12 (2020).
Choe, S. S. et al. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes 63, 3359–3371 (2014).
Poblete, J. M. S. et al. Macrophage HIF-1α mediates obesity-related adipose tissue dysfunction via interleukin-1 receptor-associated kinase M. Am. J. Physiol. Endocrinol. Metab. 318, E689–E700 (2020).
Wang, Q. & Wu, H. T. Cells in adipose tissue: critical players in immunometabolism. Front. Immunol. 9, 2509 (2018).
Tao, J.-H., Barbi, J. & Pan, F. Hypoxia-inducible factors in T lymphocyte differentiation and function. Am. J. Physiol. Cell Physiol. 309, C580–C589 (2015).
Finlay, D. K. et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J. Exp. Med. 209, 2441–2453 (2012).
Shi, L. Z. et al. HIF1α–dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011).
O’Neill, A. F. M. A. L. A. J. The role of HIF in immunity and inflammation. Cell Metab. 323, 524–536 (2020).
McGettrick, A. F. & O’Neill, L. A. Obesity and vascular dysfunction. Pathophysiology 15, 79–89 (2008).
Claret, M. & Graupera, M. Endothelial cells: new players in obesity and related metabolic disorders. Trends Endocrinol. Metab. 29, 781–794 (2018).
Huang, Y. et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl Acad. Sci. USA 109, 17561–17566 (2012).
Niecknig, H. et al. Role of reactive oxygen species in the regulation of HIF-1 by prolyl hydroxylase 2 under mild hypoxia. Free Radic. Res. 46, 705–717 (2012).
Blouin, C. C., Pagé, E. L., Soucy, G. M. & Richard, D. E. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1α. Blood 103, 1124–1130 (2004).
Ziello, J. E., Jovin, I. S. & Huang, Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 80, 51–60 (2007).
Herr, B. et al. The supernatant of apoptotic cells causes transcriptional activation of hypoxia-inducible factor–1α in macrophages via sphingosine-1-phosphate and transforming growth factor-β. Blood, J. Am. Soc. Hematol. 114, 2140–2148 (2009).
Zhou, J., Schmid, T. & Brune, B. Tumor necrosis factor-α causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB-dependent pathway. Mol. Biol. Cell 14, 2216–2225 (2003).
Hellwig-Bürgel, T., Rutkowski, K., Metzen, E., Fandrey, J. & Jelkmann, W. Interleukin-1β and tumor necrosis factor-α stimulate DNA binding of hypoxia-inducible factor-1. Blood J. Am. Soc. Hematol. 94, 1561–1567 (1999).
He, Q. et al. Regulation of HIF-1α activity in adipose tissue by obesity-associated factors: adipogenesis, insulin, and hypoxia. Am. J. Physiol. Endocrinol. Metab. 300, E877–E885 (2011).
Zheng, X. et al. Repression of hypoxia-inducible factor-1 contributes to increased mitochondrial reactive oxygen species production in diabetes. eLife 11, e70714 (2022).
Dodd, M. S. et al. Fatty acids prevent hypoxia-inducible factor-1α signaling through decreased succinate in diabetes. JACC Basic Transl. Sci. 3, 485–498 (2018).
Bento, C. & Pereira, P. Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia 54, 1946–1956 (2011).
Sun, K., Halberg, N., Khan, M., Magalang, U. J. & Scherer, P. E. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol. Cell. Biol. 33, 904–917 (2013).
Chin, C.-H. et al. YC-1 inhibits lipid droplet accumulation and induces lipolysis in lipid-laden RAW264.7 macrophages. J. Food Drug Anal. 19, 4 (2011).
Chin, C.-H. et al. YC-1, a potent antithrombotic agent, induces lipolysis through the PKA pathway in rat visceral fat cells. Eur. J. Pharmacol. 689, 1–7 (2012).
Zhang, H. et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc. Natl Acad. Sci. USA 105, 19579–19586 (2008).
Teijeiro, A., Garrido, A., Ferre, A., Perna, C. & Djouder, N. Inhibition of the IL-17A axis in adipocytes suppresses diet-induced obesity and metabolic disorders in mice. Nat. Metab. 3, 496–512 (2021).
Hu, M. et al. The role of berberine in the prevention of HIF-1α activation to alleviate adipose tissue fibrosis in high-fat-diet-induced obese mice. Evid. Based Complement. Alternat. Med. 2018, 12 (2018).
Shin, M.-K. et al. Metabolic consequences of high-fat diet are attenuated by suppression of HIF-1α. PLoS ONE 7, e46562 (2012).
Busiello, R. A., Savarese, S. & Lombardi, A. Mitochondrial uncoupling proteins and energy metabolism. Front. Physiol. 6, 36 (2015).
Argyropoulos, G. & Harper, M.-E. Invited review: uncoupling proteins and thermoregulation. J. Appl. Physiol. 92, 2187–2198 (2002).
Valle, A., Oliver, J. & Roca, P. Role of uncoupling proteins in cancer. Cancers 2, 567–591 (2010).
Machado, S. A. et al. Browning of the white adipose tissue regulation: new insights into nutritional and metabolic relevance in health and diseases. Nutr. Metab. 19, 1–27 (2022).
Enerbäck, S. et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387, 90–94 (1997).
Fleury, C. et al. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 15, 269–272 (1997).
Boss, O. et al. Uncoupling protein‐3: a new member of the mitochondrial carrier family with tissue‐specific expression. FEBS Lett. 408, 39–42 (1997).
Krauss, S., Zhang, C.-Y. & Lowell, B. B. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. cell Biol. 6, 248–261 (2005).
Arsenijevic, D. et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 26, 435–439 (2000).
Gong, D.-W. et al. Lack of obesity and normal response to fasting and thyroid hormone in mice lacking uncoupling protein-3. J. Biol. Chem. 275, 16251–16257 (2000).
López-Soriano, F. et al. Amino acid and glucose uptake by rat brown adipose tissue. Effect of cold-exposure and acclimation. Biochem. J. 252, 843–849 (1988).
Inokuma, K.-I. et al. Uncoupling protein 1 is necessary for norepinephrine-induced glucose utilization in brown adipose tissue. Diabetes 54, 1385–1391 (2005).
Giralt, M. & Villarroya, F. Mitochondrial uncoupling and the regulation of glucose homeostasis. Curr. Diabetes Rev. 13, 386–394 (2017).
Kim, J.-w, Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 (2006).
Han, J. S. et al. Adipocyte HIF2α functions as a thermostat via PKA Cα regulation in beige adipocytes. Nat. Commun. 13, 3268 (2022).
Basse, A. L. et al. Regulation of glycolysis in brown adipocytes by HIF-1α. Sci. Rep. 7, 1–15 (2017).
Viengchareun, S. et al. Prolactin potentiates insulin-stimulated leptin expression and release from differentiated brown adipocytes. J. Mol. Endocrinol. 33, 679–691 (2004).
Callapina, M. et al. Nitric oxide reverses desferrioxamine-and hypoxia-evoked HIF-1α accumulation—implications for prolyl hydroxylase activity and iron. Exp. cell Res. 306, 274–284 (2005).
Yan, H.-F., Liu, Z.-Y., Guan, Z.-A. & Guo, C. Deferoxamine ameliorates adipocyte dysfunction by modulating iron metabolism in ob/ob mice. Endocr. Connect. 7, 604 (2018).
Zhao, R. Z., Jiang, S., Zhang, L. & Yu, Z. B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 44, 3–15 (2019).
Mailloux, R. J. & Harper, M.-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 51, 1106–1115 (2011).
Seo, J. et al. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun. Biol. 3, 638 (2020).
Safran, M. et al. Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc. Natl Acad. Sci. USA 103, 105–110 (2006).
Galmozzi, A. et al. ThermoMouse: an in vivo model to identify modulators of UCP1 expression in brown adipose tissue. Cell Rep. 9, 1584–1593 (2014).
Wang, H. et al. A dual Ucp1 reporter mouse model for imaging and quantitation of brown and brite fat recruitment. Mol. Metab. 20, 14–27 (2019).
Loomis, W. F. Reversible inhibition of the coupling between phosphorylation and oxidation. J. Biol. Chem. 173, 807 (1948).
Colman, E. Dinitrophenol and obesity: an early twentieth-century regulatory dilemma. Regul. Toxicol. Pharmacol. 48, 115–117 (2007).
Cavalieri, R. et al. Activating ligands of Uncoupling protein 1 identified by rapid membrane protein thermostability shift analysis. Mol. Metab. 62, 101526 (2022).
Rial, E. et al. Retinoids activate proton transport by the uncoupling proteins UCP1 and UCP2. EMBO J. 18, 5827–5833 (1999).
Wells, C., Karamitri, A., Karamanlidis, G. & Lomax, M. The cJun kinase inhibitor SP600125 stimulates expression of PPARγ co-activator 1α (PGC-1α) and uncoupling protein 1 (UCP1) in the HIB-1B brown fat preadipocyte cell line. Proc. Nutr. Soc. 67 E393 (2008).
Acknowledgements
This work was supported by grants funded by the Ministry of Science and Technology, ICT (Information and Communications Technology), Korea (grant no. RS-2022–00144495, RS-2023–00218623, NRF-2020R1A2C2100934, NRF-2020M2D9A2092374, and NRF-2021K1A3A1A17086250); the Ministry of Education, Korea (Brain Korea 21 Four, Future Veterinary Medicine Leading Education and Research Center; grant no. A0449–20230100); the Research Institute for Veterinary Science, College of Veterinary Medicine, Seoul National University; and the Faculty Academic Supporting Grant from Seoul National University.
Author information
Authors and Affiliations
Contributions
G.S.K. and G.-O.A. designed the structure and wrote the manuscript with the co-authors' inputs. G.S.K. contributed to the HIF and stromal cells section, Y.R.L. contributed to the adipocyte section, T.O. contributed to the UCP section, and H.J.P. contributed to the general HIF biology section. H.J.J. contributed to the figure. G.-O.A. edited the manuscript and contributed to the paper revision. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kang, GS., Jo, HJ., Lee, YR. et al. Sensing the oxygen and temperature in the adipose tissues – who’s sensing what?. Exp Mol Med 55, 2300–2307 (2023). https://doi.org/10.1038/s12276-023-01113-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01113-x
- Springer Nature Limited
This article is cited by
-
The biological function of the N6-Methyladenosine reader YTHDC2 and its role in diseases
Journal of Translational Medicine (2024)