
Abstract
Ferroptosis is a non-apoptotic form of regulated cell death characterized by the lethal accumulation of iron-dependent membrane-localized lipid peroxides. It acts as an innate tumor suppressor mechanism and participates in the biological processes of tumors. Intriguingly, mesenchymal and dedifferentiated cancer cells, which are usually resistant to apoptosis and traditional therapies, are exquisitely vulnerable to ferroptosis, further underscoring its potential as a treatment approach for cancers, especially for refractory cancers. However, the impact of ferroptosis on cancer extends beyond its direct cytotoxic effect on tumor cells. Ferroptosis induction not only inhibits cancer but also promotes cancer development due to its potential negative impact on anticancer immunity. Thus, a comprehensive understanding of the role of ferroptosis in cancer is crucial for the successful translation of ferroptosis therapy from the laboratory to clinical applications. In this review, we provide an overview of the recent advancements in understanding ferroptosis in cancer, covering molecular mechanisms, biological functions, regulatory pathways, and interactions with the tumor microenvironment. We also summarize the potential applications of ferroptosis induction in immunotherapy, radiotherapy, and systemic therapy, as well as ferroptosis inhibition for cancer treatment in various conditions. We finally discuss ferroptosis markers, the current challenges and future directions of ferroptosis in the treatment of cancer.
Similar content being viewed by others

Introduction
Every living being eventually dies. Cell death is a key biological process inherent in complex organisms, serving as a crucial mechanism for the elimination of unwanted cells.1 Mammalian cell death encompasses accidental cell death, an uncontrolled biological event triggered by unexpected attacks and injuries, and regulated cell death (RCD), which is driven by a genetically encoded apparatus and can be modulated by drug or genetic interventions.2 The orderly progression of RCD in complex organisms is integral to its normal development and homeostasis,3 while the loss of controlled cell death contributes to human diseases such as cancers, characterized by the presence of abnormal cells exhibiting unlimited replication and immortality due to successful evasion of cell death regulation. Cancer treatment strategies consistently prioritize the selective eradication of cancer cells while minimizing harm to normal cells. RCD is an important channel for achieving this, as it enables the specific targeting of tumor cells and enhances the efficacy of drug-induced cell death, while simultaneously reducing adverse effects on normal cells.
Ferroptosis, a term coined by the laboratory of Brent R. Stockwell in 2012, is a distinct mode of RCD characterized by the iron-dependent lethal accumulation of membrane-localized lipid peroxides.4 Cells undergoing ferroptosis display distinct hallmarks compared to other extensively studied forms of RCD,5 such as apoptosis,6 pyroptosis,7 and necroptosis.5 Morphologically, ferroptotic cells exhibit dysmorphic small mitochondria with condensed membranes and decreased crista.4,8,9,10 Mechanically, unlike classical RCD involving specific executioner proteins of cell death (such as gasdermin D for pyroptosis, caspase for apoptosis, and mixed lineage kinase domain-like protein (MLKL) for necrosis), the identity of the cell death executioner proteins in ferroptosis remains unclear. While it is widely accepted that the execution of ferroptosis necessitates the oxidized phospholipids (PLs) containing polyunsaturated fatty acids (PUFA-PLs), the mechanisms by which these oxidized PUFA-PLs, beyond a certain threshold, lead to membrane permeabilization and cell death, as well as the downstream executioners that mediate the eventual execution event known as the ‘point of no return’ in ferroptosis, remain largely elusive.11 The process of ferroptosis involves ferrous iron accumulation, free radical production, antioxidant system dysfunction, and lipid peroxidation. Based on its distinctive features, a comprehensive panel of biomarkers and functional tests, including pharmacological inhibition, has been assembled to effectively differentiate ferroptosis from other types of RCD, providing suitable tools for investigating the pathophysiological functions of ferroptosis.12
In recent years, modulating ferroptosis to intervene in the occurrence and development of cancer has been a hotspot and focus of etiological research and treatment. Ferroptosis is tightly implicated in tumor biology. On the one hand, tumor suppressors have been found to execute part of their tumor-suppression function depending on ferroptosis induction. Ferroptosis seems to be an innate tumor-suppressive mechanism.12,13 On the other hand, cancer cells, in order to support their survival, can evolve several mechanisms to evade host ferroptosis, which provides vulnerable targets for ferroptosis-based therapy. Interestingly, mesenchymal and dedifferentiated cancer cells, which typically exhibit resistance to apoptosis and traditional treatment approaches, display a remarkable susceptibility to ferroptosis. Consequently, ferroptosis is recognized as an attractive target for cancer treatments, especially for refractory tumors. So far, a plethora of ferroptosis interventions have shown promising effectiveness in cancer treatment even overcoming resistance to traditional therapies,14,15,16 and ferroptosis is also involved in the tumor-suppressive functions of radiotherapy and immunotherapy.17,18,19,20 Combination therapy based on ferroptosis is a highly promising strategy for enhancing the effectiveness of conventional therapies, tackling resistant tumors, and preventing tumor recurrence. However, the role of ferroptosis in tumor suppression depends on the context, as it appears to have apparently paradoxical roles in different stages of some tumors. For instance, ferroptosis induction facilitates the progression of chronic liver diseases to hepatocellular carcinoma (HCC),21 while it can restrain the established HCC development.22 Moreover, a study found that ferroptosis inhibitors can effectively suppress tumor growth as long as they are administered early when the tumor is sufficiently small.23 Therefore, achieving a comprehensive and in-depth understanding of the role of ferroptosis in cancers is crucial for effectively guiding its application in cancer treatment.
Given the vigorous growth in ferroptosis, it is imperative to gain iterative insights into ferroptosis. Here, we review the major milestones and molecular machinery of ferroptosis, including drivers and defenses two systems. Then, we decipher the functions of ferroptosis in tumor biology, the classic cancer-related ferroptosis regulatory pathways and ferroptosis-mediated crosstalk between cancers and immune cells. Lastly, we summarize the potential ferroptosis-based therapy and ferroptosis markers and discuss the current limitations and future directions of ferroptosis in the treatment of cancer.
Major milestone of ferroptosis
In the past decade, we have witnessed a significant surge in research on ferroptosis24 (Fig. 1). Although the term ferroptosis was coined in 2012,4 the clues to ferroptosis date back much earlier. Iron-induced toxicity was first observed in 1908.25 The importance of cystine in the viability and growth of mouse fibroblast strain L and the HeLa cell was reported in 1955.26,27 Dietary cystine and selenium in 1959 were further found to significantly reduce peroxidation in the liver and muscle of vitamin E-deficient chicks.28 In 1977, Shiro Bannai and colleagues observed that withdrawal of cystine-induced cell death accompanied by glutathione (GSH) depletion could be rescued by antioxidant vitamin E supplementation.29 They further reported in 1980 that cystine could be taken up from the environment by the system xc- in exchange for glutamate.30 The identification of glutathione peroxidase 4 (GPX4), a selenoprotein, in 1982 as a GSH-dependent peroxidase to counteract lipid peroxidation in membranes, marked a significant milestone.31 Over the following decade, GPX4 was found to counteract cell death associated with lipid peroxidation.32 In 1989, it was observed that glutamate-induced cytotoxicity resulted from the cystine uptake inhibition, leading to decreased GSH levels, oxidative stress, and ultimate cell death.33 Antioxidant treatments, such as alpha-tocopherol (α-toc), as well as inhibition of iron-containing lipid dioxygenase arachidonate lipoxygenase 12 (ALOX12), in 1992 and 1997, respectively, were found to prevent this form of cell death.34,35 Remarkedly, in 2001, the concept of “oxytosis” was coined to characterize the non-apoptotic cell death in neurons that is induced by oxidative stress in response to glutamate toxicity.36 While both oxytosis and ferroptosis involve reactive oxygen species (ROS) production, ALOXs and GSH depletion,36 and could be suppressed by iron chelators and enhanced by various sources of iron,37 some special features in oxytosis including cyclic guanosine monophosphate (cGMP)-gated channels, mitochondrial swelling and DNA fragmentation,38 highlighted ferroptosis as a distinct form of RCD.

History of research on the discovery and development of ferroptosis. The term ferroptosis was coined in 2012, but the understanding of ferroptosis can be traced back as early as 1908. Since 2012, there has been a flourishing development in the research of ferroptosis and its regulatory mechanisms. ACSL4 acyl-CoA synthetase long-chain family member 4, DHODH dihydroorotate dehydrogenase, Fer-1 ferrostatin-1, FSP1 ferroptosis suppressor protein 1, GCH1 GTP cyclohydrolase 1, GPX4 glutathione peroxidase 4, HSC hematopoietic stem cells, MBOAT1/2 membrane-bound O-acyltransferase domain-containing 1 and 2, PL phospholipid, PMN-MDSC polymorphonuclear myeloid-derived suppressor cell, PUFA polyunsaturated fatty acid, VK vitamin K
The discovery of ferroptosis stemmed from the high-throughput screening of small molecules aimed at targeting oncogenic RAS mutations. In 2003, erastin was identified as a selective inducer of non-apoptotic cell death in cancer cells dependent on ST- and RASG12V,39 along with the involvement of the RAS/BRAF/MEK/MAPK pathway and voltage-dependent anion channel (VDAC) that mediate oxidative stress and mitochondrial dysfunction, respectively.40 Another small molecule compound, RSL3, was discovered in 2008 through the same screening system, which could activate an iron-dependent form of cell death.41 In the same year, the inactivation of GPX4 was reported to induce a non-apoptotic cell death that could be suppressed by alpha-tocopherol and ALOX12/15 inhibitors.42 It was only in 2012 that the term “ferroptosis” was coined to describe this form of cell death, due to its dependence on iron, unique morphology, biochemical traits, and genetic features that distinguish it from other forms of regulated cell death.4 Erastin was discovered to block cystine uptake by inhibiting system xc- to induce ferroptosis, while ferrostatin-1 was identified as a powerful inhibitor of ferroptosis in cancer cells.4 In the following decades, breakthroughs in ferroptosis yielded a comprehensive insight into the mechanisms responsible for the execution and regulation of this process.
In 2014, GPX4 was identified as the central regulator of RSL3- and erastin-induced ferroptosis, and RSL3 directly inactivates GPX4, leading to lipid peroxidation and ultimately ferroptosis.43 Moreover, knockout of GPX4 causes cell death, while liproxstatin-1, a potent spiroquinoxalinamine derivative, is reported to suppress ferroptosis.8 In 2015, through the extensive use of massive insertional mutagenesis on haploid KBM7 cells, the inactivation of acyl-coenzyme A (CoA) synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) was shown to render these cells resistant to ferroptosis. Meanwhile, the most commonly mutated tumor suppressor protein, p53, suppresses solute carrier family 7 members 11 (SLC7A11) expression and cystine uptake, sensitizing cells to ferroptosis.13 In 2016, ferroptosis was found to rely on PUFA oxidation by ALOXs via a phosphorylase kinase G2 (PHKG2)-dependent iron pool and the covalent inhibition of the catalytic selenocysteine in GPX4 hinders the removal of PUFA hydroperoxides.44 Simultaneously, it has been reported that FIN56 not only induces the degradation of GPX4 but also depletes ubiquinone (CoQ10) through the mevalonate pathway to enhance ferroptosis sensitivity.45 In 2017, ACSL4 was further identified as an essential component for ferroptosis execution by promoting arachidonic acid (AA) or adrenic acid (AdA) esterification into phosphatidylethanolamines (PEs).46 A further study in 2018 underlined the requirement for selenium utilization by GPX4 to inhibit hydroperoxide-induced ferroptosis.45 In 2019, CD8+ T cells were shown to induce tumor ferroptosis during cancer immunotherapy.19 In the meantime, E-cadherin-mediated intercellular contacts control ferroptosis sensitivity through the Merlin/Hippo/Yes-associated protein 1(YAP) pathway to regulate the expression of ACSL4 and transferrin receptor (TFR1) in response to cell-cell contacts.47 Moreover, using unbiased genetic screens, ferroptosis suppressor protein (FSP1), previously named apoptosis-inducing factor mitochondria-associated 2 (AIFM2), was independently discovered as a novel ferroptosis resistance gene capable of complementing the loss or inhibition of GPX4.48,49
Two groups in 2020 independently identified GTP cyclohydrolase-1 (GCH1) as a suppressor of ferroptosis.50,51 Mechanistically, GCH1 suppresses ferroptosis through two main mechanisms. First, it produces the lipophilic antioxidant tetrahydrobiopterin (BH4), which aids in the prevention of lipid peroxidation. Second, GCH1 increases the abundance of the reducing agent CoQ10, which further protects against ferroptosis. This dual action of GCH1 contributes to the suppression of lipid peroxidation and the maintenance of cellular redox balance.50,51 Moreover, Zou et al. identified the oxidative organelles peroxisomes as a crucial factor in driving susceptibility to and evasion from ferroptosis through the synthesis of polyunsaturated ether PLs (PUFA-ePLs), which serve as substrates for lipid peroxidation.52 The administration of the engineered enzyme cyst(e)inase demonstrated a viable method to trigger ferroptosis in pancreatic ductal adenocarcinoma (PDAC) by depleting cysteine and cystine.53 Additionally, Ubellacker et al. found that melanoma cells in the lymph are prone to forming metastases in blood because lymph protects metastasizing melanoma cells from ferroptosis.54 In 2021, dihydroorotate dehydrogenase (DHODH) was discovered to be a mitochondrial suppressor of ferroptosis through its ability to decrease mitochondrial CoQ10 levels, and DHODH inhibitors had ferroptosis-sensitizing effects which was argued by Mishima et al. that DHODH inhibitors enhance sensitivity to ferroptosis through the inhibition of FSP1.55 In 2022, it was discovered that pathologically activated neutrophils, known as polymorphonuclear (PMN) myeloid-derived suppressor cells (MDSCs), undergo spontaneous ferroptosis, which contributes to immune suppression in cancer, highlighting the role of ferroptosis in immune regulation within the tumor microenvironment.23 Furthermore, FSP1 was further discovered to efficiently reduce vitamin K to its hydroquinone, providing protection against harmful lipid peroxidation and ferroptosis.56 Further studies in 2023 demonstrated that FSP1-dependent phase separation is crucial for ferroptosis induction.57 Besides, Zhao et al. found that low protein synthesis rates increased the susceptibility of hematopoietic stem cells to ferroptosis.58 Strikingly, through a whole-genome CRISPR activation screen, sex hormone-driven membrane-bound O-acyltransferase domain-containing 1 and 2 (MBOAT1/2) expressions were reported to prevent ferroptosis in cancer cells lacking the two main ferroptosis defense systems GPX4 and FSP1.59 Collectively, these studies contribute to a deeper understanding of the mechanisms and regulation of ferroptosis, highlighting its significance in the potential for therapeutic interventions.
Molecular mechanisms of ferroptosis
Ferroptosis reflects a redox imbalance between its drivers and defenses system.24,60 Here, we briefly outline its core mechanisms, with a specific emphasis on its driving and defense mechanisms (Fig. 2). For a more comprehensive and detailed understanding of the molecular pathways and intricate mechanisms underlying ferroptosis, we recommend referring to several recent reviews in the field.12,24,60,61,62,63,64

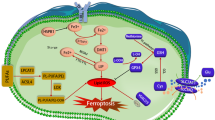
Molecular mechanisms of ferroptosis. Ferroptosis is driven by PUFA-PLs synthesis, lipid peroxidation and iron toxicity. Major defense systems of ferroptosis include the GPX4 antioxidant system, FSP1/ubiquinol (CoQH2), DHODH/CoQH2, GCH1/tetrahydrobiopterin (BH4) systems, monounsaturated fatty acid (MUFA)-PLs synthesis, and the ESCRT-III-mediated membrane repair systems. When ferroptosis-promoting activities significantly surpass the detoxification capabilities provided by the defense systems, a fatal accumulation of lipid peroxides on the cellular membranes ultimately results in membrane rupture and ferroptotic cell death. ABCB7 ATP binding cassette subfamily B member 7, ACC acetyl-CoA carboxylase, ALOX lipoxygenase, CISD1 CDGSH iron sulfur domain 1, CoQ coenzyme Q, Cys cysteine, Cys2 cystine, FTMT ferritin mitochondrial, GCL glutamate-cysteine ligase, GSH glutathione, GSSG oxidized glutathione, iPLA2b phospholipase A2 group VI, LIP labile iron pool, LPCAT3 lysophosphatidylcholine acyltransferase 3, NAD(P)H nicotinamide adenine dinucleotide phosphate, POR cytochrome P450 oxidoreductase, SCD1 stearoyl-CoA desaturase 1, SFA saturated fatty acid, SLC25A37, solute carrier family 25 member 37, SLC25A28 solute carrier family 25 member 28, SLC40A1 solute carrier family 40 member 1, STARD7 StAR-related lipid transfer domain containing 7, TF transferrin, TFR1 transferrin receptor, VDAC voltage-dependent anion channel. This figure was created with BioRender.com
Drivers of ferroptosis
PUFA-PLs synthesis
PUFAs are highly prone to lipid peroxidation due to the presence of weak C-H bonds at the bis-allylic positions.44,65 Recent studies mainly focus on ω-6 PUFAs, such as linoleic acid (18:2), gamma-linolenic acid (18:3), dihomo-gamma-linolenic acid (20:3), AA (20:4) and AdA (22:4), as well as ω-3 PUFAs, including alpha-linolenic acid (18:3), eicosapentaenoic acid (20:5) and docosahexaenoic acid (22:6).66,67 Among them, AA (20:4) and AdA (22:4), are the primary substrates of lipid peroxidation during ferroptosis.68 Notably, free PUFAs are not the direct drivers of ferroptosis, and they need to be esterified into membrane PLs to exhibit lethality after peroxidation.46,68,69 ACSL4 and LPCAT3 are responsible for the biosynthesis and esterification of PUFA-PLs. Taking AA (20:4) as a case in point, ACSL4 catalyzes the combination of free AA (20:4) and CoA to form a CoA-AA (20:4) intermediate, which is subsequently esterified into PEs by LPCAT3 to generate AA (20:4)-PE (PE-AA),46,68,69 which are necessary for the execution of ferroptosis. Consistently, malonyl-CoA generated by acetyl-CoA carboxylase (ACC)-catalyzed carboxylation of acetyl-CoA is critical for the synthesis of certain PUFAs and is therefore necessary for ferroptosis.60 The peroxisome-mediated biosynthesis of plasmalogens has been suggested as an additional pathway for the production of PUFAs involved in lipid peroxidation, favoring ferroptosis onset.52,70 On the contrary, phospholipase A2 group VI (iPLA2β) cleaves oxidized PUFA tails from PLs to suppress p53-driven ferroptosis.71 Remarkedly, deuterated PUFAs (D-PUFAs) at bis-allylic position retards the radical chain reaction of lipid peroxidation to protect against RSL3- or erastin-induced ferroptosis,44 suggesting the significance of the structure of PUFAs in its activity.
Lipid peroxidation
Lipid peroxidation is the hallmark of ferroptosis.4 PUFA-PLs are highly susceptible to peroxidation because of the presence of bis-allylic moieties in PUFAs. The oxidation of PUFA-PLs occurs through both enzymatic reactions and non-enzymatic autoxidation driven by the Fenton reaction.44,72 Enzymatic lipid peroxidation of PUFA-PLs primarily involves the action of ALOXs and cytochrome P450 oxidoreductase (POR).73 ALOXs are enzymes containing nonheme iron, which directly introduce oxygen to PUFAs and PUFA-containing lipids within biological membranes. For example, ALOX12 is essential for p53-dependent ferroptosis, while ALOX15 is involved in erastin- or RSL3-induced ferroptosis through complexing with PE binding protein 1 (PEBP1), specifically recognizing stearoyl-AA-PE to generate lipid peroxides.74 Moreover, ALOXE3, ALOX5, ALOX12B, and ALOX15B have been implicated in ferroptosis induction.75,76,77,78 Several ALOX inhibitors have been shown to possess antioxidant properties, effectively shielding cells from lipid peroxidation.35,42 However, genetic deletion of Alox15 in Gpx4 knockout mice failed to avert ferroptosis in vivo,8 and Alox12/15 failed to restore the viability of Gpx4 deficient T cells,79 suggesting the existence of alternative mechanisms in certain contexts of ferroptosis. As expected, POR directly supplies electrons to the P450 enzyme, which catalyzes the peroxidation of PUFA-PLs in an ALOX-independent manner.73,80 These studies suggested that several iron-dependent enzymes can promote lipid peroxidation and ferroptosis. Future investigations are needed to determine the potential involvement of other oxygenases, such as cyclooxygenases and peroxygenases, in lipid peroxidation. Non-enzymatic lipid peroxidation of PUFA-PLs is driven by the Fenton reaction, with iron serving as a catalyst.72,81 In this process, once the initial phospholipid hydroperoxides (PLOOHs) are generated (via enzymatic reactions or other cellular metabolic processes) and are not promptly reduced by GPX4, they can interact with ferrous iron to yield alkoxyl and peroxyl radicals (Fenton reaction), initiating PLOOHs production.82,83
Iron metabolism and toxicity
As noted above, lipid peroxidation requires both iron-dependent enzymes and iron-mediated Fenton reactions, thereby at least partly providing the iron-dependent nature of ferroptosis. Thus, interventions targeting iron metabolism have an impact on the vulnerability to ferroptosis. Ferric iron is the primary form of iron in circulation and binds to transferrin (TF).84 It is delivered into cells and localized in endosomes with the assistance of TFR1, a membrane protein.85 Within the endosome, ferric iron is reduced to ferrous iron by the six-transmembrane epithelial antigen of the prostate 3 (STEAP3).86 The endocytosed ferrous iron is later released into the cytoplasm via solute carrier family 11 member 2 (SLC11A2), forming the labile iron pool (LIP), which catalyzes the generation of hydroxyl radicals and triggers ferroptosis.87 Excess intracellular iron is typically sequestered within the ferritin protein, which consists of two subunits: ferritin heavy chain 1 (FTH1) and ferritin light chain (FTL).41 Ferritin undergoes degradation via ferritinophagy, facilitated by nuclear receptor coactivator 4 (NCOA4), resulting in the release of substantial amounts of iron.88,89 Additionally, excess cytoplasmic ferrous iron can be exported from the cell through solute carrier family 40 member 1 (SLC40A1).90 Consistently, deletion of TF, TFR1, SLC11A2 and NCOA1, and overexpression of FTH1, FTL and SLC40A1 suppresses ferroptosis by decreasing the LIP.91 Therefore, interventions that modulate the import, storage, and export of iron in the cytoplasm contribute to an increase in susceptibility to ferroptosis.
In addition to the cytoplasm, mitochondria, which is the primary site of iron utilization and the main source of ROS, plays a major role in modulating redox-active reactions and ferroptosis.92,93 To reach the mitochondria, iron must traverse both the outer and inner mitochondrial membranes to enter the matrix through SLC11A2,94 and solute carrier family 25 member 37 (SLC25A37) or solute carrier family 25 member 28 (SLC25A28), respectively.95,96,97 Moreover, recent studies highlighted the key role of CDGSH iron sulfur domain 1 (CISD1) in regulating iron homeostasis in mitochondria.98,99 CISD1 knockdown significantly increases the content of erastin-induced mitochondrial ferrous irons and promotes mitochondrial lipid peroxidation and ferroptosis.100 CISD1 can also bind with VDAC proteins and regulate their gating in a redox-dependent manner.101 Inhibiting VDAC proteins can prevent mitoCISD1-dependent mitochondrial iron accumulation and erastin-induced ferroptosis.40,101,102,103 Ferritin mitochondrial (FTMT) serves as the iron-storage protein in mitochondria, inhibiting ferroptosis by reducing total and chelatable iron levels.104,105,106 ATP binding cassette subfamily B member 7 (ABCB7) is involved in the transfer of iron from mitochondria to cytosol.107,108,109 Although mitochondrial iron accumulation can be observed in the absence of ABCB7,107 ABCB7 loss does not lead to an increase in mitochondrial ROS and ferroptosis.110 On the contrary, ABCB8 can facilitate mitochondrial iron export.111,112 Overexpression of ABCB8 reduces mitochondrial iron and protects against ferroptosis-related I/R damage and doxorubicin-induced cardiomyopathy.113,114 Collectively, these results provide strong evidence that diverse factors controlling iron metabolism regulate susceptibility to ferroptosis.
Defenses of ferroptosis
GPX4 antioxidant system
Erastin and RSL3 are the representative two types of ferroptosis inducers (FINs) through directly inhibiting the activity of xc- system and GPX4, respectively.4,43 The system xc- containing subunits SLC7A11 and solute carrier family 3 member 2 (SLC3A2) mediates the exchanges of intracellular glutamate for extracellular cystine.115,116 Intracellular cystine is quickly converted to cysteine, playing a vital role as a cellular antioxidant and acting as the limiting factor for the synthesis of glutamate-cysteine ligase (GCL)-mediated GSH synthesis.117,118 The availability of cellular GSH closely regulates the cellular GPX4 activity.119,120 Thus, the inactivation of GPX4 by both erastin and RSL3, either directly or indirectly, underscores the significance of GPX4 as a key repressor of ferroptosis.
GPX4 is the sole member of the GPX family that acts as a phospholipid hydroperoxidase, directly reducing PLOOH to their corresponding phospholipid alcohols (PLOH).31,121 GPX4’s catalytic reaction operates according to a ping-pong mechanism, where the enzyme’s active site shuttles between oxidation and reduction states. Firstly, the PLOOH oxidizes the active site selenol in GPX4 (GPX4-SeH) to form the selenenic acid intermediate (GPX4-SeOH). Secondly, this intermediate undergoes a reaction with GSH, resulting in the formation of the selenium-glutathione adduct (GPX4-Se-SG). Thirdly, through a reaction with a second GSH molecule, GPX4-Se-SG undergoes conversion to GPX4-SeH, generating oxidized glutathione (GSSG).122,123 By examining the crystal structure of seleno-GPX4, researchers observed the existence of seleninic acid (GPX4-Se-OO-) within the enzyme’s active site.124 This finding implies the possibility of an alternative reaction mechanism that encompasses three distinct redox states (GPX4-SeH, GPX4-SeOH, GPX4-Se-OO-) of the catalytically active selenocysteine. These studies have also emphasized the essential role of selenocysteine in the expression and activity of GPX4, which is in line with Ingold et al.’s findings that the substitution of a cysteine residue for selenocysteine (U46C) in GPX4 is required to prevent hydroperoxide-induced ferroptosis.125
GPX4 exists in three isoforms: mitochondrial, cytosolic, and nuclear GPX4. While derived from the same GPX4 gene, the isoforms of GPX4 have distinct transcription initiation sites.126,127,128,129 Early embryonic lethality occurs when the cytosolic GPX4 gene is genetically ablated or expresses an inactive form.130 The rescue of the lethal phenotype in Gpx4-null mutant mice was achieved by re-expression of cytoplasmic GPX4, rather than mitochondrial or nuclear GPX4, indicating the crucial role of cytosolic GPX4 in preventing embryonic lethality.131,132 Disruption of mitochondrial GPX4 in mice does not result in lethality but instead causes male infertility due to abnormal sperm development.130,133 It seems that only cytosolic GPX4 could suppress ferroptosis, which is challenged by recent studies that mitochondrial GPX4, but not cytoplasmic GPX4, could potently suppress lipid peroxidation and ferroptosis in DHODH or glycerol-3-phosphate dehydrogenase 2 (GDP2) knockout cells.134,135 Therefore, these organelle-specific forms of GPX4 may independently inhibit local lipid hydroperoxides and ferroptosis, although the potential role of nuclear GPX4 requires further investigation. Notably, GPX4 depletion also mediates apoptosis, necroptosis and pyroptosis in mice, suggesting that GPX4 depletion-induced lipid peroxidation occupies a central position at the intersection of these forms of RCD. Thus, the detection of multiple markers is essential for definitively identifying ferroptosis in addition to lipid peroxidation.
Radical-trapping antioxidant system
As mentioned above, GPX4 is a central suppressor of ferroptosis, and other mechanisms that regulate the activity or expression of GPX4 also control susceptibility to ferroptosis.43,45,136,137,138 However, some cancer cells survived GPX4 inhibition, suggesting the existence of alternative mechanisms of ferroptosis resistance. In recent years, three GPX4-independent systems that capture free radicals to exert their antioxidative effects and suppress ferroptosis have been identified. These systems include FSP1/ ubiquinol (CoQH2), DHODH/CoQH2, and GCH1/BH4. Coenzyme Q (CoQ) is an endogenous antioxidant and exists in three forms: CoQ10, semiquinone, and CoQH2, wherein CoQH2 traps lipid peroxyl radicals to protect cells from ferroptosis.15,49 The synthesis and cellular distribution of CoQ10 are linked to StAR related lipid transfer domain containing 7 (STARD7), which transports CoQ10 from mitochondria, where it is synthesized, to the plasma membrane.139 In 2020, two independent teams found that N-myristylation-dependent recruitment of FSP1 to the plasma membrane resists ferroptosis in the plasma membrane.48,49,140 Mechanistically, FSP1 suppresses lipid peroxidation by catalyzing the reduction of CoQ10 to CoQH2 with the consumption of nicotinamide adenine dinucleotide phosphate (NAD(P)H).48 Moreover, FSP1 was identified as a vitamin K reductase to generate its associated hydroquinone, which inhibits lipid peroxidation at the expense of NAD(P)H.56 A recent study also suggested that phase separation of FSP1 plays a role in promoting ferroptosis which requires N-terminal myristoylation, as well as specific amino acid residues and essentially disordered, low-complexity regions in FSP1.57 Analogous to the function of FSP1 in the plasma membranes, DHODH detoxifies lipid peroxides by reducing CoQ10 to CoQH2, thereby inhibiting ferroptosis specifically in the mitochondria.134 Furthermore, GCH1 has been identified as a suppressor of ferroptosis through a two-pronged mechanism.48,50 On one hand, GCH1 produces the lipophilic antioxidant BH4 preventing lipid peroxidation; on the other hand, GCH1 induces lipid remodeling as a protective measure against ferroptosis by selectively safeguarding PLs with two polyunsaturated fatty acyl tails from depletion.48,50 However, the subcellular compartments wherein the GCH1/BH4 system functions still need further investigation.
MUFA-PLs synthesis
Different from PUFAs, monounsaturated fatty acids (MUFAs) are less susceptible to peroxidation owing to a lack of bis-allylic positions. Exogenous MUFAs can prevent ferroptosis by displacing PUFAs from membrane lipids.44,141 The biosynthesis of anti-ferroptosis MUFA-PLs is mainly regulated by stearoyl-CoA desaturase 1 (SCD1) and acyl-CoA synthetase long-chain family member 3 (ACSL3).141,142 SCD1 introduces a double bond in the cis-Δ9 position of the de novo synthesized saturated fatty acids (SFAs), particularly palmitic acid (C16:0) and stearic acid (C18:0), resulting in the formation of palmitoleic acid (C16:1) and oleic acid (C18:1), respectively.142 As a result, overexpression of SCD1 enhances MUFA synthesis and protects cells from ferroptosis, while inhibition of SCD1 enhances the sensitivity to ferroptosis.59,142 Moreover, MUFAs are reported to enhance both the number of lipid droplets and the number/function of peroxisomes, leading to a reduction in ether lipids and lipid oxidation.143 However, oleic acid does not lower the propensity of cells to succumb to ferroptosis in ACSL3-depleted cells.54 ACSL3 converts MUFAs into their acyl-CoA esters, facilitating their incorporation into membrane PLs.141 Thus, similar to PUFAs, MUFAs need to be inserted into the membrane to exhibit antioxidant properties. Interestingly, MBOAT1/2 has recently been identified to selectively transfer MUFAs into lyso-PE, resulting in an increase in cellular PE-MUFA and a corresponding decrease in cellular PE-PUFA, ultimately resisting ferroptosis.59 On all accounts, the anti-ferroptosis role of MBOAT1/2 operates independently of GPX4 and FSP1 through a surveillance mechanism mediated by PL remodeling.
Membrane repair system
The rupture of the plasma membrane is involved at the terminal stage of ferroptosis. Membrane damages in ferroptosis cover a loss of plasma membrane integrity,88 and rupture of the outer mitochondria membrane.4,8 Consequently, membrane repair systems have been proposed and demonstrated to prevent ferroptosis. Among these systems, the Endosomal Sorting Complex Required for Transport-III (ESCRT-III) has gained attention as a common mechanism for membrane repair, acting as a defense against various forms of RCD, including ferroptosis.144,145,146 Mechanistically, ferroptosis leads to an elevation in cytosolic Ca2+ levels due to an osmotic imbalance triggered by the opening of small nanopores.147 In response to the influx of Ca,2+ subunits of ESCRT-III known as charged multivesicular body proteins (CHMPs), specifically CHMP5 and CHMP6, are recruited and assembled at the location of damage to facilitate membrane repair processes. Erastin and RSL3, which are known as ferroptosis activators, lead to the buildup of CHMP5 and CHMP6 in the plasma membrane of pancreatic cancer cells, and the knocking out of CHMP5 or CHMP6 intensifies the susceptibility of cancer cells to ferroptosis.146 Furthermore, in certain cases, FSP1 inhibits ferroptosis by promoting the accumulation of CHMP5 and CHMP6 on the plasma membrane.148 Overall, these findings highlight the critical role of ESCRT-III activation in preventing ferroptosis.
Functions of ferroptosis in cancer biology
Oxygen (O2)-driven metabolism is vital for the survival of organisms and the execution of biological activities, achieved via a sequence of redox reactions.149 The transition metal iron is the key element to catalyze these redox processes, leading to the generation of ROS, which encompasses various oxygen derivatives, including ferroptosis divers PLOOHs. Accumulating evidence indicates ferroptosis in tumor biology.
Ferroptosis induction in tumor suppression
Ferroptosis appears to function as an innate mechanism for tumor suppression, mediating the anticancer activity of several tumor suppressor genes. Tumor suppressors such as p53, BRCA1-associated protein 1 (BAP1), fumarate hydratase (FH), Kelch-like ECH-associated protein 1 (KEAP1), and the epigenetic regulator MLL4 have been shown to exert their tumor-suppressive functions, at least partially, by inducing ferroptosis in tumor cells.
The tumor suppressor TP53, widely regarded as the most critical barrier to cancer development, effectively exerts its ferroptosis-mediated tumor-suppression function by suppressing the cystine transporter SLC7A11 in an ALOX12-dependent manner.13,150,151 While the acetylation-defective mutant p533KR (K117R, K161R, K162R) loses its conventional functions of promoting cell-cycle arrest, apoptosis, and senescence, it still retains its tumor-suppressive ability by promoting ferroptosis.13 By contrast, the mutant p534KR (K98R+3KR) lack ferroptosis regulatory activity and consequently lose their tumor-suppressive functions,152,153 suggesting the significance of acetylation in ferroptosis. Moreover, the TP53 single-nucleotide polymorphism P47S found in many people of pre-menopausal African-American women has an increased risk of breast cancer.152,154 Mechanistically, p53P47S is defective in promoting ferroptosis and repressing tumor development through increasing the cellular levels of CoA and GSH.154 These findings indicate that ferroptosis is at least partly responsible for TP53-mediated tumor suppression.
BAP1 encodes a deubiquitinase responsible for removing ubiquitin from histone 2A and frequently exhibits inactivating mutations and deletions in various sporadic cancers.155 Interestingly, BAP1 suppresses tumorigenesis partly through ferroptosis by repressing SLC7A11 via reducing histone 2A ubiquitination (H2Aub) occupancy on the SLC7A11 promoter.156 Deletions and mutations of BAP1 result in the loss of its ability to repress SLC7A11, enabling cells to evade ferroptosis and promoting tumor formation.156 BAP1 re-expression in a BAP1-deficient background significantly inhibited tumor development with condensed mitochondria and increased 4-hydroxynonenal (4-HNE) protein expression, which could be partially restored by the ferroptosis inhibitor liproxstatin-1,156 suggesting that ferroptosis at least partly contribute to BAP1’s tumor suppression in vivo.
FH is an enzyme involved in the tricarboxylic acid (TCA) cycle, which has been confirmed as a bona fide tumor suppressor in renal cancer.157 Genetic mutation of FH has been detected in both benign and malignant renal cancer lesions.157,158,159 Notably, renal cancer cells with FH mutations display resistance to ferroptosis and maintain their viability and ability to proliferate even when deprived of cystine. In contrast, wild-type FH cancer cells are unable to proliferate under these conditions.160 These findings confer the tumorigenic advantage of the loss of FH function under oxidative stress through suppressing ferroptosis, supporting the notion that ferroptosis may serve as a physiologically relevant mechanism to suppress tumors.
KEAP1, a ubiquitinated enzyme, is commonly mutated or inactivated in lung cancers.161,162 KEAP1 binds to nuclear factor erythroid 2-related factor 2 (NRF2) and triggers its proteasomal degradation, thereby inhibiting tumor development.163 Loss of KEAP1 function leads to increased tumor burden and accelerates tumor growth,163,164 because its mutants or deficiency in lung cancers upregulate the expression of FSP1 by stabilizing NRF2 proteins, resulting in ferroptosis resistance.165 Moreover, KEAP1 knockdown protects glioma cells from ferroptosis and promotes their proliferation by upregulating NRF2-mediated expression of SLC7A11.166 These findings indicate that the ferroptosis-promoting role of KEAP1 potentially at least partly accounts for its tumor-suppressive function.
The epigenetic regulator MLL4 is one of the most commonly mutated genes in cancer biology.167,168 It can activate key ALOXs genes, such as ALOX12, promoting epidermal differentiation and barrier formation and, in turn, inhibiting cutaneous squamous cell carcinomas through ferroptosis.169 Epidermal MLL4 deficiency results in impaired skin differentiation, the development of precancerous neoplasms and resistance to ferroptosis, accompanied by downregulation of pro-ferroptosis genes ALOXs (ALOX12, ALOX12B, and ALOXE3) and the upregulation of anti-ferroptosis genes (GPX4, SLC7A11, and SCD1).169 This suggests that MLL4-mediated ferroptosis serves as a critical natural mechanism in promoting epidermal differentiation, maintaining skin homeostasis, and preventing cutaneous carcinomas formation.
Ferroptosis evasion in tumor progression
Despite the presence of the ferroptosis-mediated tumor suppression mechanism, tumors inevitably arise and progress uncontrollably, indicating the existence of the mechanism of ferroptosis evasion in tumor. Building upon the core driver and defense mechanism of ferroptosis. We will briefly discuss the mechanisms through which tumor cells have evolved to evade ferroptosis and support tumor development.
Tumor cells exhibit heightened antioxidant capacity as an adaptive response to increased levels of ROS caused by metabolic and signaling abnormalities.170 Stabilizing and overexpressing the anti-ferroptotic systems are crucial mechanisms evolved by tumor cells to avert ferroptosis and promote tumor progression. The upregulation of the SLC7A11/GSH/GPX4 axis, a key ferroptosis defense system, is a significant evasion mechanism evolved by tumor cells. SLC7A11 is overexpressed in multiple cancers, and it is one of the extensively studied mechanisms by which tumor cells evade ferroptosis.53,171,172 For instance, its upregulation by the inactivation of tumor suppressors like TP53, BAP1, and ARF confers ferroptosis evasion and promotes tumor growth.13,156,173 Moreover, Oncogenic KRAS activation has also been shown to upregulate SLC7A11 expression, defending against ferroptosis and promoting lung adenocarcinoma (LUAD) development.174 GSH, an antioxidant that functions as the cofactor of GPX4, is frequently elevated in tumors, accelerating tumor progression and therapy resistance.170,175,176,177,178,179 GPX4, the essential antioxidant peroxidase of ferroptosis, has also been found to be highly expressed in various tumors.180,181 Several cancer phenotypes characterized by stem cell-like or dedifferentiated states exhibit highly dependent GPX4 for survival, indicating its crucial role in evading ferroptosis and supporting tumor cell surviva.15,182 The radical-trapping antioxidant system mechanisms of ferroptosis, which are mediated by the FSP1 and GCH1, are also upregulated in some cancers and contribute to ferroptosis evasion and tumor development.50,165 Additionally, NRF2, a master regulator of antioxidant defense, which is upregulated in multiple cancers and is considered a driver of cancer progression, metastasis, and therapy resistance,183 regulates components of the ferroptosis cascade, including SLC7A11, GPX4 and FSP1, to defend against ferroptosis contribute to tumor progression and therapy resistance.165,184,185,186,187,188
Tumor cells also employ mechanisms that limit pro-ferroptotic systems to evade ferroptosis. Downregulation of peroxidized PUFA-PLs and reduction of the LIP within cancer cells have been associated with ferroptosis evasion and tumor progression.189 For instance, iPLA2β, which cleaves and detoxifies peroxidized lipids to avert ferroptosis, is overexpressed in some human cancers and is involved in inhibiting p53-mediated ferroptosis and tumor suppression.190 Sterol regulatory element-binding protein 2 (SREBP2)-driven iron homeostatic pathways are overexpressed in melanoma circulating tumor cells, reducing intracellular iron pools and conferring resistance to ferroptosis, contributing to cancer progression, metastasis, and drug resistance.191 Proteins involved in the iron-sulfur clusters (ISCs) synthesis and assembly, such as NFS1, Frataxin, and CISD2, have been found to be highly expressed in tumors, enabling cancer cells to evade ferroptosis and contribute to tumor progression by reducing the LIP.192,193,194 Breast cancer cells detaching from the extracellular matrix increase the expression of prominin 2, stimulating iron export and reducing the LIP to evade ferroptosis.195
Strikingly, tumor microenvironment functions in ferroptosis evasion in tumor progression. For example, the lymphatic environment, which contains an abundant amount of oleic acid, can enhance the synthesis of MUFA-phospholipids (MUFA-PLs) in melanoma cells through an ACSL3-dependent pathway, leading to resistance to oxidative stress and ferroptosis. This facilitates the migration and survival of cancer cells in lymphatics and enhances their ability to survive during subsequent metastasis via the bloodstream.54 Moreover, mammary adipocytes provide protection for triple-negative breast cancer (TNBC) cells against ferroptosis by secreting oleic acid,196 providing a unique micro-environment for cancer cell survival. Additionally, sex hormones could regulate ferroptosis surveillance.59 MBOAT1 and MBOAT2 could be regulated by estrogen receptors (ER) and androgen receptors (AR), respectively. Both of them could catalyze the incorporation of MUFAs into PL to mediate the ferroptosis defense mechanism independently of GPX4 and the radical-trapping antioxidant system, suggesting the potential role of ferroptosis suppression in specific contexts related to sex hormone signaling. These findings also indicate that sex differences need to be considered, and ER or AR antagonists should combine with FINs to inhibit the ER+ cancer or AR+ prostate tumor growth, respectively.59,197
Cancer-related pathway in ferroptosis
RAS signaling
The RAS family are the most frequently mutated oncogenes in human cancers, comprising three major mutation variants: KRAS, HRAS, and NRAS.198,199 RAS is the first oncogene associated with ferroptosis, due to erastin and RSL3 were initially discovered from RAS synthetic lethal screen.39,40,41 Inhibiting RAS or the downstream RAF/MEK/MAPK axis can reverse the erastin or RSL3-induced selective cytotoxicity in engineered RAS-mutant tumor cells, possibly because mutant RAS signaling enhances the cellular basal iron by modulating the expression of iron metabolism-related genes.40,41 Mutations in epidermal growth factor receptor (EGFR), the RAS signaling upstream, increase the sensitivity of ferroptosis in non-small cell lung cancer (NSCLC) cells and human mammary epithelial cells.200 Notably, mutant KRAS has been reported to evade ferroptosis, establishing a targetable vulnerability in KRAS-mutant lung cancer174,201 (Fig. 3a). Mutant KRAS upregulate the NRF2/SLC7A11 axis, resulting in the selective SLC7A11 inhibition killing in KRAS-mutant cancer cells.174 Mutant KRAS can also elevate FSP1 by activating MAPK and NRF2 pathways to protect KRAS-mutant cells from ferroptosis during tumor initiation.202 Combining FSP1 in ferroptosis-inducing therapy represents an effective strategy for treating KRAS-mutant tumors.202 Moreover, mutant KRAS lung cancer has been found to upregulate fatty acid synthase (FASN) and escape from ferroptosis by promoting the synthesis and availability of SFA/MUFA, potentially through an ACSL3-dependent mechanism.201 ACSL3, downstream of FASN, is essential for tumorigenesis in mutant KRAS lung cancer.203 Targeting FASN represents an effective therapeutic strategy for inducing ferroptosis in mutant KRAS lung cancer.

Cancer-related pathways in ferroptosis. a RAS signaling governs upregulation of SCL7A11, FASN, and FSP1 to evade ferroptosis, establishing a targetable vulnerability. b NRF2 protects cancer cells from ferroptosis primarily through transcriptional regulation of downstream target genes involved in iron metabolism, GSH metabolism and ROS detoxification enzymes. c mTOR signaling primarily inhibits the sensitivity to ferroptosis through autophagy, promoting GPX4 protein synthesis, and upregulating the SREBP1/SCD and KEAP1/NRF2 axis. d Hypoxia plays a dual role in regulating ferroptosis by inducing the expression of its primary regulators HIF1α and HIF2α. e EMT reshapes the metabolic status granting mesenchymal tumor cells vulnerability to ferroptosis. f p53 transcriptionally suppresses SLC7A11 expression or modulates metabolism-related genes to promote ferroptosis. g The YAP/TAZ pathway plays a crucial role in regulating cell density-mediated and D-lactate-induced ferroptosis. h Ferroptosis serves as a type of autophagy-dependent cell death involving ferritinophagy, lipophagy, mitophagy, clockophagy, and chaperone-mediated autophagy. i Mitochondrial TCA cycle, ETC and glutamate are required for cystine deprivation-induced ferroptosis. PPP generate NADPH to implicate in ferroptosis process. Energy stresses facilitate tumor defense against ferroptosis by activating AMPK to enhance ACC-mediated MUFA formation. 4EBP 4E (eIF4E)-binding proteins, α-KG α-Ketoglutaric acid, ACSL5 acyl-CoA synthetase long chain family member 5, AKT AKT serine/threonine kinase, ASS1 argininosuccinate synthase 1, AKR1C1 aldo-keto reductase family 1 member C1, ANGPTL4 angiopoietin-like 4, ARNTL aryl hydrocarbon receptor nuclear translocator like, AMPK protein kinase AMP-activated catalytic subunit alpha 1, ATM ataxia-telangiectasia mutated, BAMBI BMP and activin membrane bound inhibitor, BRAF B-Raf proto-oncogene, serine/threonine kinase, CDKN1A cyclin dependent kinase inhibitor 1A, CDK7 cyclin dependent kinase 7, CHAC1 ChaC glutathione specific gamma-glutamylcyclotransferase 1, DPP4 dipeptidyl peptidase 4, DPP9 dipeptidyl peptidase 9, EGLN2 egl-9 family hypoxia inducible factor 2, EMP1 epithelial membrane protein 1, EMT epithelial-mesenchymal transition, FABP3/7 fatty acid binding protein 3/7, FASN fatty acid synthase, FTH1 ferritin heavy chain 1, GCLC glutamate-cysteine ligase catalytic subunit, GCLM glutamate-cysteine ligase modifier subunit, GFPT1 glutamine--fructose-6-phosphate transaminase 1, GINS4 GINS complex subunit 4, GLS glutaminase, GLUD1 glutamate dehydrogenase 1, HDAC Type-2 histone deacetylase 2, HIF1α hypoxia inducible factor 1 subunit alpha, HIF2α hypoxia inducible factor 2 subunit alpha, HILPDA hypoxia inducible lipid droplet associated, HMOX1 heme oxygenase 1, HSP90 heat shock protein 90, HSC70 heat shock cognate 71 kDa protein, Keap1 Kelch-1ike ECH- associated protein l, KDM5A lysine demethylase 5A, *KRAS mutant KRAS, KRAS, KRAS proto-oncogene, GTPase, LATS1 large tumor suppressor kinase 1, LAMP2A lysosomal-associated membrane protein 2, LC3 MAP1LC3A microtubule associated protein 1 light chain 3 alpha, LDHD lactate dehydrogenase D, LKB1 Lkb1 kinase, MEK MAP kinase-ERK kinase, MDM2 proto-oncogene, MDMX MDM4 regulator of p53, MEX3A mex-3 RNA binding family member A, mTOR rapamycin target protein, MT1G metallothionein 1G, MPC1 mitochondrial pyruvate carrier 1, MST macrophage stimulating, MYC MYC proto-oncogene, bHLH transcription factor, NCOA4 nuclear receptor coactivator 4, NRF2 nuclear factor erythroid 2-related factor 2, NF2 neurofibromin 2, NOX2 NADPH oxidase 2, NOX4 NADPH oxidase 4, OXPHOX oxidative phosphorylation, PI3K phosphoinositide 3-kinase, PPARGC1A PPARG coactivator 1 alpha, PRMT5 protein arginine methyltransferase 5, RAB7A member RAS oncogene family, SCD5 stearoyl-Coenzyme A desaturase 5, SREBP1 sterol regulatory element-binding protein 1, SESN2 sestrin 2, E-cad E-cadherin, SLC40A1 solute carrier family 40 member 1, SLC7A11 solute carrier family 7 member 11, SOD1 superoxide dismutase 1, SQSTM1 sequestosome 1, TAZ Tafazzin, TXNRD1 thioredoxin reductase 1, TCA cycle tricarboxylic acid cycle, WTAP WT1 associated protein, YAP1 Yes1 associated transcriptional regulator, ZEB1 zinc finger E-box binding homeobox 1, ZNF498 zinc finger and SCAN domain containing 25. This figure was created with BioRender.com
NRF2 signaling
NRF2 is a crucial transcription factor involved in cellular defense against oxidative and electrophilic stress.204 NRF2 acts as a suppressor of tumor initiation in the early stages of cancer.205,206,207 However, once oncogenic driver mutations occur, the high expression status of NRF2 in cancer cells may promote tumor progression and therapeutic resistance,208,209 partly through its ability to defend against ferroptosis. NRF2 primarily defends against ferroptosis through the transcriptional regulation of downstream target genes involved in iron metabolism (including SLC40A1, metallothionein 1G (MT1G), heme oxygenase 1 (HMOX1), and FTH1), GSH metabolism (including SLC7A11, glutamate-cysteine ligase catalytic subunit (GCLC), glutamate-cysteine ligase modifier subunit (GCLM), and ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1)) and ROS detoxification enzymes (including thioredoxin reductase 1 (TXNRD1), aldo-keto reductase family 1 member C 1/2/3 (AKR1C1/2/3), sestrin 2 (SESN2), glutathione S-transferase pi 1 (GSTP1), and NAD(P)H quinone dehydrogenase 1(NQO1)), thus suppressing oxidative damage induced by ferroptotic stress210,211 (Fig. 3b). This transcriptional regulation mechanism relies heavily on the stability of NRF2, which is negatively regulated by the ubiquitin ligase scaffold protein KEAP1, a tumor suppressor frequently mutated in NSCLC, via the ubiquitin-proteasome pathway.212 Protein arginine methyltransferase 5 (PRMT5) inhibits NRF2 by methylating and stabilizing KEAP1.213 p62 and dipeptidyl peptidase 9 (DPP9) disrupt the interaction between KEAP1 and NRF2 by competitively binding to KEAP1 to maintain the NRF2 stability and promote the transcription of its downstream iron metabolism and antioxidant genes, resulting in ferroptosis-mediated sorafenib resistance.187,214 mTORC1 and disulfiram/copper (DSF/Cu)-activated p62 phosphorylation, along with mitochondrial translocator protein (TSPO)-mediated p62 accumulation by inhibiting its autophagy, enhance the competitive inhibition of p62 on KEAP1, resulting in increased NRF2 accumulation and ferroptosis resistance.215,216,217 In addition to KEAP1-mediated degradation, other oncogenes such as KRASG12D, BRAFV619E and MycERT2 can transcriptionally induce NRF2 expression, ensuring the maintenance of stable cellular antioxidant programs to reduce intracellular ROS accumulation and potentially provide defense against ferroptosis.208 Nevertheless, a recent study has revealed that the regulatory function of NRF2 in ferroptosis is influenced by cellular ferrous ions in cancer cells.213 Overexpression of NRF2 can promote RSL3-induced cell death in TNBC cells, which harbor high levels of ferrous ions.213 Further research is needed to investigate the role of NRF2 in mediating ferroptosis under different conditions.
mTOR signaling
The mammalian target of rapamycin (mTOR), a serine/threonine protein kinase,218 is a key target in cancer research due to its involvement in the PI3K/AKT/mTOR signaling pathway, which is frequently activated in human cancers and is often associated with therapeutic resistance.219 Both mTORC1 and mTORC2 have been implicated in ferroptosis in human cancers. mTORC2 inhibits the cystine-glutamate reverse transport activity and promotes ferroptosis by phosphorylating serine at position 26 of SLC7A11.20 Nevertheless, mTORC1 primarily inhibits ferroptosis sensitivity through three mechanisms (Fig. 3c): inhibition of autophagy, promotion of GPX4 protein synthesis, and upregulation of the sterol regulatory element-binding protein 1 (SREBP1)/SCD axis.220 mTORC1 acts as a potent autophagy inhibitor via the phosphorylation-dependent inhibition of autophagy-related gene (ATG) complexes. Large tumor suppressor 1/2 (LATS1/2) kinases, core components of the Hippo pathway, are activated under high cell density conditions, leading to mTORC1 phosphorylation and subsequent inhibition of autophagy-induced degradation of SLC7A11, ultimately suppressing ferroptosis.221 Cysteine, mediated by SLC7A11, participates not only in GSH biosynthesis but also activates Rag/mTORC1/eukaryotic initiation factor 4E (eIF4E)-binding proteins (4EBPs) signaling pathway to promote GPX4 protein synthesis, revealing a novel mechanism of ferroptosis resistance through GPX4 metabolism.222 Oncogenic mutations in the PI3K/AKT pathway activate mTORC1, but not mTORC2, to promote SREBP1 expression, which in turn induces SCD1-mediated MUFAs synthesis and inhibit ferroptosis.217 We also reported that lorlatinib sensitizes ferroptosis by inhibiting PI3K/AKT/mTOR-mediated SREBP1/SCD1 signaling axis via targeting insulin-like growth factor 1 receptor (IGF1R) and synergizes with RSL3 to inhibit melanoma.223 Argininosuccinate synthase 1 (ASS1), a key enzyme in the urea cycle, can activate the mTORC1/SREBP1/SCD5 signaling pathway to promote the synthesis of MUFAs, thereby suppressing ferroptosis.224 Notably, mTORC1 can modulate the KEAP1/NRF2 signaling pathway by promoting the binding of p62 and KEAP1, indicating a crosstalk between the PI3K/AKT/mTOR and the KEAP1/NRF2 signaling pathways.217
Hypoxia signaling
Hypoxia, a common characteristic of cancer, is present in approximately 90% of solid tumors, and it promotes tumor progression and therapy resistance.225,226,227 The hypoxic response is mainly mediated by hypoxia-inducible factors (HIFs) that are widely upregulated in human cancers and play a critical role in enabling cancer cells to adapt to hypoxic environments.228,229,230 HIFs seem to play a dual role in modulating ferroptosis and subsequently affecting therapeutic efficacy in cancers (Fig. 3d). In human fibrosarcoma and lung cancer cells, hypoxia pretreatment has been demonstrated to limit RSL3/FIN56-induced ferroptosis by inducing HIF1α expression.231 Mechanistically, hypoxia-induced HIF1α expression transcriptionally upregulates fatty acid-binding proteins 3 and 7 (FABP3/7), promoting lipid droplet formation via enhancing fatty acid uptake and lipid storage to evade ferroptosis.231 Hypoxia can enhance intracellular lactate accumulation and increase cystine uptake by promoting HIF1α-mediated transcription of lactate dehydrogenases (LDH) and SLC7A11, ultimately promoting resistance to ferroptosis in solid tumors in a lactate/GPX4-dependent manner.232 Additionally, under hypoxic conditions, WTAP-mediated m6A modification modulates the PPARGC1A/BAMBI/ACSL5 axis, suppressing ROS production and subsequent lipid peroxidation to inhibit ferroptosis.233 FASN, which is significantly upregulated in cancers with treatment-resistant features, can bind to HIF1α and inhibit its ubiquitination and degradation, facilitating the nuclear translocation of HIF1α and subsequently promoting the transcription of SLC7A11, leading to resistance to ferroptosis and sorafenib treatment in HCC.234 Therefore, inhibiting hypoxia-activated HIF1α signaling may be an effective strategy to reverse drug resistance by enhancing ferroptosis. Notably, hypoxia also can confer ferroptosis susceptibility to colorectal cancer cells by increasing the expression of lipid and iron-regulatory genes in a HIF2α-dependent manner.235 Similarly, the activation of HIF2α increases hypoxia-inducible lipid droplet-associated protein (HILPDA) expression, driving the accumulation of PUFAs and subsequent lipid peroxidation, which contributes to the vulnerability of clear-cell carcinomas to ferroptosis.236 This evidence reveals the complex mechanisms through which hypoxia regulates ferroptosis in cancer and emphasizes the importance of targeting hypoxia signaling as a crucial approach in anticancer therapies.
Epithelial-mesenchymal transition
Epithelial-mesenchymal transition (EMT) is a major driver of cancer progression, as it involves the reorganization of cellular cytoskeleton, acquisition of mesenchymal features, and distant metastasis.237,238,239,240 Notably, EMT not only promotes the colonization of tumor cells in distant sites through a metastatic cascade but also renders these mesenchymal-like cells resistant to multiple treatment strategies.241,242,243 Interestingly, tumor cells with mesenchymal characteristics are more sensitive to ferroptosis compared to epithelial cells, partly due to the upregulation of zinc finger E-box binding homeobox 1 (ZEB1).20,240,244,245 ZEB1 is an EMT-related transcription factor that promotes the maintenance of mesenchymal phenotype which can be induced by TGFβ.246 ZEB1 enhance PUFA-PLs accumulation partially via direct transcriptional activation of the lipid biology regulator peroxisome proliferator-activated receptor gamma (PPARG), endowing susceptibility to ferroptosis.247 (Fig. 3e). Moreover, iron metabolism reprogramming may also contribute to the ferroptosis vulnerability of mesenchymal cells. CD44-mediated hyaluronate-dependent iron endocytosis pathway is enhanced during EMT. Endocytosed iron acts as a catalyst to relieve epigenetic suppression of mesenchymal-related proteins, thereby sustaining cellular mesenchymal characteristics and supporting ferroptosis vulnerability.248 This finding reveals the connection between epigenetic regulation of EMT and ferroptosis vulnerability. Notably, EMT could be induced by histone deacetylase inhibitor with increased intracellular iron accumulation and reduced expression of the iron export protein ferroportin, thereby enhancing vulnerability to ferroptosis.249 Erlotinib-tolerant persistent cancer cells also maintain mesenchymal characteristics with increased glutaminolysis induced by histone lysine demethylase 5 A (KDM5A) mediated mitochondrial pyruvate carrier 1 (MPC1) inhibition.250 Consequently, these cells become susceptible to ferroptosis. These findings shed light on the potential to selectively eliminate multidrug-resistant cancer cells with mesenchymal-like phenotypes using ferroptosis-inducing drugs, which may lay the foundation for significant advances in the field of cancer therapy resistance.
TP53 signaling
As noted above, P53 mediates tumor suppression partly through SLC7A11 inhibition-induced ferroptosis13,150,152,153,154 (Fig. 3f). Consistently, cell cycle promoter GINS4 suppresses ferroptosis in LUAD via inhibiting p53 acetylation and promoting SLC7A11 expression.251 MDM2/MDMX, MEX3A, and ZNF498 inhibit p53’s transcriptional activity through post-translational modifications in different subtype cancer cells, thereby suppressing p53-mediated ferroptosis.252,253,254 However, p53 can also inhibit ferroptosis in a context-dependent manner. Upon cystine deprivation, p53 induces cyclin-dependent kinase inhibitor 1 A (CDKN1A)/p21 expression and reduces the ferroptosis sensitivity of tumor cells in a GSH-dependent manner by affecting cysteine metabolism.255,256 Additionally, p53 directly binds to dipeptidyl-peptidase-4 (DPP4), blocking its activity, thus inhibiting DPP4/ NADPH oxidase 1 (NOX1) complex-mediated lipid peroxidation and erastin-induced ferroptosis.257 Notably, TP53 null cancer cells can still undergo ferroptosis via p53-independent pathways,173,258 which may indicate the potential limitation of p53 as a regulator of ferroptosis.
YAP/TAZ signaling
Cancer cells show density-dependent vulnerability to ferroptosis, with increased resistance observed in spheroids,47 suggesting the impact of cell density and cell-cell connections on ferroptosis sensitivity independent of genetic factors. The Hippo pathway, the primary regulator of intercellular communication and mechanical forces, plays a critical role in modulating density-mediated ferroptosis susceptibility.259 (Fig. 3g). Specifically, high cell density induces E-cadherin-mediated recruitment of NF2 and activation of the MST1/2-LATS1/2 cascade, which phosphorylates and retains YAP/TAZ in the cytoplasm, inhibiting their transcriptional activation of ACSL4 and TFR1, ultimately contributing to ferroptosis resistance.47,260 Consistently, various post-transcriptional modifications regulate the expression and activity of YAP protein, impacting ferroptosis susceptibility. Cyclin-dependent kinase 7 (CDK7) independently promotes nuclear YAP phosphorylation at the S127 and S397 sites, inducing downstream LDHD protein expression and D-lactate-induced ferroptosis resistance in esophageal squamous cell carcinoma (ESCC).261 Glutamine-fructose-6-phosphate transaminase (GFPT1) maintains YAP stability through o-GlcNAcylation, countering Hippo pathway suppression. Inhibition of system xc- impairs this process, reducing ferritin levels, increasing intracellular iron, and enhancing ferroptosis sensitivity.262 Furthermore, TAZ can activate NOX2/4 to promote ferroptosis by upregulating angiopoietin-like 4 (ANGPTL4) in ovarian cancers or epithelial membrane protein 1 (EMP1) in renal cancers.259,263,264 However, the ferroptosis sensitivity of the Burkitt lymphoma cell lines, which do not express YAP or its homolog TAZ, can still be influenced by cell density, indicating an alternative mechanism and the limited role of YAP/TAZ in cell density-mediated ferroptosis vulnerability.47
Autophagy pathway
Ferroptosis exhibits a dependence on autophagy in various induction mechanisms,265 referring to ferritinophagy, lipophagy, mitochondrial autophagy, clockophagy, and chaperone-mediated autophagy (CMA) (Fig. 3h). Ferritinophagy involves autophagic degradation of ferritin, facilitated by NCOA4 binding and subsequent delivery to lysosomes.266 Ataxia telangiectasia mutated (ATM) phosphorylates NCOA4 to enhance ferritinophagy, promoting ferroptosis by increasing intracellular labile iron.267 Conversely, tripartite motif-containing protein 7 (TRIM7) ubiquitinates and degrades NCOA4, inhibiting ferritinophagy and tumor cell sensitivity to ferroptosis.268 Lipophagy targets lipid droplets for lysosomal degradation, providing substrates for lipid peroxidation.269 RAB7A, member of the RAS oncogene family, enhances lipophagy-mediated ferroptosis by promoting autophagosome formation.269,270 Progesterone receptor membrane component 1 (PGRMC1) enhances ferroptosis susceptibility through silent information regulator 1 (SIRT1) activation-mediated lipophagy.271 Mitophagy and clockophagy selectively degrade mitochondria and ARNTL, respectively, promoting ferroptosis by inducing mitochondrial depletion and inhibiting fatty acid uptake and lipid storage.231,272,273 CMA, a highly selective autophagy pathway independent of vesicles, relies on chaperone proteins and lysosome-associated membrane protein 2a (LAMP2A) to deliver ferroptosis-related proteins to lysosomes for degradation, regulating ferroptosis in tumor cells.274 GPX4, a common substrate protein, undergoes CMA degradation facilitated by heat shock cognate 71 kDa protein (HSC70) and heat shock protein 90 (HSP90), enhancing the sensitivity of tumor cells to ferroptosis.275,276 Creatine kinase B (CKB) inhibits CMA-mediated GPX4 degradation by phosphorylating GPX4 and preventing its interaction with HSP70, providing protection against ferroptosis.276 However, most studies supporting the autophagy dependence of ferroptosis focus on the late stages of the ferroptosis process. This adds uncertainty to the concept of autophagy dependence in ferroptosis, as, in the late stages of oxidative damage, the mixed forms of cell death involving autophagy may become more common.277,278,279,280 Therefore, further research is required to clarify the permissive or regulatory role of autophagy in the process of ferroptosis.
Metabolism pathway
Energy metabolism is responsible for sustaining fundamental biological activities. Mitochondria serves as the primary energy production and acts as the main regulator for ROS stress and antioxidant defense.281,282 Glutaminolysis, TCA cycle and electron transport chain (ETC) are crucial for cysteine starvation-induced ferroptosis160 (Fig. 3i). Glutaminolysis metabolism fuels the TCA cycle by converting intracellular glutamine to glutamate via glutaminase (GLS), which is further metabolized to alpha-ketoglutarate (αKG) in mitochondria via glutamate dehydrogenase 1 (GLUD1). Glutaminolysis inhibition disrupts cystine deprivation-induced ferroptosis, whereas TCA metabolites downstream of αKG, including succinate, fumarate and malate can restore the role of glutamine in ferroptosis.160,283,284 Inhibiting the mitochondrial ETC also attenuates ferroptosis induced by cystine deprivation, as does depletion of mitochondria,160 partly due to the less leakage of electrons that produce superoxide and H2O2, which can then react with ferrous iron to drive Fenton chemistry and lipid peroxidation.12 The pentose phosphate pathway (PPP) contributes to ferroptosis by generating NADPH, which is involved in various defense mechanisms against ferroptosis, including GSH reduction,285 and the synthesis of thioredoxin and CoQ10.116,286 Inhibition of PPP-related enzymes impedes erastin-induced ferroptosis.4 Furthermore, glucose deprivation-induced energy stress activates AMP-activated protein kinase (AMPK), inhibiting PUFA biosynthesis and conferring ferroptosis resistance via ACC phosphorylation.287 The activation of AMPK in response to energy stress could also be regulated by liver kinase B1 (LKB1), which negatively regulates ferroptosis through AMPK/ACC-mediated PUFA inhibition.287,288
Lipid, amino acid, and vitamin metabolism also regulate ferroptosis sensitivity. As noted above, lipid droplet degradation, PUFA/MUFA phospholipid activation/synthesis, and PUFA-PL oxidation are essential in ferroptosis.4,44,141,269,270,271 Moreover, high-fat diet downregulates ACSL4 and promotes tumor cell invasiveness and resistance to ferroptosis.289 Chronic exposure to 27-hydroxycholesterol enhances GPX4 expression in ER-breast cancer cells, counteracting metabolic stress and leading to ferroptosis resistance.290 Adipokine inhibits ferroptosis by suppressing fatty acid oxidation and maintaining lipid levels via HIF2α activation.291 Additionally, cysteine starvation and glutamine supplementation induce or promote ferroptosis.292,293 Interestingly, prolonged methionine deprivation prevents GSH depletion from ferroptosis, whereas short-term methionine starvation promotes ferroptosis by stimulating CHAC1 transcription.294 Tryptophan facilitates cancer cells to escape from ferroptosis through its metabolites serotonin and 3-hydroxyanthranilic acid as radical-trapping antioxidants.295 Kynurenine, a product of tryptophan oxidation, also inhibits ferroptosis by scavenging ROS and activating NRF2 activity.296 Vitamin E is a well-known inhibitor of ferroptosis, both in vivo and in vitro, due to its powerful antioxidant properties.191,297,298 Vitamin K also inhibits ferroptosis by reducing to hydroquinone via FSP1 and vitamin K epoxide reductase complex subunit 1 like 1 (VKORC1L1).56,299
Ferroptosis-mediated crosstalk within the tumor microenvironment (TME)
The TME is a dynamic and complex ecosystem comprising cancer cells, stromal cells, diverse subpopulations of immune cells, the blood and lymphatic vasculature, and various acellular components.300 In the TME, bidirectional communication between cancer cells and their microenvironment is critical for tumor growth.301 In particular, dying cancer cells communicate with immune cells through the exposure or release of multiple signals during ferroptosis, thus modulating the anti-tumor immune responses. Simultaneously, mediators released by immune cells also have a crucial impact on regulating the susceptibility of cancer cells to ferroptosis. Pharmacologic screening identifies that CD8+ T cells exhibited a higher sensitivity to FINs than cancer cells,302 suggesting that pro-ferroptotic stimuli could elicit ferroptosis not only in cancer cells but also in tumor-infiltrating immune cells. Correspondingly, the occurrence of ferroptosis in immune cells will affect their survival and immunomodulatory function, ultimately reprograming tumor progression in the TME. Therefore, the versatile and complex roles of ferroptosis in the crosstalk between tumor cells and nonmalignant cells, particularly immune cells, within the TME are discussed below.
Immunomodulatory role of ferroptotic cancer cells
The emission of immunomodulatory signals by ferroptotic cancer cells, such as damage-associated molecular patterns (DAMPs), MHC class I molecules, cytokines, and lipid metabolites, exerts a significant and multifaceted impact on tumor growth by activating distinct immune responses (Fig. 4a).

Ferroptosis-mediated crosstalk in the tumor microenvironment (TME). a Ferroptotic cancer cells in the TME exhibit dual immunoregulatory effects, encompassing both immunostimulatory and immunosuppressive roles. The emission of various immunomodulatory signals by ferroptotic cancer cells activates different immune responses regulating tumor development. b The pro-ferroptotic and anti-ferroptotic impact on cancer cells mediated by immune cells and adipocytes in the TME. c The mechanisms and tumor-modulating effects of ferroptotic immune cells in the TME, including CD8+ T cells, dendritic cells (DCs), natural killer (NK) cells, tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs). AA arachidonic acid, AGER advanced glycosylation end product-specific receptor, CAF cancer-associated fibroblast, CRT calreticulin, FATP2 fatty acid transport protein 2, FIN ferroptosis inducer, HMGB1 high-mobility group box 1, IFNγ interferon gamma, 8-OHG 8-hydroxy-2-deoxyguanosine, oxLDL oxidized low-density lipoproteins, STING stimulator of interferon genes, TGF-β, transforming growth factor beta, TLR2 Toll-like receptors 2, ULBP UL16 binding protein. This figure was created with BioRender.com
Immunostimulatory activities of ferroptotic cancer cells
Cancer cells undergoing RCD, including ferroptosis, could elicit protective anticancer immunity by emitting a series of endogenous adjuvant signals that are generally referred to as DAMPs.303 Under pro-ferroptotic stress, the intracellular DAMPs, including double-stranded DNA and mitochondrial DNA, can activate the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) pathway, leading to the release of interferon β (IFNβ).304,305,306 Subsequently, IFNβ enhances dendritic cell (DC) maturation, macrophage phagocytosis, and the infiltration of cytotoxic CD8+ T cells, thereby resulting in tumor regression in preclinical cancer models.304,305,306 Additionally, oxidative stress triggers the upregulation and translocation of the calreticulin on the surface of ferroptotic cancer cells.306,307,308 Calreticulin serves as an ‘eat-me’ signal, promotes DC maturation, and increases the infiltration of cytotoxic CD8+ T cells into tumors, thus boosting anti-tumor immune responses.306,307,308 Likewise, oxidized PE, 1-steaoryl-2-15-HpETE-sn-glycero-3-phosphatidylethanolamine (SAPE-OOH), another ‘eat-me’ signal, accumulates on the membranes of ferroptotic cancer cells and could be directly recognized by its counterpart, the macrophage Toll-like receptors 2 (TLR2).309 This process promotes macrophage-mediated phagocytosis and elimination of ferroptotic cancer cells, thereby inhibiting tumor growth.309 Ferroptotic tumor cells also secrete high-mobility group box 1 (HMGB1) and ATP, the best-characterized DAMPs involved in immunogenic cell death.306,308,310,311 Notably, only early (1-3 hours), but not late (24 hours) ferroptotic cells release sufficient ATP and HMGB1 to stimulate DC maturation and elicit a vaccination-like anti-tumor immune response.310 Hence, further investigation is required to understand the immunostimulatory properties of these signals at different stages of ferroptotic cancer cells, which may contribute to the advancement of cancer vaccines based on ferroptosis.
In addition to DAMPs, pro-ferroptotic stimulation can upregulate other immunoregulatory molecules on the surface of ferroptotic tumor cells or regulate the secretion of cytokines into the TME, thereby boosting anti-tumor immune responses. For instance, inhibition of alpha 1,3-mannosyltransferase (ALG3) stimulates ferroptosis in cancer cells, which leads to the upregulation of MHC class I molecules on the cell surface,312 facilitating the infiltration of cytotoxic CD8+ T cells and subsequent tumor reduction.312 Furthermore, the interaction between the NK cells activating receptor NKG2D and its ligand UL16 binding protein (ULBP) is implicated in ferroptosis-mediated anti-tumor surveillance.313 Mechanistically, pro-ferroptosis nanoparticle promotes the upregulation of ULBP on the tumor cell surface, further activate NK cells with increased IFNγ secretion and lytic degranulation, and thus inhibit tumor growth in vivo.313 Cytokines secreted by cancer cells play a significant role in manipulating immune functions and guiding cancer progression.314 The cytokine transforming growth factor β (TGFβ) is responsible for cancer-associated fibroblast (CAF) formation and the establishment of an immunosuppressive TME after tumorigenesis.315,316 Recently, gastrointestinal cancer cells with anoctamin 1 (ANO1) high expression can release TGFβ through inhibiting ferroptosis to facilitate CAF recruitment and cripple CD8+ T cell-mediated anti-tumor immunity.317 Inhibition of ANO1 promotes an immune-activated TME with impaired TGFβ secretion, which can be restored by ferroptosis inhibitors in vivo, highlighting the significant role of ferroptosis in regulating cytokine release and orchestrating the TME.317 In summary, tumor cells could modulate the exposure or release of DAMPs, immunostimulatory molecules, and cytokines upon stimulation by ferroptosis, ultimately enhancing anti-tumor immunity and leading to tumor suppression.
Immunosuppressive activities of ferroptotic cancer cells
Intriguingly, the release of DAMPs triggered by cancer cells undergoing ferroptosis is a double-edged sword that not only boosts anti-tumor immune cell function but also enhances tumor-promoting responses of immunosuppressive cells in specific contexts.318 Ferroptotic damage induces the release of 8-OHG from pancreatic cells, which is a marker of oxidative DNA damage and serves as a DAMP.319,320 The released 8-OHG activates the STING-dependent DNA sensor pathway that enhances the infiltration and M2 polarization of macrophages, facilitating pancreatic carcinogenesis.319 In addition, the KRAS oncoprotein with G12D mutation is also released as a DAMP by ferroptotic pancreatic cancer cells and can be engulfed by macrophages via the advanced glycosylation end product-specific receptor (AGER) and promotes fatty acid oxidation driven by signal transducer and activator of transcription 3 (STAT3)-dependent in macrophages.321 Activation of the AGER-STAT3 pathway ultimately leads to pro-carcinogenic M2 macrophage polarization, and blocking this pathway or ferroptosis with ferrostatin-1 could inhibit TAM-mediated pancreatic tumor growth.321 In a hepatocellular tumorigenic model, GPX4 deletion-induced ferroptosis results in the release of high levels of HMGB1, thereby promoting the recruitment of immunosuppressive MDSCs.322 GPX4-deficient liver tumors also increase the expression of programmed cell death ligand 1 (PD-L1). Thus, MDSC infiltration and the concomitant PD-L1 upregulation counteract the cytotoxic CD8+ T cell response elicited by ferroptotic liver tumor cells, ultimately leading to no significant tumor suppression.322
Ferroptotic cancer cells could also release immunosuppressive lipid mediators that favor immunosuppressive responses. It is well-established that PTGS2, a gene that encodes cyclooxygenase-2 and determines the production of prostaglandin E2 (PGE2), is upregulated during ferroptosis in cancer cells.43 PGE2 is an immunosuppressive prostanoid lipid that could impair the anti-tumor activity of conventional type 1 DC (cDC1), NK cells, and effector T cells.323,324,325 Meanwhile, PGE2 can activate immunosuppressive cells such as MDSCs and regulatory T cells (Tregs), contributing to immune escape.325,326 Overall, ferroptotic tumor cells could emit multiple immunosuppressive signals, especially DAMPs and lipid metabolites, thereby facilitating tumorigenesis and tumor growth.
Effect of immune cells on cancer cells ferroptosis
Anti-tumor immune cells exert their functions partially by releasing mediators such as cytokines, which can enhance the susceptibility of tumor cells to ferroptosis (Fig. 4b). IFNγ released by cytotoxic CD8+ T cells binds to its receptor and activates the Janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1) pathway in cancer cells, leading to the suppression of transcription and expression of SLC3A2 and SLC7A11, the two subunits of the cystine antiporter system xc.-19,327,328 Hence, system xc- downregulation dampens the import of cystine and enhances lipid peroxidation, thereby rendering cancer cells vulnerable to ferroptosis triggered by pharmacological manipulations.19,329 Subsequent research has shown that CD8+ T cell-released IFNγ cooperates with AA to directly cause cancer cell ferroptosis in an ACSL4-dependent manner.330 Mechanistically, IFNγ activates the JAK/STAT1/interferon regulatory factor 1 (IRF1) signaling pathway and promotes IRF1 to bind to the IFN-stimulated response elements in the ACSL4 promoter region, ultimately leading to ACSL4 transcriptional upregulation in cancer cells.330,331 ACSL4 functions by facilitating the incorporation of PUFAs (including AA) into PLs on the plasm membrane.46 Therefore, it is not surprising that IFNγ released by activated cytotoxic CD8+ T cells reprograms lipid patterns in the presence of AA via ACSL4, thereby inducing and enhancing ferroptosis in cancer cells and resulting in tumor reduction.332,333 These studies suggest that IFNγ and AA induce ferroptosis in cancer cells.19 In addition to activated CD8+ T cells, NK cells are also significant producers of IFNγ.334 A recent study has indicated that chimeric antigen receptor (CAR)-modified NK cells, genetically engineered immune cells, could promote cancer cell ferroptosis by releasing IFNγ. Similar to CD8+ T cells, IFNγ produced by CAR NK cells enhances ferroptosis seemingly through downregulating the system xc- subunits (SLC3A2 and SLC7A11) in cancer cells.335 Nevertheless, the precise mechanism by which CAR NK cells inhibit their expression requires further investigation. In summary, tumor-infiltrating activated CD8+ T cells and CAR-modified NK cells can kill cancer cells by enhancing their vulnerability to ferroptosis through the production and release of IFNγ.
Immunosuppressive TAMs have been reported to hinder the ferroptosis of tumor cells by secreting cytokines and microRNAs into the TME, thereby supporting tumor growth. For instance, TAMs can release the cytokine TGFβ1, which binds to its receptor and promotes SMAD family member 3 (SMAD3) on the promoter region of hepatic leukemia factor (HLF) in TNBC cells.336 Increased HLF transcription, in turn, enhances the transcription of gamma-glutamyltransferase 1 (GGT1), an enzyme that increases intracellular cysteine availability for GSH synthesis.337 Accordingly, TAM-derived TGFβ1 suppresses ferroptosis by boosting the GGT1/GSH/GPX4 axis in TNBC cells. Intriguingly, in addition to GGT1, HLF also induces interleukin 6 (IL6) transcription, activating the JAK2/STAT3 axis to augment TGFβ1 secretion by TAMs, ultimately constituting a feedforward circuit to promote TNBC tumor growth.336 In addition, TAMs package the miRNA-660-5p into exosomes, which are secreted into the TME and internalized by cervical cancer cells to interfere with ALOX15 expression.338 Downregulation of ALOX15 is involved in cervical cancer cell resistance to ferroptosis.338 Furthermore, the role of cancer-associated adipocytes in fueling cancer has gained increasing attention, and this pro-tumor effect may be associated with ferroptosis resistance.196,339 Mammary adipocytes secrete oleic acid, a MUFA, which impairs lipid peroxidation and ferroptosis in TNBC cells through an ACSL3-dependent mechanism.196 Taken together, TAMs and cancer-associated adipocytes could secrete cytokines, microRNAs, and lipid metabolites to shield cancer cells from ferroptosis and promote tumor progression.
Effects of ferroptotic immune cells on cancer cells
Emerging evidence has revealed the occurrence of ferroptosis in immune cells within the TME, in addition to cancer cells. This ferroptotic process affects not only the survival of tumor-infiltrating immune cells but also their immunoregulatory properties, ultimately regulating cancer behavior. Herein, a comprehensive understanding of the role of ferroptotic immune cells, including CD8+ T cells, B cells, DCs, NK cells, TAMs, Tregs, and MDSCs, in cancer progression will help develop ferroptosis-targeted immunotherapeutic strategies (Fig. 4c).
CD8+ T cells are essential for effective anti-tumor immune responses, and ferroptosis-associated lipid metabolism reprogramming contributes to the impairment of CD8+ T cells in the TME.340 Fatty acids, particularly AA from the TME, facilitate lipid peroxidation and ferroptosis in tumor-infiltrating CD8+ T cells through the fatty acid transporter CD36.341 This CD36-mediated ferroptosis hampers cytotoxic cytokine production and anti-tumor function of CD8+ T cells, with decreased levels of IFNγ, TNFα, and perforin.341 Adoptive transfer of CD8+ T cells treated with the ferroptosis inhibitor ferrostatin-1 improves the survival of tumor-bearing mice and reduces tumor burden, suggesting that targeting ferroptosis in CD8+ T cells could enhance anti-tumor efficacy in vivo.341 In addition to AA, CD36 can enhance the absorption of oxidized low-density lipoproteins (OxLDL) into intratumoral CD8+ T cells, which induces lipid peroxidation and dysfunction in CD8+ T cells.342 Importantly, compared to Tc1 cells, IL-9-secreting CD8+ Tc9 cells could activate the IL-9/STAT3/fatty acid oxidation pathway, which protects against tumor- or ROS-induced lipid peroxidation and ferroptosis within the TME.343 Consistently, STAT3 inhibitor-treated Tc9 cells exhibit increased lipid peroxidation and compromised anti-tumor ability, whereas ferroptosis inhibitor-treated Tc9 cells display reduced iron levels and lipid peroxidation, as well as stronger anti-tumor ability in vivo.343 These findings suggest that inhibiting ferroptosis in CD8+ T cells can augment anti-tumor immunity and kill tumor cells better.
B cells exhibit remarkable heterogeneity and play complex roles within the TME.344 Up to now, the role of ferroptosis in regulating the homeostasis and immune responses of tumor-infiltrating B cells has not been extensively reported. Emerging evidence indicates that marginal zone B cells and B1 cells, but not follicular B2 cells, are susceptible to GPX4 inhibition-induced ferroptosis due to their high expression of CD36 and consequent fatty acid uptake.345 Moreover, ferroptosis has been observed in B cells from both systemic lupus erythematosus patients and mice, suggesting that it may regulate B cell differentiation and plasma cell formation to participate in the pathogenesis of lupus.346 These studies highlight the significance of ferroptosis in the survival and function of B cells, but the impact of ferroptosis on tumor-infiltrating B cells and B cell-mediated tumor immunity remains to be further investigated.
DCs are recognized as antigen-presenting cells and powerful initiators of T-cell responses that eliminate tumor cells within the TME.347 Recent evidence indicates that pro-ferroptotic regulators can impair the anti-tumor function of tumor-infiltrating DCs. Damaging molecules, including ROS and lipid peroxidation byproduct 4-HNE, the marker of ferroptosis, accumulate in ovarian cancer-associated DCs.348,349 This accumulation promotes endoplasmic reticulum stress response and X-box binding protein 1 (XBP1) activation, ultimately impairing the ability of tumor-associated DCs to present antigens and initiate anti-tumor T-cell responses.348,350 Intriguingly, GPX4 inhibitor, but not SLC7A11 inhibitor, could trigger ferroptosis in DCs in a PPARG-dependent manner.351 Genetic inhibition of PPARG significantly restores the impaired anti-tumor activities of ferroptotic DCs in vivo.351 Besides, in an inflammatory model, the enzymatic production of lipid peroxides mediated by ALOX12/15 disrupts the maturation and activation of DCs via NRF2, but the exact role of ALOX12/15-triggered dysfunction of DCs in anti-tumor immunity within the TME requires further clarification.352 Collectively, ferroptosis can occur in DCs and cripple their normal anti-tumor function.
Dysfunction of NK cells, a subset of natural cytotoxic lymphocytes, in the TME due to lipid peroxidation-associated oxidative stress favors tumor growth.353 L-kynurenine (L-KYN), a tryptophan metabolite in gastric cancer TME, has been reported to trigger lipid peroxidation and ferroptosis in NK cells, thereby facilitating tumor growth in vivo.354 Overexpression of GPX4 confers resistance of NK cells to ferroptosis induced by L-KYN within the TME and augments NK cell-mediated tumoricidal effects in vivo.354 These findings suggest that TME can render NK cells susceptible to ferroptosis and lead to their dysfunction, highlighting the therapeutic potential of inhibiting NK cell ferroptosis within the TME for cancer therapy.
TAMs exhibit strong plasticity and can differentiate into either immunostimulatory M1 phenotype or immunosuppressive M2 phenotype.355 Notably, M1 macrophages display increased resilience against ferroptosis compared to the M2 phenotype despite similar expression levels of GPX4, ACSL4, and LPCAT3 between the two subtypes.356 This resistance is attributed to the elevated levels of inducible nitric oxide synthase (iNOS) and NO• in M1 macrophages, which can substitute for GPX4 and inhibit ALOX15-mediated lipid peroxidation and ferroptosis.356 GPX4 inhibitor RSL3 effectively induces ferroptosis in M2 macrophages while sparing M1 macrophages.356 In addition, pro-ferroptotic stimuli can re-educate TAMs into an anti-tumorigenic M1 phenotype through multiple reprogramming pathways during ferroptosis, thereby inhibiting tumor progression. For instance, inhibition of apolipoprotein C1 (APOC1) or SLC7A11 promotes ferroptosis in TAMs, which is characterized by increased iron content, downregulated anti-ferroptosis mediators (GPX4, NRF2, SLC7A11, and GSH), and significant ferroptosis-associated mitochondrial changes.357,358 These pro-ferroptosis modifications by APOC1 and SLC7A11 further increase CD86 expression of the M1 phenotype and decrease the expression of CD206, CD163, and ARG1 of M2 phenotype in TAMs, thus inhibiting pro-tumoral M2 polarization and the development of HCC.357,358 Additionally, several pro-ferroptosis nanoparticles, such as iron-based metal-organic frameworks loaded with FINs (RSL3 or dihydroartemisinin), drive multiple signaling pathways to shift TAMs from the M2 to M1 phenotype.359,360,361 Ultimately, the shift from M2 to M1 phenotype provokes strong anti-tumor activities of TAMs with phagocytic killing and metastasis inhibition.359,360,362 These studies highlight that targeting ferroptosis in TAMs is promising to eliminate pro-tumorigenic M2 macrophages or reprogram TAMs towards a tumoricidal M1 type, thereby inhibiting tumor progression.
Activated Tregs represent a crucial barrier against autoimmunity as well as anti-tumor immunity.363 Gpx4-deficient activated Tregs are susceptible to ferroptosis and exhibit enhanced production of the proinflammatory cytokine IL-1β, leading to a promotion of T helper cell 17 (Th17) responses. This process compromises the immunosuppressive function of Tregs within the TME and limits tumor growth in vivo.364 Ferroptosis inhibitor administration indeed restores tumor burden in mice with Treg-specific deletion of GPX4. Collectively, targeting ferroptosis by inhibiting GPX4 in intratumoral Tregs seems to be a promising strategy for reprogramming the TME and treating cancer. However, it is worth mentioning that non-selective deletion of GPX4 in Tregs not only elicits anti-tumor immunity but also detrimental autoimmunity, such as significant inflammation in the colon.364 Therefore, future studies should further determine how to selectively target tumor-infiltrating Tregs without affecting Tregs in healthy tissues when inducing ferroptosis to avoid systemic loss of immune tolerance.
MDSCs are pathologically activated immature cells with potent immunosuppressive effects and great heterogeneity.365 They can be identified as two subgroups: PMN- and monocytic (M)-MDSCs.366 Tumor-infiltrating PMN-MDSCs, but not M-MDSCs, are vulnerable to ferroptosis or even experience spontaneous ferroptosis within the TME.23 Increased AA uptake through the fatty acid transport protein 2 (FATP2) and hypoxia-mediated downregulation of GPX4 both contribute to this susceptibility to ferroptosis.23 Although ferroptosis reduces the number of PMN-MDSCs, the increased release of immunosuppressive molecules, such as PGE2 and oxidized lipids, from ferroptotic PMN-MDSCs promotes tumor growth by restricting anti-tumor T cells and supporting the suppressive activity of TAMs.23,367 These findings suggest ferroptosis induction could decrease the viability of MDSCs, but the complex immunoregulatory nature of ferroptosis in MDSCs, as well as TME, must be considered in further research.
Therapeutic strategies of ferroptosis in cancer
As noted above, ferroptosis is tightly interwoven with cell metabolic and oxidative burdens, suggesting the possibility that cancer cells may have higher predispositions to FINs for their overall more active metabolism, higher ROS levels and iron requirements.11,368,369 Intriguingly, mesenchymal and dedifferentiated cancer cells, which are usually resistant to apoptosis and traditional therapies, are exquisitely vulnerable to ferroptosis.182,247,370 Therefore, FINs hold promise in cancer treatment.371 However, due to the immunosuppressive regulatory role and the tissue-damaging ability of ferroptosis, ferroptosis inhibition also proves to be an effective strategy to prevent tumor initiation, inhibit tumor progression, improve tissue damage-mediated cachexia in advanced tumors, and alleviate the side effects of traditional therapies. For these reasons, a comprehensive understanding of the current applications of both ferroptosis induction and inhibition in cancer will pave the way for their clinical implementation.
Ferroptosis induction
In addition to immunotherapy and radiotherapy,19,372 a diverse range of systemic drugs, including but not limited to targeted therapy,214 chemotherapy,138,373 lipid-lowering drugs,374,375 and anti-inflammatory drugs,376,377 have been identified as FINs and possess tumor-suppressive abilities.60 (Table 1). Here, we will describe in detail FINs that have previously entered clinical trials and briefly discuss the main tool compounds used to induce ferroptosis (Fig. 5) (Table 2).

Ferroptosis induction for cancer therapy. a Radiotherapy induces ferroptosis to suppress tumor through the following three mechanisms: ① Radiotherapy-induced DNA damage activates the ATM and cGAS/STING/ATF3 axis, leading to SLC7A11 inhibition and subsequent triggering of ferroptosis.② Radiotherapy upregulates the expression of ACSL4, facilitating the PUFA-PLs formation and inducing ferroptosis. ③ RT-MPs induce ferroptosis in neighboring unirradiated cells relying on the bystander effect. After immunotherapy treatment, activated CD8+ T cells release IFNγ, sensitizing tumor cells to ferroptosis by inhibiting SLC7A11, and promoting ACSL4-mediated PUFA-PLs formation, ultimately triggering ferroptosis. Immunotherapy and radiotherapy synergistically inhibit tumors by suppressing SLC7A11. b, c Major systemic drugs and experimental tool compounds for effective treatment of tumors through ferroptosis induction. ATF3 activation transcription factor 3, cGAS cyclic GMP-AMP synthase, cGAMP cyclic 2’,3’-GMP-AMP, DHA dihydroartemisinin, DHODH dihydroorotate dehydrogenase, FSP1 ferroptosis suppressor protein 1, FTH1 ferritin heavy chain 1, GCL glutamate-cysteine ligase, IFNγ interferon gamma, IKE imidazole ketone erastin, RT-MPs irradiated tumor cell-derived microparticles, STING stimulator of interferon genes. This figure was created with BioRender.com
Immunotherapy
Immunotherapy with immune checkpoint inhibitors (ICIs) is a promising approach that targets explicitly dysfunctional immune systems and mainly activates CD8+ T cells to eradicate tumor cells effectively.378 The advent of ICIs, specifically anti-CTLA4 and anti-PD-1/PD-L1 antibodies, have brought about a paradigm shift in cancer therapy and represent a significant breakthrough in oncology.379 As described above, on one hand, IFNγ derived from CD8+ T cells activates the JAK/STAT1 pathway to downregulate SLC7A11 and SLC3A2, thereby sensitizing tumor cells to ferroptosis19,380; on the other hand, IFNγ can transcriptionally stimulate ACSL4 expression to promote the integration of TME-associated AA into PLs by STAT1/IRF1 signaling, ultimately inducing ferroptosis in tumor cells.330 Therefore, ferroptosis induction contributes to the anti-tumor effects of CD8+ T cells, and immunotherapy can promote cancer cell ferroptosis in vivo. As expected, inhibiting ferroptosis by liproxstatin-1 diminishes the effectiveness of ICIs in controlling tumor growth.19 Additionally, the resistance of tumor cells to ferroptosis is associated with unresponsiveness to ICIs. Restoring their sensitivity to ferroptosis could enhance immunotherapy efficacy. TYRO3high tumors, which are resistant to ICIs, can be re-sensitized to anti-PD1 therapy by restoring ferroptosis via inhibiting TYRO3-mediated AKT/NRF2 pathway.381
Owing to the immunomodulatory effect of ferroptotic cells and the involvement of ferroptosis in ICIs anti-tumor effect, ferroptosis induction holds promise as an anti-tumor strategy to enhance the efficacy of ICIs. A growing body of evidence has demonstrated that combining ICIs and ferroptosis-inducing agents synergistically inhibits tumor growth in vitro and vivo.19,309,382 For example, the combined treatment of GPX4 inhibitors and anti-PD-1 blockade significantly suppressed tumor growth and induced a pronounced immune response with increased proportions of activated CD8+ T cells in TNBC tumor-bearing immunocompetent mice.382 IL-1β sustains Fe-S cluster maintenance to repress iron accumulation and ferroptosis. The combination of IL-1β blockade and anti-PD-1 antibody leads to enhanced tumor inhibition compared to monotherapy, but this effect could be reversed by liproxstatin-1, indicating the involvement of ferroptosis.383 Moreover, we also found that bromodomain containing 4 (BRD4) is upregulated in ICB-resistant melanoma patients, and inhibiting BRD4/AKR1C2 axis by bromodomain and extra-terminal motif (BET) inhibitors has been shown to enhance the susceptibility of melanoma to ferroptosis and immunotherapy.384 Nevertheless, the intricate impact of ferroptosis on the TME limits the applicability of this combination strategy due to immunosuppressive activities triggered by ferroptosis. For example, although GPX4 inhibition-induced ferroptosis in HCC cells increased CD8+ T cell infiltration, this effect was counteracted by PD-L1 upregulation on tumor cells.322 Synchronously, ferroptosis triggered immunosuppressive MDSC infiltration through increased release of HMGB1 from hepatocytes. The triple combination of pharmacological FINs, checkpoint blockade, and MDSC suppression effectively inhibits primary liver tumors and liver metastasis.322 Hence, the specific components of multidrug combination therapy based on ferroptosis-inducing agents and immunotherapy could be customized to counteract the immunosuppression triggered by ferroptosis, so as to evoke robust anti-tumor immune responses and enhance the efficacy of anti-tumor treatment.
Radiation therapy
Radiotherapy, a widely employed cancer treatment modality, involves precisely administering ionizing radiation (IR) to target and eliminate tumor cells selectively.385,386 Radiotherapy directly induces diverse forms of DNA damage and is capable of inducing ferroptosis to inhibit tumor growth.327,387 The mechanisms through which radiotherapy induces ferroptosis are multifaceted.18 Firstly, radiotherapy-induced DNA double-strand breaks (DSBs) downregulate SLC7A11expression in an ATM-dependent manner, resulting in reduced cystine uptake, and enhanced ferroptosis.327 Moreover, radiotherapy-induced DNA damage also can activate the cGAS/STING pathway to trigger tumor ferroptosis via activating transcription factor 3 (ATF3) /SLC7A11/GPX4 axis.304 Secondly, radiotherapy induces the expression of ACSL4 to promote the integration of PUFAs into PLs, resulting in the formation of PUFA-PLs, ultimately resulting in ferroptosis.372 Thirdly, irradiated tumor cell-derived microparticles (RT-MPs) induce a bystander effect via inducing ferroptosis, which causes the generation of oxidative stress and DNA damage in neighboring unirradiated cells.388 The mechanism by which RT-MPs induce ferroptosis is not yet fully understood, but the colocalization of RT-MPs membranes with lysosomes and mitochondria provides a direction for understanding its potential mechanisms.388
The occurrence of radioresistance, which leads to the failure of radiotherapy, is undeniably linked to metastasis, cancer recurrence, and unfavourable prognosis.389 Ferroptosis has also been implicated in radioresistance. IR can induce SLC7A11 and GPX4 upregulated as an adaptive response to safeguard cells against ferroptosis and contribute to radioresistance,372,390,391 suggesting that inhibiting SLC7A11 or GPX4 sensitizes radioresistant cancers to IR. As expected, the combination of class I FINs that inhibit SLC7A11 or class II or III FINs that inhibit or deplete GPX4 with IR demonstrated synergistic effects in inducing lipid peroxidation and ferroptosis.372 Moreover, suppressors of cytokine signaling 2 (SOCS2) were screened out as a potential biomarker predicting radiosensitivity of HCC. Mechanistically, SOCS2 transfers the attached ubiquitin to SLC7A11 and promotes K48-linked polyubiquitination degradation of SLC7A11.391 Conversely, stanniocalcin 2 (STC2) activate PRMT5, leading to upregulation of SLC7A11 and resistance to ferroptosis, thus playing a role in ESCC radioresistance.390 Other mechanisms that inhibit ferroptosis also contribute to radioresistance. For example, KEAP1 mutant lung cancers, which are refractory to most therapies, including radiotherapy, exhibit the upregulation of the NRF2/FSP1 anti-ferroptosis axis, resulting in resistance to ferroptosis and radiotherapy.165 Targeting FSP1 confers vulnerability to ferroptosis and enhances radiosensitivity in KEAP1 mutant lung cancers.165 IR-induced downregulation of copper metabolism MURR1 domain 10 (COMMD10) contributes to the radioresistance because COMMD10 inhibition represses ferroptosis through reducing iron concentration and facilitating HIF1α/SLC7A11 axis.392 These findings suggested that ferroptosis inhibition contributes to radioresistance, and combining FINs and IR synergistically induces ferroptosis.
Systemic drugs
Sorafenib, the first tyrosine kinase inhibitor authorized for the treatment of patients with unresectable HCC, advanced renal cell carcinoma, and differentiated thyroid cancer,210 has been shown to trigger ferroptosis by inhibiting system xc- and increasing intracellular iron levels.393,394,395 Therefore, increased SLC7A11 expression and the inhibition of ferritin autophagy contribute to resistance against sorafenib.234,396,397 For example, YAP/TAZ maintains the protein stability, nuclear localization, and transcriptional activity of ATF4, synergistically encouraging the expression of SLC7A11 and resulting in resistance against sorafenib-induced ferroptosis.397 Activation of ATF2 can inhibit protein degradation of SLC7A11 and mediate resistance to sorafenib-induced ferroptosis in gastric cancer.396 Moreover, CISD2 dissociates Beclin-1 from the PI3K-III complex and therefore leads to the inhibition of autophagy and resistance against sorafenib-induced ferroptosis.398 Depletion of PTBP1 leads to resistance against sorafenib-induced ferroptosis through disrupting NCOA4 translation and avoiding ferritin autophagy.399 However, sorafenib-induced ferroptosis is context-dependent, because sorafenib fails to trigger ferroptosis in various tumor cell lines,400 and whether these cells have acquired resistance against sorafenib-triggered ferroptosis needs to be clarified.
Lapatinib and neratinib are both tyrosine kinase inhibitors approved for the treatment of breast cancer. In lapatinib-resistant NSCLC cells, the activation of mTORC1 leads to the upregulation of GPX4 expression and inhibits lapatinib-induced ferroptosis. Therefore, inhibition of GPX4 or mTOR can overcome lapatinib resistance and facilitate lapatinib-induced ferroptosis.401 Moreover, the combination of lapatinib and siramesine, a lysosomal destabilizing lysosomotropic drug, synergistically induces ferroptosis through regulating iron homeostasis.402,403 Neratinib facilitates ferroptosis and suppresses brain metastasis in human epidermal growth factor receptor 2 (HER2)-positive breast cancer as a neoadjuvant therapy through the elevation of intracellular iron levels.404 Moreover, neratinib inhibits acute myeloid leukemia cell proliferation by activating autophagy-dependent ferroptosis.405 A recent study revealed that neratinib effectively tackled resistance to RSL3 in non-HER2 amplified luminal breast cancer, and combination treatment with RSL3 and neratinib enhances ferroptosis by increasing mitochondrial iron-dependent ROS production and lipid peroxidation.406
Cisplatin is a platinum-based chemotherapeutic agent approved by the Food and Drug Administration (FDA) for oncology use in 1978.407 Cisplatin-based regimens remain the mainstay of treatment for a wide range of solid tumors.408 The anti-tumor mechanism of cisplatin is mainly mediated by the production of nuclear DNA adducts, which ultimately leads to apoptosis.409 However, recent studies have shown that cisplatin can also induce ferroptosis by depleting GSH and inactivating GPX4, providing an alternative mechanism for inhibiting tumor growth.373 Consistently, NRF2/SLC7A11 signaling pathway is activated in cisplatin-resistant cancer cells, and inhibiting this pathway could trigger ferroptosis and overcome cisplatin resistance.410,411,412,413 Combining cisplatin with FINs also may be a better strategy to improve the therapeutic effect of cisplatin.414,415 Notably, cisplatin-mediated tumor cell ferroptosis can promote the anti-tumor efficacy of ICI therapy by reprogramming TME characterized by the N1 neutrophil polarization and increased T-cell infiltration and Th1 differentiation in NSCLC.416
Gemcitabine (GEM) undergoes a complex intracellular conversion into gemcitabine diphosphate and triphosphate nucleotides, which induce DNA chain termination and interfere with DNA synthesis, conferring potent anti-tumor activity across a wide spectrum of tumors.417 The anticancer activity of GEM is associated with the Hsp70 member 5 (HSPA5)/GPX4 pathway-mediated ferroptosis induction.138,254 Epigallocatechine gallate or sulfasalazine enhances the sensitivity of gemcitabine in PDAC by inhibiting the HSPA5/GPX4 pathway, thereby disinhibiting ferroptosis.138 Consistently, the combination of GEM and IKE shows a synergistic antiproliferative effect on LUAD.418 Moreover, PDAC-associated fibroblasts can secrete exosome-derived miR-3173-5p, which inhibits ferroptosis and promotes gemcitabine resistance by targeting ACSL4.419
Sulfasalazine (SAS), a clinical anti-inflammatory drug used in rheumatoid arthritis,420 induces ferroptosis by inhibiting cystine/glutamate antiporter SLC7A11.376,377 Similar to classical system xc- inhibitors such as erastin/IKE, SAS effectively induces ferroptotic cell death in chemotherapy-resistant cells and improves chemotherapy response.410 SAS also enhances the therapeutic effectiveness of front-line therapies, such as anthracycline daunorubicin, in acute myeloid leukemia,421 as well as paclitaxel in ovarian clear cell carcinoma.422 Moreover, SAS, as a radiosensitizer, enhances the therapeutic efficacy of radiotherapy by promoting ferroptosis.327,372,423 Notably, a specially designed injectable hydrogel drug delivery system loaded with SAS demonstrates remarkable therapeutic efficacy in combating peritoneal dissemination and malignant ascites in advanced HCC, which is resistant to systemic therapies,424,425 particularly when combined with anti-PD-1 immunotherapy.426
Statins, a class of clinical drugs aimed at reducing blood cholesterol levels,427 including fluvastatin,428 atorvastatin,429 pravastatin,430 lovastatin,431 and simvastatin,375 are considered attractive FINs in daily practice due to their favorable safety profile.430 Statins induce ferroptosis by inhibiting the GSH/GPX4 and FSP1/CoQ10/NAD(P)H axes via the mevalonate pathway.247,375 Simvastatin inhibits the expression of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR) to downregulate the mevalonate pathway and GPX4, thereby inducing cancer cell ferroptosis.375 Lovastatin induces ferroptosis and converts the immuno-cold phenotype to an inflammatory phenotype in NSCLC by downregulating PD-L1 expression in lung cancer cells, making the tumors more responsive to immunotherapy.431
Artemisinin, derived from the Chinese herb Artemisia annua,432 has been found to have anti-tumor effects in various types of tumors through ferroptosis.433 Mechanistically, artemisinin promotes ferritinophagy and increases intracellular free iron levels, and finally leads to ferroptosis and tumor inhibition.434,435 Artesunate, as an artemisinin derivative, has demonstrated efficacy in suppressing sunitinib-resistant renal cell carcinoma cells through ferroptosis and cell cycle arrest.435 Expectedly, artesunate synergistizes with sorafenib to induce ferroptosis in HCC and non-Hodgkin lymphoma cells.436,437 Consistently, dihydroartemisinin, the metabolite of artemisinin,438 inhibits lung cancer cells by suppressing the PRIM2/SLC7A11 axis,439 and enhances the cytotoxicity of gefitinib in LUAD cells,440 sorafenib in HCC,441 and cisplatin in PDAC.442
Haloperidol, a specific antagonist of dopamine receptor D2 (DRD2) extensively used for the treatment of psychiatric disorders,443 exhibits inhibitory effects in cancers by inducing ferroptosis.444,445 However, the precise mechanism by which it induces ferroptosis remains unclear, and there may be a potential association with autophagy.446 Haloperidol demonstrates synergistic activity with temozolomide in the growth inhibition of glioblastoma multiforme (GBM) through enhancing temozolomide-induced autophagy-mediated ferroptosis by inhibiting DRD2.444 Haloperidol can also sensitize the erastin- and sorafenib-induced ferroptosis in HCC by inhibiting the sigma 1 receptor (S1R).447
Zalcitabine, also known as 2’, 3’-dideoxycytidine, is used for the treatment of patients infected with the human immunodeficiency virus (HIV) by targeting mitochondrial DNA polymerase gamma (POLG).448 Zalcitabine induces autophagy-mediated ferroptosis in pancreatic cancer cells by activating mitochondrial DNA stress and the STING1/TMEM173-dependent DNA sensing pathway.449
β-Elemene (β-ELE), derived from Curcuma wenyujin, is widely used to treat NSCLC in clinical settings in China.450 β-ELE binds to transcription factor (TFEB), which is the key regulator of lysosome biogenesis, and notably activates TFEB-mediated lysosome degradation of GPX4, thus inducing NSCLC ferroptosis and resulting tumor suppression.451
Withaferin A (WA) is a bioactive compound derived from the ashwagandha plant, Withania somnifera.452 WA eradicates high-risk neuroblastoma tumors and suppresses relapse rates by inducing ferroptosis via GPX4 targeting and inactivation.453 WA also attenuated sorafenib resistance and metastatic potential by KEAP1/NRF2-associated EMT and ferroptosis.454 Moreover, a triple combination of withaferin A, the CXCR2 inhibitor and anti-PD-1 immunotherapy greatly improves the survival of wild-type mice with liver tumors and reduces liver metastasis of colorectal cancer. This effect may be attributed to the triggering of GPX4-associated ferroptotic hepatocyte death, leading to an adaptive immune response characterized by the activation of CD8+ T cells, upregulation of PD-L1 on tumor cells, and infiltration of immunosuppressive MDSCs.322
Buthionine sulfoxide amine (BSO) activates ferroptosis by targeting GCL for de novo GSH elimination.455 BSO induces ferroptotic cell death in lung cancer and HCC.192,456 BSO enhances the therapeutic effect of traditional chemotherapy regimens455 and radiotherapy in TNBC by promoting ferroptosis.457
Brequinar, a DHODH inhibitor, serves as a potential agent to treat GPX4low cancers by triggering ferroptosis,134 whereas combined administration of brequinar and sulfasalazine synergistically suppresses GPX4high tumors growth.134 Moreover, AMPK activation enhances the assembly of pyrimidinosomes, rendering cancer cells more reliant on DHODH-mediated ferroptosis defense to counteract AMPK-associated stresses.458 The combination of brequinar and AMPK activators exhibit synergistic efficacy in tumor suppression through ferroptosis.458
Curcumenol, an effective compound found in Wenyujin, has been discovered to inhibit the growth of lung cancer tumors by inducing ferroptosis through lncRNA H19/miR-19b-3p/FTH1 axis.459
Others
In addition to FINs that have previously entered clinical trials, a wide range of experimental tool compounds have been employed in preclinical studies of ferroptosis (Table 2). These compounds could be classified into four classes.12,460,461,462 Classical I FINs are identified through the depletion of GSH to trigger ferroptosis, such as erastin, its derivatives imidazole ketone erastin (IKE),460,463,464 and cyst(e)inase, which depletes GSH by degrading cysteine and cystine.465 Classical II FINs directly inhibit GPX4 to induce ferroptosis, such as RSL3, ML162 and ML210.43,269,382 Classical III FINs deplete the GPX4 protein and CoQ10 to induce ferroptosis, such as FIN56.45,466,467,468,469 Class IV FINs induce ferroptosis by augmenting the LIP, such as FINO2.136 These tool compounds have made significant contributions to the understanding of the mechanism of ferroptosis due to their specificity towards ferroptosis.
Moreover, the drug delivery system has gained increasing interest in tumor treatment due to its effective delivery and precise control of drug release. Nowadays, three delivery systems, including nanoparticles, hydrogels, and liposomes, have been utilized to improve the efficiency and selectivity of FINs in targeting tumors while minimizing toxicity to normal organs. For example, the acidity-activatable dynamic nanoparticles BNP@R were developed to specifically deliver RSL3 to tumors and enable acid-activatable photodynamic therapy, thereby promoting RSL3-induced ferroptosis and ultimately inhibiting tumor growth.470 The tumor-suppressing effect of RSL3 in vivo was also potentiated when delivered in an injectable alginate hydrogel RTFG@SA.471 Brequinar-loaded mitochondrial-targeted liposomes BQR@MLipo were employed to enhance brequinar-mediated mitochondrial ferroptosis, effectively inhibiting bladder cancer growth.306 The continuous advancements in drug delivery systems targeting ferroptosis contribute to the clinical applications of FINs in cancer treatment.
Ferroptosis inhibition
The heterogeneity of tumors and the immune microenvironment complicates the role of ferroptosis in tumor suppression. Under some conditions, ferroptosis is even conducive to tumor initiation and progression: 1) The inflammation resulting from ferroptosis-induced tissue damage contributes the onset of necroinflammation-driven tumors.21,148; 2) The vulnerability of immune cells to ferroptosis compromises their anti-tumor function or enhances their pro-tumor effect, leading to tumor growth.23,354,449; 3) The immunosuppressive activities of ferroptotic cancer cells promotes the tumor progression.321 Moreover, it is worth noting that ferroptosis inhibition could also be considered an effective means to suppress the side effects mediated by traditional therapy-induced ferroptosis, due to the tissue-damaging ability of ferroptosis. Thus, ferroptosis inhibition seems to be a potential context-dependent strategy for cancer treatment.
Inhibition of ferroptosis is advantageous in suppressing the initiation of necroinflammation-driven tumors. Liver-related diseases, including steatohepatitis, can trigger the initiation of hepatocyte stress, excessive cell death, subsequent necroinflammation, and compensatory proliferation, which are considered to be the aetiologies of hepatocellular carcinogenesis.472,473 A recent study has revealed that ferroptosis stands out as the most pertinent form of hepatocyte death, leading to HCC-promoting necroinflammation and compensatory proliferation.21 By contrast, the activation transcription factor 4 (ATF4) attenuates the progression from steatohepatitis to HCC by upregulating SLC7A11 to block stress-related ferroptosis, thereby blunting HCC onset. Moreover, high-iron diets or depletion of Gpx4-induced ferroptosis promotes pancreatitis and pancreatic tumorigenesis. Inhibiting ferroptosis through the administration of liproxstatin-1 reduces the formation of spontaneous pancreatic cancer and reverses the promotion of PDAC development by high-iron diets or GPX4 depletion.319 The initiation of ferroptosis-driven pancreatic cancer may be associated with macrophage infiltration and activation, which is mediated by the release of 8-OHG caused by ferroptotic damage and subsequent activation of the STING-dependent DNA sensor pathway in macrophages.319
Ferroptosis inhibition rescues immune cells from undergoing ferroptosis and affects their immune regulatory ability, thereby impeding tumor development. CD36-mediated ferroptosis hampers the effector function of intratumoral CD8+ T cells and diminishes their ability to combat tumors. Consequently, genetic deletion of CD36 or inhibition of ferroptosis with ferrostatin-1 in CD8+ T cells can effectively restore their anti-tumor effects.449 NK cells also undergo ferroptosis induced by various triggers in the TME, including L-KYN released by tumor cells and CAF-derived follistatin-like protein 1 (FSTL1) and iron.354,474 Overexpression of GPX4 in NK cells effectively inhibits ferroptosis and prevents their reduction within the TME, thereby suppressing tumor growth.354 Consistently, the combination of FSTL1-neutralizing antibody and deferoxamine significantly inhibits NK cell ferroptosis, enhancing the cytotoxicity of NK cells against tumor cells.474 In addition, tumor-associated PMN-MDSCs undergo ferroptosis spontaneously.23 Intriguingly, although this ferroptotic process decreases PMN-MDSC numbers, their immunosuppressive activity is enhanced due to the increased release of immunosuppressive molecules.23 Inhibition of ferroptosis by liproxstatin-1 could alleviate the immune suppression mediated by PMN-MDSCs and reduce tumor growth, especially when combined with immunotherapy.23 These studies indicate that ferroptosis in several immune cell types exerts tumor-supportive effects, raising the possibility for future investigation into selectively inhibiting ferroptosis in immune cells through cell-specific delivery to inhibit tumor growth effectively.
Ferroptosis inhibition counteracts the immunosuppressive activities of ferroptotic cancer cells to restrict tumor progression. For example, mutated KRAS protein derived from ferroptotic PDAC cells is packaged into exosomes, which are then engulfed by adjacent macrophages, promoting their subsequent polarization into an M2 tumor-promoting state.321 Administration of ferrostatin-1 can inhibit the tumor-promoting growth arising from peripheral blood mononuclear cell-derived macrophages (PBMCMs) in immunodeficient mice.321 However, the impact of ferroptosis inhibition on PDAC tumor progression has not been investigated in immunocompetent mice. It is crucial to consider that inhibiting ferroptosis may also suppress the tumoricidal effect of cancer cell ferroptotic death and affect the immunoregulatory role of immune cells within the TME. Therefore, the application of ferroptosis-based approaches in cancer treatment should be applied with caution. More studies are needed to fully understand the complex interplay between ferroptosis, tumor progression, and immune response, to make informed decisions regarding the application of ferroptosis modulation in cancer therapy.
Because of the link between the anti-tumor effects of traditional therapy and ferroptosis induction, as well as the potential tissue-damaging properties of ferroptosis, ferroptosis inhibition is considered an effective approach to suppress the side effects mediated by traditional therapy-induced ferroptosis. For instance, cisplatin-induced ferroptosis was implicated in chemotherapy-induced ovarian damage, and the administration of antioxidant NAC can alleviate cisplatin-induced toxicity in normal ovarian cells by inhibiting ferroptosis and oxidative stress.475 Cisplatin-induced acute kidney injury and doxorubicin-induced cardiomyopathy also could be inhibited by ferroptosis inhibitor ferrostatin-1.476,477 The FDA-approved iron chelator dexrazoxane protects against doxorubicin-induced cardiotoxicity by chelating mitochondrial iron.114 Moreover, ferroptosis is also implicated in radiation-induced intestinal injury, which can be alleviated by administering ferrostatin-1.478 Ferroptosis inhibition appears to be a potential target for mitigating treatment side effects, potentially enhancing treatment tolerance, and prolonging the life quality of patients. However, it also raises concerns regarding whether the administration of ferroptosis inhibitors to mitigate the ferroptosis-related side effects induced by traditional therapy might also inhibit its therapeutic effect. In addition, the occurrence of ferroptosis was observed in wasting tissues during advanced tumors cachexia. It is reported that tissue-infiltrating neutrophils-secreted LCN2 induces ferroptosis and wasting tissues in lung cancer cachexia. Liproxstatin-1 has been shown to mitigate tissue wasting in lung cancer cachexia, improve symptoms, and extend the survival of cachectic mice by inhibiting ferroptosis.479
Collectively, ferroptosis inhibition holds promise as a strategy for suppressing tumor growth, attenuating adverse effects of traditional therapies and improving cachexia. Ferroptosis inhibitors generally encompass four classes120: 1) Radical-trapping antioxidants including ferrostatin-1, liproxstatin-1 and vitamin E.4,8,298 2) Iron chelators such as deferoxamine, cyclipirox, and deferiprone.4,120 3) Inhibitors of ferroptosis-promoting enzymes, such as ACSL4 inhibitor (thiazolidinediones and triacsin C.46,68 4) Inhibitors of protein degradation in the ferroptosis defense system, such as 5-(tetradecyloxy)-2-furoic acid (TOFA) and dopamine for preventing GPX4 protein degradation.45,480 Additionally, compounds, like CoQ10, N-acetylcysteine (NAC) and β-mercaptoethanol (2ME) that disrupt the pathways involved in ferroptosis excitation can also be potential inhibitors.120 Several clinical trials are pending to assess the effectiveness of ferroptosis inhibitors, such as deferoxamine (DFO) and deferasirox (DFX) in anti-tumor treatment (Table 3). However, further investigations are required to ascertain whether their anti-tumor potential is dependent on their ability to inhibit ferroptosis.
Markers of ferroptosis
A standardized set of identification hallmarks for ferroptosis enables us to determine its occurrence in physiological and pathological conditions, facilitating further investigation into its role in cancer. Currently, four classes of markers, including lipid peroxidation, mitochondria morphological alteration, gene expression change, and TFR1 re-localization, have been considered suitable for detecting and accurately distinguishing ferroptosis from other forms of RCD.12 First, the core of ferroptosis occurrence is the peroxidation of membrane-localized lipids and their lethal accumulation. There are five methods available to detect lipid peroxidation, including BODIPY 581/591 C11 fluorescent probes, lipidomics, thiobarbituric acid reactive substances (TBARS), malondialdehyde (MDA) and 4-HNE staining. Among those, flow cytometry following BODIPY 581/591 C11 staining is a sensitive and convenient method for detecting lipid peroxidation. Second, multiple organelles are involved in ferroptosis. Endoplasmic reticulum-related oxidative stress,481,482,483,484,485,486,487 mitochondria-induced cysteine starvation,134,160 lysosome dysfunction,138,275,488,489,490,491 peroxisomes-mediated ether lipids peroxidation,52 and Golgi stress-related lipid peroxidation all contribute to ferroptosis induction.492,493 Among these organelles, the morphological alterations in mitochondria, characterized by shrinkage, increased density, and decreased cristae, are considered the morphological features of ferroptosis and can be observed using transmission electron microscopy. Nevertheless, these mitochondria alterations are not specific, as they can be observed in oxidative stress and mitochondrial stress, and mitochondria are not indispensable for ferroptosis induction in some conditions.12,481 Third, specific gene expression changes, such as increased CHAC1, PTGS2, SLC7A11 and ACSL4 can be detected in cells undergoing ferroptosis. However, these changes may not be universally observed in all contexts of ferroptosis.12,494 Moreover, a recent study has identified hyperoxidized peroxiredoxin 3 (PRDX3) protein as a novel marker of ferroptosis in vitro and in vivo, specifically detectable in ferroptosis, rather than mitochondrial oxidative stress or other forms of RCD, such as apoptosis, necroptosis and cuproptosis.495 Fourth, the re-localization of TFR1, which imports extracellular ferric into cells by endocytosis, contributing to the liable iron pool required for ferroptosis, has been demonstrated as a marker of ferroptosis.86 The relocation of TFR1 from the region surrounding Golgi to the plasma membrane can be observed through staining with the 373-FMA antibody. This method provides a potential avenue for selectively staining ferroptotic cells in tissue sections.
To accurately determine the occurrence of ferroptosis, multiple markers are needed, which must be detected before cell demise. The selection of an appropriate time point is important in detecting ferroptosis markers. Among these markers, lipid peroxidation is essential, while other indicators may not be fully detected. Additionally, pharmacological rescue experiments using ferroptosis inhibitors are also crucial. In evaluating the efficacy of ferroptosis-based therapies, appropriate markers for detecting ferroptosis within tumor tissue are necessary. Although staining for 4-HNE, hyperoxidized PRDX3, MDA and TFR1 shows relatively promising prospects in their applicability for ferroptosis detection in tumor tissue sections, these markers could not be used in a living organism. Blood, urine, and feces are regularly checked in clinic practice, and whether or not using these samples to detect ferroptosis levels in patients is worth exploring, because ferroptotic cells could release some specific substances into their microenvironment. Deciphering the changes in iron, lipids, metabolites, and immune mediators may provide guidance for the development of these techniques.
Conclusions and perspectives
Ferroptosis is a distinct form of cell death characterized by iron-dependent phospholipid peroxidation, which is strictly controlled at multiple levels. Pharmacologically targeting ferroptosis holds great promise as an anticancer strategy. However, to realize the prospects of ferroptosis drugs in clinical practice, several additional challenges remain to be overcome in future research.
Firstly, there is a lack of well-established animal models to evaluate cancer ferroptosis in vivo. Current models mainly rely on the use of FINs, such as IKE and cyst(e)inase,43,53,192,465,496 to treat xenograft tumors. However, the timing and frequency of administration vary in practice, and the side effects of long-term administration and potential drug resistance are not well understood. Additionally, generating CRISPR/Cas9-mediated GPX4 knockout cancer cells for xenograft models fails to evaluate the efficacy and safety effects of ferroptosis drugs. Therefore, standardized animal models are needed to improve our understanding of ferroptosis biology in cancer and ease the comparison of studies between research laboratories and clinicians.
Secondly, the complex biological effects of ferroptosis in cancer present another challenge. In some cases, ferroptosis induction initially promotes tumor formation but later leads to tumor cell demise and suppression.21,319 Tumor formation is a complex process involving metabolic disorders. For example, our team first put forward that melanoma is a metabolically driven and metabolically remodeled cancer.497,498 Given the coexistence of established tumor cells and cells transitioning into tumor cells,23 solely using ferroptosis inhibitors or inducers may not effectively control early-stage tumors. Therefore, it is crucial to determine the appropriate therapeutic time window for FINs.
Thirdly, the lack of effective and specific drugs that can safely induce ferroptosis in cancer cells poses an additional challenge. Although several compounds have been discovered to induce ferroptosis, their in vivo potential is limited due to poor bioavailability and insufficient targeting. Developing small molecules compatible with in vivo conditions and exploring targeted protein degradation technologies, such as proteolysis-targeting chimaeras (PROTACs)499,500 and lysosome-targeting chimaera,499,501 offer promising strategies. Moreover, reducing the toxicity of FINs remains a challenge in clinical oncology. Shifting the focus of developing ferroptosis-targeting drugs from completely abrogating master regulators, such as GPX4, to other controlling complexes with lower toxicity, and developing combination treatment strategies based on ferroptosis are all viable approaches to mitigate its toxicity.
Fourthly, ferroptosis induction may have negative impacts on anti-tumor immunity, posing a challenge in achieving complete tumor elimination. It is essential to promptly neutralize the factors that contribute to the immunosuppression induced by ferroptotic cancer cells. Moreover, FINs could potentially kill anti-tumor immune cells. Therefore, the development of cell-specific precision targeting strategies is crucial for maximizing the efficacy of ferroptosis-induced therapy.
Lastly, identifying the patient population that would benefit most from ferroptosis therapy is crucial for successful clinical trials. The sensitivity of different cancer types to ferroptosis varies based on tumor origin and genotype. Integrating genetic information from the cancer genome can aid in predicting tumor response to specific ferroptosis drugs.
In conclusion, we are on the verge of an exciting era in the realm of ferroptosis research. Overcoming the challenges above will pave the way for successful translation into clinical cancer treatment, enabling the development of personalized ferroptosis-related anticancer strategies. We anticipate that novel ferroptosis-based therapies, guided by standardized animal models and precise evaluation of therapeutic time windows, will be developed and implemented in the near future.
References
Hotchkiss, R. S., Strasser, A., McDunn, J. E. & Swanson, P. E. Cell death. N. Engl. J. Med 361, 1570–1583 (2009).
Galluzzi, L. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 22, 58–73 (2015).
Green, D. R. The coming decade of cell death research: five riddles. Cell 177, 1094–1107 (2019).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012).
Kung, G., Konstantinidis, K. & Kitsis, R. N. Programmed necrosis, not apoptosis, in the heart. Circ. Res. 108, 1017–1036 (2011).
Feldmann, G. Liver apoptosis. J. Hepatol. 26, 1–11 (1997).
Fang, Y. et al. Pyroptosis: A new frontier in cancer. Biomed. Pharmacother. 121, 109595 (2020).
Friedmann Angeli, J. P. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014).
Koskenkorva-Frank, T. S., Weiss, G., Koppenol, W. H. & Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med 65, 1174–1194 (2013).
Cao, J. Y. & Dixon, S. J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 73, 2195–2209 (2016).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Stockwell, B. R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421 (2022).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Zhang, C. et al. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol. Cancer 21, 47 (2022).
Friedmann Angeli, J. P., Krysko, D. V. & Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 19, 405–414 (2019).
Chen, J. J. & Galluzzi, L. Fighting resilient cancers with iron. Trends Cell Biol. 28, 77–78 (2018).
Beretta, G. L. & Zaffaroni, N. Radiotherapy-induced ferroptosis for cancer treatment. Front Mol. Biosci. 10, 1216733 (2023).
Lei, G. et al. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell 12, 836–857 (2021).
Wang, W. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Zhao, L. et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 42, 88–116 (2022).
He, F. et al. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J. Hepatol. 79, 362–377 (2023).
Tang, D., Kroemer, G. & Kang, R. Ferroptosis in hepatocellular carcinoma: from bench to bedside. Hepatology https://doi.org/10.1097/HEP.0000000000000390 (2023).
Kim, R. et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature 612, 338–346 (2022).
Tang, D., Chen, X., Kang, R. & Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125 (2021).
Moore, B. & Hawkes, J. L. An investigation of the toxic actions of dilute solutions of the salts of certain heavy metals (viz.: Copper, Iron, Nickel, Cobalt, Manganese, Zinc, Silver, and Lead) upon the Bacillus Typhosus, with a view to practical application in the Purification of Shell-fish. Biochem J. 3, 313–345 (1908).
Eagle, H. The specific amino acid requirements of a human carcinoma cell (Stain HeLa) in tissue culture. J. Exp. Med. 102, 37–48 (1955).
Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 122, 501–514, (1955).
Bieri, J. G. An effect of selenium and cystine on lipide peroxidation in tissues deficient in vitamin E. Nature 184, 1148–1149 (1959).
Bannai, S., Tsukeda, H. & Okumura, H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochem Biophys. Res. Commun. 74, 1582–1588 (1977).
Bannai, S. & Kitamura, E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J. Biol. Chem. 255, 2372–2376 (1980).
Ursini, F. et al. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 710, 197–211 (1982).
Geiger, P. G., Thomas, J. P. & Girotti, A. W. Lethal damage to murine L1210 cells by exogenous lipid hydroperoxides: protective role of glutathione-dependent selenoperoxidases. Arch. Biochem. Biophys. 288, 671–680 (1991).
Murphy, T. H. et al. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558 (1989).
Schubert, D., Kimura, H. & Maher, P. Growth factors and vitamin E modify neuronal glutamate toxicity. Proc. Natl Acad. Sci. USA 89, 8264–8267 (1992).
Li, Y., Maher, P. & Schubert, D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 19, 453–463 (1997).
Tan, S., Schubert, D. & Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med Chem. 1, 497–506 (2001).
Kang, Y. et al. Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat. Commun. 5, 3672 (2014).
Albrecht, P. et al. Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc- as a neuroprotective drug target. CNS Neurol. Disord. Drug Targets 9, 373–382 (2010).
Dolma, S., Lessnick, S. L., Hahn, W. C. & Stockwell, B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296 (2003).
Yagoda, N. et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 (2007).
Yang, W. S. & Stockwell, B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245 (2008).
Seiler, A. et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248 (2008).
Yang, W. S. et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014).
Yang, W. S. et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl Acad. Sci. USA 113, E4966–E4975 (2016).
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Doll, S. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Kraft, V. A. N. et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 6, 41–53 (2020).
Soula, M. et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat. Chem. Biol. 16, 1351–1360 (2020).
Zou, Y. et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608 (2020).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89 (2020).
Ubellacker, J. M. et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118 (2020).
Mishima, E. et al. DHODH inhibitors sensitize to ferroptosis by FSP1 inhibition. Nature 619, E9–E18 (2023).
Mishima, E. et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783 (2022).
Nakamura, T. et al. Phase separation of FSP1 promotes ferroptosis. Nature 619, 371–377 (2023).
Zhao, J. et al. Human hematopoietic stem cell vulnerability to ferroptosis. Cell 186, 732–747 e716 (2023).
Liang, D. et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 186, 2748–2764 e2722 (2023).
Lei, G., Zhuang, L. & Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 22, 381–396 (2022).
Sun, S. et al. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target Ther. 8, 372 (2023).
Dixon, S. J. & Pratt, D. A. Ferroptosis: A flexible constellation of related biochemical mechanisms. Mol. Cell 83, 1030–1042 (2023).
Dos Santos, A. F., Fazeli, G., Xavier da Silva, T. N., & Friedmann Angeli, J. P. Ferroptosis: mechanisms and implications for cancer development and therapy response. Trends Cell Biol. 33, 1062–1076 (2023).
Pope, L. E. & Dixon, S. J. Regulation of ferroptosis by lipid metabolism. Trends Cell Biol. 33, 1077–1087 (2023).
Porter, N. A., Wolf, R. A., Yarbro, E. M. & Weenen, H. The autoxidation of arachidonic acid: formation of the proposed SRS-A intermediate. Biochem Biophys. Res Commun. 89, 1058–1064 (1979).
Gill, I. & Valivety, R. Polyunsaturated fatty acids, Part 2: Biotransformations and biotechnological applications. Trends Biotechnol. 15, 470–478 (1997).
Gill, I. & Valivety, R. Polyunsaturated fatty acids, Part 1: Occurrence, biological activities and applications. Trends Biotechnol. 15, 401–409 (1997).
Kagan, V. E. et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017).
Dixon, S. J. et al. Human Haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609 (2015).
Cui, W., Liu, D., Gu, W. & Chu, B. Peroxisome-driven ether-linked phospholipids biosynthesis is essential for ferroptosis. Cell Death Differ. 28, 2536–2551 (2021).
Chen, D. et al. iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 12, 3644 (2021).
Shah, R., Shchepinov, M. S. & Pratt, D. A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 4, 387–396 (2018).
Zou, Y. et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 16, 302–309 (2020).
Wenzel, S. E. et al. PEBP1 Wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641 e626 (2017).
Wang, J. et al. PM(2.5) caused ferroptosis in spermatocyte via overloading iron and disrupting redox homeostasis. Sci. Total Environ. 872, 162089 (2023).
Yang, X. et al. miR-18a promotes glioblastoma development by down-regulating ALOXE3-mediated ferroptotic and anti-migration activities. Oncogenesis 10, 15 (2021).
Wang, M. et al. ALOX5 promotes autophagy-dependent ferroptosis by activating the AMPK/mTOR pathway in melanoma. Biochem Pharm. 212, 115554 (2023).
Li, C. et al. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 17, 948–960 (2021).
Matsushita, M. et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 212, 555–568 (2015).
Ghosh, M. K., Mukhopadhyay, M. & Chatterjee, I. B. NADPH-initiated cytochrome P450-dependent free iron-independent microsomal lipid peroxidation: specific prevention by ascorbic acid. Mol. Cell Biochem. 166, 35–44 (1997).
Gaschler, M. M. & Stockwell, B. R. Lipid peroxidation in cell death. Biochem. Biophys. Res Commun. 482, 419–425 (2017).
Milne, G. L., Dai, Q. & Roberts, L. J. 2nd The isoprostanes-25 years later. Biochim Biophys. Acta 1851, 433–445 (2015).
Conrad, M. & Pratt, D. A. The chemical basis of ferroptosis. Nat. Chem. Biol. 15, 1137–1147 (2019).
Andrews, N. C. & Schmidt, P. J. Iron homeostasis. Annu Rev. Physiol. 69, 69–85 (2007).
Richardson, D. R. & Ponka, P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys. Acta 1331, 1–40 (1997).
Feng, H. et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 30, 3411–3423 e3417 (2020).
El Hout, M., Dos Santos, L., Hamaï, A. & Mehrpour, M. A promising new approach to cancer therapy: Targeting iron metabolism in cancer stem cells. Semin. Cancer Biol. 53, 125–138 (2018).
Gao, M. et al. Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016).
Yambire, K. F. et al. Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife 8, e51031 (2019).
Donovan, A. et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 1, 191–200 (2005).
Chen, X., Yu, C., Kang, R. & Tang, D. Iron metabolism in ferroptosis. Front. Cell Dev. Biol. 8, 590226 (2020).
Battaglia, A. M. et al. Ferroptosis and cancer: Mitochondria Meet the “Iron Maiden” cell death. Cells 9, 1505 (2020).
Galy, B., Conrad, M. & Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nature reviews. Molecular cell biology. https://doi.org/10.1038/s41580-023-00648-1, (2023).
Wolff, N. A. et al. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 8, 211 (2018).
Shaw, G. C. et al. Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100 (2006).
Paradkar, P. N. et al. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell Biol. 29, 1007–1016 (2009).
Wu, J. R., Tuo, Q. Z. & Lei, P. Ferroptosis, a recent defined form of critical cell death in neurological disorders. J. Mol. Neurosci. 66, 197–206 (2018).
Nechushtai, R. et al. The balancing act of NEET proteins: Iron, ROS, calcium and metabolism. Biochim. Biophys. Acta Mol. Cell Res. 1867, 118805 (2020).
Tamir, S. et al. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim. Biophys. Acta 1853, 1294–1315 (2015).
Yuan, H. et al. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res Commun. 478, 838–844 (2016).
Lipper, C. H. et al. Redox-dependent gating of VDAC by mitoNEET. Proc. Natl Acad. Sci. USA 116, 19924–19929 (2019).
Yang, Y. et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 11, 433 (2020).
Huang, F. et al. Hedyotis diffusa injection induces ferroptosis via the Bax/Bcl2/VDAC2/3 axis in lung adenocarcinoma. Phytomedicine 104, 154319 (2022).
Fuhrmann, D. C. et al. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 36, 101670 (2020).
Wang, Y. Q. et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front. Aging Neurosci. 8, 308 (2016).
Wang, X. et al. Mitochondrial Ferritin deficiency promotes osteoblastic ferroptosis via mitophagy in Type 2 diabetic Osteoporosis. Biol. Trace Elem. Res 200, 298–307 (2022).
Cavadini, P. et al. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood 109, 3552–3559 (2007).
Li, P. et al. Structures of Atm1 provide insight into [2Fe-2S] cluster export from mitochondria. Nat Commun 13, 4339 (2022).
Srinivasan, V., Pierik, A. J. & Lill, R. Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 343, 1137–1140 (2014).
Lehrke, M. J. et al. The mitochondrial iron transporter ABCB7 is required for B cell development, proliferation, and class switch recombination in mice. Elife 10, e69621 (2021).
Ichikawa, Y. et al. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl Acad. Sci. USA 109, 4152–4157 (2012).
Chang, H. C. et al. Augmenter of liver regeneration regulates cellular iron homeostasis by modulating mitochondrial transport of ATP-binding cassette B8. Elife 10, e65158 (2021).
Chang, H. C. et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO Mol. Med. 8, 247–267 (2016).
Ichikawa, Y. et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Invest. 124, 617–630 (2014).
Bannai, S., Sato, H., Ishii, T. & Sugita, Y. Induction of cystine transport activity in human fibroblasts by oxygen. J. Biol. Chem. 264, 18480–18484 (1989).
Liu, X. et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 22, 476–486 (2020).
Lu, S. C. Regulation of glutathione synthesis. Mol. Asp. Med. 30, 42–59 (2009).
Parker, J. L. et al. Molecular basis for redox control by the human cystine/glutamate antiporter system xc. Nat. Commun. 12, 7147 (2021).
Ursini, F. & Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 152, 175–185 (2020).
Chen, X. et al. Ferroptosis: machinery and regulation. Autophagy 17, 2054–2081 (2021).
Seibt, T. M., Proneth, B. & Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 133, 144–152 (2019).
Forcina, G. C. & Dixon, S. J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19, e1800311 (2019).
Brigelius-Flohé, R. & Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303 (2013).
Borchert, A. et al. Crystal structure and functional characterization of selenocysteine-containing glutathione peroxidase 4 suggests an alternative mechanism of peroxide reduction. Biochim Biophys. Acta Mol. Cell Biol. Lipids 1863, 1095–1107 (2018).
Ingold, I. et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422 e421 (2018).
Pushpa-Rekha, T. R. et al. Rat phospholipid-hydroperoxide glutathione peroxidase. cDNA cloning and identification of multiple transcription and translation start sites. J. Biol. Chem. 270, 26993–26999 (1995).
Moreno, S. G. et al. Testis-specific expression of the nuclear form of phospholipid hydroperoxide glutathione peroxidase (PHGPx). Biol. Chem. 384, 635–643 (2003).
Maiorino, M. et al. Distinct promoters determine alternative transcription of gpx-4 into phospholipid-hydroperoxide glutathione peroxidase variants. J. Biol. Chem. 278, 34286–34290 (2003).
Pfeifer, H. et al. Identification of a specific sperm nuclei selenoenzyme necessary for protamine thiol cross-linking during sperm maturation. FASEB J. 15, 1236–1238 (2001).
Schneider, M. et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 23, 3233–3242 (2009).
Liang, H. et al. Short form glutathione peroxidase 4 is the essential isoform required for survival and somatic mitochondrial functions. J. Biol. Chem. 284, 30836–30844 (2009).
Yant, L. J. et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med 34, 496–502 (2003).
Conrad, M. et al. The nuclear form of phospholipid hydroperoxide glutathione peroxidase is a protein thiol peroxidase contributing to sperm chromatin stability. Mol. Cell Biol. 25, 7637–7644 (2005).
Mao, C. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021).
Wu, S. et al. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proc. Natl Acad. Sci. USA 119, e2121987119 (2022).
Gaschler, M. M. et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 14, 507–515 (2018).
Muller, T. et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol. Life Sci. 74, 3631–3645 (2017).
Zhu, S. et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 77, 2064–2077 (2017).
Deshwal, S. et al. Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat. Cell Biol. 25, 246–257 (2023).
Zeng, F., Chen, X. & Deng, G. The anti-ferroptotic role of FSP1: current molecular mechanism and therapeutic approach. Mol. Biomed. 3, 37 (2022).
Magtanong, L. et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem. Biol. 26, 420–432 e429 (2019).
Tesfay, L. et al. Stearoyl-CoA Desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 79, 5355–5366 (2019).
Papsdorf, K. et al. Lipid droplets and peroxisomes are co-regulated to drive lifespan extension in response to mono-unsaturated fatty acids. Nat. Cell Biol. 25, 672–684 (2023).
Gong, Y. N. et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169, 286–300 e216 (2017).
Ruhl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 (2018).
Dai, E. et al. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys. Res. Commun. 522, 415–421 (2020).
Pedrera, L. et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 28, 1644–1657 (2021).
Dai, E. et al. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys. Res. Commun. 523, 966–971 (2020).
David, L. A. & Alm, E. J. Rapid evolutionary innovation during an Archaean genetic expansion. Nature 469, 93–96 (2011).
Wang, Y. et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 20, e47563 (2019).
Chu, B. et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 21, 579–591 (2019).
Jennis, M. et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 30, 918–930 (2016).
Wang, S. J. et al. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 17, 366–373 (2016).
Leu, J. I., Murphy, M. E. & George, D. L. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc. Natl Acad. Sci. USA 116, 8390–8396 (2019).
Jensen, D. E. et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 16, 1097–1112 (1998).
Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 20, 1181–1192 (2018).
Tomlinson, I. P. et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410 (2002).
Alam, N. A. et al. Missense mutations in fumarate hydratase in multiple cutaneous and uterine leiomyomatosis and renal cell cancer. J. Mol. Diagn. 7, 437–443 (2005).
Chuang, G. S. et al. Germline fumarate hydratase mutations and evidence for a founder mutation underlying multiple cutaneous and uterine leiomyomata. J. Am. Acad. Dermatol. 52, 410–416 (2005).
Gao, M. et al. Role of Mitochondria in Ferroptosis. Mol. Cell 73, 354–363 e353 (2019).
Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Scalera, S. et al. KEAP1-mutant NSCLC: The catastrophic failure of a cell-protecting hub. J. Thorac. Oncol. 17, 751–757 (2022).
Romero, R. et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med 23, 1362–1368 (2017).
Koppula, P. et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 13, 2206 (2022).
Fan, Z. et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 6, e371 (2017).
Bartha, I., di Iulio, J., Venter, J. C. & Telenti, A. Human gene essentiality. Nat. Rev. Genet 19, 51–62 (2018).
Lee, J. E. et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2, e01503 (2013).
Egolf, S. et al. MLL4 mediates differentiation and tumor suppression through ferroptosis. Sci. Adv. 7, eabj9141 (2021).
Gorrini, C., Harris, I. S. & Mak, T. W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013).
Koppula, P., Zhang, Y., Zhuang, L. & Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 38, 12 (2018).
Koppula, P., Zhuang, L. & Gan, B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12, 599–620 (2021).
Chen, D. et al. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 68, 224–232 e224 (2017).
Hu, K. et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Invest. 130, 1752–1766 (2020).
Harris, I. S. et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015).
Xiong, Y., Xiao, C., Li, Z. & Yang, X. Engineering nanomedicine for glutathione depletion-augmented cancer therapy. Chem. Soc. Rev. 50, 6013–6041 (2021).
Anasagasti, M. J. et al. Glutathione protects metastatic melanoma cells against oxidative stress in the murine hepatic microvasculature. Hepatology 27, 1249–1256 (1998).
Estrela, J. M. et al. Glutathione in metastases: From mechanisms to clinical applications. Crit. Rev. Clin. Lab. Sci. 53, 253–267 (2016).
Yang, C. et al. A self-amplified ferroptosis nanoagent that inhibits the tumor upstream glutathione synthesis to reverse cancer chemoresistance. J. Control. Rel. 357, 20–30 (2023).
Yu, L. et al. WIPI2 enhances the vulnerability of colorectal cancer cells to erastin via bioinformatics analysis and experimental verification. Front. Oncol. 13, 1146617 (2023).
Zhang, L. et al. Hypersensitivity to ferroptosis in chromophobe RCC is mediated by a glutathione metabolic dependency and cystine import via solute carrier family 7 member 11. Proc. Natl Acad. Sci. USA 119, e2122840119 (2022).
Hangauer, M. J. et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017).
Rojo de la Vega, M., Chapman, E. & Zhang, D. D. NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43 (2018).
Liu, S. et al. Tubastatin A potently inhibits GPX4 activity to potentiate cancer radiotherapy through boosting ferroptosis. Redox Biol. 62, 102677 (2023).
Dodson, M., Castro-Portuguez, R. & Zhang, D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019).
Sun, R. et al. AADAC protects colorectal cancer liver colonization from ferroptosis through SLC7A11-dependent inhibition of lipid peroxidation. J. Exp. Clin. Cancer Res. 41, 284 (2022).
Chang, K. et al. DPP9 Stabilizes NRF2 to Suppress Ferroptosis and Induce Sorafenib resistance in clear cell renal cell carcinoma. Cancer Res. 83, 3940–3955 (2023).
Wang, X. et al. Mitochondrial calcium uniporter drives metastasis and confers a targetable cystine dependency in pancreatic cancer. Cancer Res. 82, 2254–2268 (2022).
Sun, W. Y. et al. Phospholipase iPLA(2)β averts ferroptosis by eliminating a redox lipid death signal. Nat. Chem. Biol. 17, 465–476 (2021).
Chen, D. et al. iPLA2β-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat. Commun. 12, 3644 (2021).
Hong, X. et al. The Lipogenic Regulator SREBP2 induces transferrin in circulating melanoma cells and suppresses ferroptosis. Cancer Discov. 11, 678–695 (2021).
Alvarez, S. W. et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017).
Du, J. et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 32, 101483 (2020).
Kim, E. H. et al. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 432, 180–190 (2018).
Brown, C. W. et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev. Cell 51, 575–586 e574 (2019).
Xie, Y. et al. Mammary adipocytes protect triple-negative breast cancer cells from ferroptosis. J. Hematol. Oncol. 15, 72 (2022).
Belavgeni, A., Tonnus, W. & Linkermann, A. Cancer cells evade ferroptosis: sex hormone-driven membrane-bound O-acyltransferase domain-containing 1 and 2 (MBOAT1/2) expression. Signal Transduct. Target Ther. 8, 336 (2023).
Murugan, A. K., Grieco, M. & Tsuchida, N. RAS mutations in human cancers: Roles in precision medicine. Semin Cancer Biol. 59, 23–35 (2019).
Prior, I. A., Hood, F. E. & Hartley, J. L. The frequency of Ras Mutations in cancer. Cancer Res 80, 2969–2974 (2020).
Poursaitidis, I. et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 18, 2547–2556 (2017).
Bartolacci, C. et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat. Commun. 13, 4327 (2022).
Muller, F. et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 30, 442–456 (2023).
Padanad, M. S. et al. Fatty acid oxidation mediated by Acyl-CoA Synthetase Long Chain 3 is required for mutant KRAS Lung Tumorigenesis. Cell Rep. 16, 1614–1628 (2016).
Itoh, K. et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86 (1999).
Tao, S. et al. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol. Carcinog. 57, 182–192 (2018).
Alam, J. et al. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078 (1999).
Liby, K. et al. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 67, 2414–2419 (2007).
DeNicola, G. M. et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011).
Chio, I. I. C. et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 166, 963–976 (2016).
Chen, X., Kang, R., Kroemer, G. & Tang, D. Broadening horizons: the role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 18, 280–296 (2021).
Yang, Z. et al. ACTL6A protects gastric cancer cells against ferroptosis through induction of glutathione synthesis. Nat. Commun. 14, 4193 (2023).
Kobayashi, A. et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 24, 7130–7139 (2004).
Wang, Z. et al. PRMT5 reduces immunotherapy efficacy in triple-negative breast cancer by methylating KEAP1 and inhibiting ferroptosis. J. Immunother. Cancer 11, e006890 (2023).
Sun, X. et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016).
Zhang, D. et al. Mitochondrial TSPO Promotes Hepatocellular Carcinoma Progression through Ferroptosis Inhibition and Immune Evasion. Adv. Sci. 10, e2206669 (2023).
Ren, X. et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 46, 102122 (2021).
Yi, J. et al. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl Acad. Sci. USA 117, 31189–31197 (2020).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017).
Glaviano, A. et al. PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol. Cancer 22, 138 (2023).
Lei, G., Zhuang, L. & Gan, B. mTORC1 and ferroptosis: Regulatory mechanisms and therapeutic potential. Bioessays 43, e2100093 (2021).
Gan, W. et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat. Cell Biol. 22, 246–256 (2020).
Zhang, Y. et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 12, 1589 (2021).
Zeng, F. et al. Inhibiting SCD expression by IGF1R during lorlatinib therapy sensitizes melanoma to ferroptosis. Redox Biol. 61, 102653 (2023).
Hu, Q. et al. ASS1-mediated reductive carboxylation of cytosolic glutamine confers ferroptosis resistance in cancer cells. Cancer Res. 83, 1646–1665 (2023).
Ye, Y. et al. Characterization of Hypoxia-associated molecular features to aid hypoxia-targeted therapy. Nat. Metab. 1, 431–444 (2019).
Bristow, R. G. & Hill, R. P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 8, 180–192 (2008).
Gilkes, D. M., Semenza, G. L. & Wirtz, D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat. Rev. Cancer 14, 430–439 (2014).
Rohwer, N. & Cramer, T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist. Updat 14, 191–201 (2011).
Gordan, J. D., Thompson, C. B. & Simon, M. C. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12, 108–113 (2007).
Chen, Z. et al. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct. Target Ther. 8, 70 (2023).
Yang, M. et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5, eaaw2238 (2019).
Yang, Z. et al. HIF-1alpha drives resistance to ferroptosis in solid tumors by promoting lactate production and activating SLC1A1. Cell Rep. 42, 112945 (2023).
Zhang, Q. et al. Hypoxia-responsive PPARGC1A/BAMBI/ACSL5 axis promotes progression and resistance to lenvatinib in hepatocellular carcinoma. Oncogene 42, 1509–1523 (2023).
Li, Y. et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J. Exp. Clin. Cancer Res. 42, 6 (2023).
Singhal, R. et al. HIF-2alpha activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J. Clin. Invest 131, e143691 (2021).
Zou, Y. et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 10, 1617 (2019).
Yang, J. et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 21, 341–352 (2020).
Dongre, A. & Weinberg, R. A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 20, 69–84 (2019).
Pastushenko, I. et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018).
Lee, J., You, J. H., Kim, M. S. & Roh, J. L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 37, 101697 (2020).
Zheng, X. et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015).
Fischer, K. R. et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476 (2015).
Marcucci, F., Stassi, G. & De Maria, R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat. Rev. Drug Discov. 15, 311–325 (2016).
Vucetic, M. et al. Together we stand, apart we fall: how cell-to-cell contact/interplay provides resistance to ferroptosis. Cell Death Dis. 11, 789 (2020).
Sun, L. et al. Lipid Peroxidation, GSH depletion, and SLC7A11 inhibition are common causes of EMT and ferroptosis in A549 cells, but different in specific mechanisms. DNA Cell Biol. 40, 172–183 (2021).
Joseph, J. V. et al. TGF-beta is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 5, e1443 (2014).
Viswanathan, V. S. et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457 (2017).
Muller, S. et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 12, 929–938 (2020).
Oliveira, T. et al. HDAC inhibition induces EMT and alterations in cellular iron homeostasis to augment ferroptosis sensitivity in SW13 cells. Redox Biol. 47, 102149 (2021).
You, J. H., Lee, J. & Roh, J. L. Mitochondrial pyruvate carrier 1 regulates ferroptosis in drug-tolerant persister head and neck cancer cells via epithelial-mesenchymal transition. Cancer Lett. 507, 40–54 (2021).
Chen, L. et al. GINS4 suppresses ferroptosis by antagonizing p53 acetylation with Snail. Proc. Natl Acad. Sci. USA 120, e2219585120 (2023).
Hassin, O. & Oren, M. Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov. 22, 127–144 (2023).
Wang, C. K. et al. MEX3A mediates p53 degradation to suppress ferroptosis and facilitate ovarian cancer tumorigenesis. Cancer Res. 83, 251–263 (2023).
Zhang, X. et al. ZNF498 promotes hepatocellular carcinogenesis by suppressing p53-mediated apoptosis and ferroptosis via the attenuation of p53 Ser46 phosphorylation. J. Exp. Clin. cancer Res. 41, 79 (2022).
Maddocks, O. D. et al. Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 (2013).
Tarangelo, A. et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 22, 569–575 (2018).
Xie, Y. et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 20, 1692–1704 (2017).
Gan, Y. et al. UTP11 deficiency suppresses cancer development via nucleolar stress and ferroptosis. Redox Biol. 62, 102705 (2023).
Sun, T. & Chi, J. T. Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes Dis. 8, 241–249 (2021).
Magesh, S. & Cai, D. Roles of YAP/TAZ in ferroptosis. Trends Cell Biol. 32, 729–732 (2022).
Lv, M. et al. CDK7-YAP-LDHD axis promotes D-lactate elimination and ferroptosis defense to support cancer stem cell-like properties. Signal Transduct. Target Ther. 8, 302 (2023).
Zhang, X. et al. Endogenous glutamate determines ferroptosis sensitivity via ADCY10-dependent YAP suppression in lung adenocarcinoma. Theranostics 11, 5650–5674 (2021).
Yang, W. H. et al. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol. Cancer Res. 18, 79–90 (2020).
Yang, W. H. et al. The Hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 28, 2501–2508 e2504 (2019).
Zhou, B. et al. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 66, 89–100 (2020).
Mancias, J. D. et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109 (2014).
Wu, H. et al. ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy 19, 2062–2077 (2023).
Li, K. et al. TRIM7 modulates NCOA4-mediated ferritinophagy and ferroptosis in glioblastoma cells. Redox Biol. 56, 102451 (2022).
Bai, Y. et al. Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res Commun. 508, 997–1003 (2019).
Schroeder, B. et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 61, 1896–1907 (2015).
You, J. H., Lee, J. & Roh, J. L. PGRMC1-dependent lipophagy promotes ferroptosis in paclitaxel-tolerant persister cancer cells. J. Exp. Clin. Cancer Res. 40, 350 (2021).
Chang, L. C. et al. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 416, 124–137 (2018).
Rademaker, G. et al. Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol. 53, 102324 (2022).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Wu, Z. et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl Acad. Sci. USA 116, 2996–3005 (2019).
Wu, K. et al. Creatine kinase B suppresses ferroptosis by phosphorylating GPX4 through a moonlighting function. Nat. Cell Biol. 25, 714–725 (2023).
Ott, M., Gogvadze, V., Orrenius, S. & Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 12, 913–922 (2007).
Shindo, R. et al. Critical contribution of oxidative stress to TNFalpha-induced necroptosis downstream of RIPK1 activation. Biochem. Biophys. Res Commun. 436, 212–216 (2013).
Zhang, Z. et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 16, 1482–1505 (2020).
Li, J. et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 17, 3361–3374 (2021).
Lee, W. C., Guntur, A. R., Long, F. & Rosen, C. J. Energy metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 38, 255–266 (2017).
Chen, Y. R. & Zweier, J. L. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 114, 524–537 (2014).
Gao, M. et al. Glutaminolysis and Transferrin regulate ferroptosis. Mol. Cell 59, 298–308 (2015).
Wang, Y. Q. et al. Sirtuin5 contributes to colorectal carcinogenesis by enhancing glutaminolysis in a deglutarylation-dependent manner. Nat. Commun. 9, 545 (2018).
Oka, S., Hsu, C. P. & Sadoshima, J. Regulation of cell survival and death by pyridine nucleotides. Circ. Res. 111, 611–627 (2012).
Llabani, E. et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat. Chem. 11, 521–532 (2019).
Lee, H. et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 22, 225–234 (2020).
Li, C. et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct. Target Ther. 5, 187 (2020).
Zhang, Y. et al. High-fat diet impairs ferroptosis and promotes cancer invasiveness via downregulating tumor suppressor ACSL4 in lung adenocarcinoma. Biol. Direct 16, 10 (2021).
Liu, W. et al. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 12, 5103 (2021).
Tan, S. K. et al. Obesity-dependent adipokine chemerin suppresses fatty acid oxidation to confer ferroptosis resistance. Cancer Discov. 11, 2072–2093 (2021).
Shi, Z., Naowarojna, N., Pan, Z. & Zou, Y. Multifaceted mechanisms mediating cystine starvation-induced ferroptosis. Nat. Commun. 12, 4792 (2021).
Jin, J., Byun, J. K., Choi, Y. K. & Park, K. G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 55, 706–715 (2023).
Xue, Y. et al. Intermittent dietary methionine deprivation facilitates tumoral ferroptosis and synergizes with checkpoint blockade. Nat. Commun. 14, 4758 (2023).
Liu, D. et al. Tryptophan Metabolism Acts as a New Anti-Ferroptotic Pathway to Mediate Tumor Growth. Adv. Sci. 10, e2204006 (2023).
Fiore, A. et al. Kynurenine importation by SLC7A11 propagates anti-ferroptotic signaling. Mol. Cell 82, 920–932 e927 (2022).
Panda, S. K. et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity 56, 797–812 e794 (2023).
Hu, Q. et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 12, 706 (2021).
Yang, X. et al. Regulation of VKORC1L1 is critical for p53-mediated tumor suppression through vitamin K metabolism. Cell Metab. 35, 1474–1490 e1478 (2023).
Dai, E. et al. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 20, 171 (2021).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013).
Drijvers, J. M. et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8(+) T Cells. Cancer Immunol. Res. 9, 184–199 (2021).
Kroemer, G., Galassi, C., Zitvogel, L. & Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 23, 487–500 (2022).
Shen, D. et al. PARPi treatment enhances radiotherapy-induced ferroptosis and antitumor immune responses via the cGAS signaling pathway in colorectal cancer. Cancer Lett. 550, 215919 (2022).
Liang, J. L. et al. Specific activation of cGAS-STING pathway by nanotherapeutics-mediated ferroptosis evoked endogenous signaling for boosting systemic tumor immunotherapy. Sci. Bull. 68, 622–636 (2023).
Ding, Q. et al. Mitochondrial-targeted brequinar liposome boosted mitochondrial-related ferroptosis for promoting checkpoint blockade immunotherapy in bladder cancer. J. Control Rel. 363, 221–234 (2023).
Yu, B., Choi, B., Li, W. & Kim, D. H. Magnetic field boosted ferroptosis-like cell death and responsive MRI using hybrid vesicles for cancer immunotherapy. Nat. Commun. 11, 3637 (2020).
Yu, Y. et al. Iron-based nanoscale coordination polymers synergistically induce immunogenic ferroptosis by blocking dihydrofolate reductase for cancer immunotherapy. Biomaterials 288, 121724 (2022).
Luo, X. et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 28, 1971–1989 (2021).
Efimova, I. et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J. Immunother. Cancer 8, e001369 (2020).
Bianchi, M. E. et al. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 280, 74–82 (2017).
Liu, P. et al. Inhibition of ALG3 stimulates cancer cell immunogenic ferroptosis to potentiate immunotherapy. Cell Mol. Life Sci. 79, 352 (2022).
Kim, K. S. et al. Enhanced natural killer cell anti-tumor activity with nanoparticles mediated ferroptosis and potential therapeutic application in prostate cancer. J. Nanobiotechnol. 20, 428 (2022).
Hinshaw, D. C. & Shevde, L. A. The tumor microenvironment innately modulates cancer progression. Cancer Res. 79, 4557–4566 (2019).
Peng, D. et al. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol. Cancer 21, 104 (2022).
Shi, X. et al. TGF-beta signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 15, 135 (2022).
Jiang, F. et al. ANO1-mediated inhibition of cancer ferroptosis confers immunotherapeutic resistance through recruiting cancer-associated fibroblasts. Adv. Sci. 10, e2300881 (2023).
Hernandez, C., Huebener, P. & Schwabe, R. F. Damage-associated molecular patterns in cancer: a double-edged sword. Oncogene 35, 5931–5941 (2016).
Dai, E. et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat. Commun. 11, 6339 (2020).
Fang, C. et al. Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell Mol. Immunol. 18, 2211–2223 (2021).
Dai, E. et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 16, 2069–2083 (2020).
Conche, C. et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut 72, 1774–1782 (2023).
Bottcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037 e1014 (2018).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 (2015).
Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28 (2012).
Wang, D. & DuBois, R. N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 36, 1085–1093 (2015).
Lang, X. et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 9, 1673–1685 (2019).
Zitvogel, L. & Kroemer, G. Interferon-gamma induces cancer cell ferroptosis. Cell Res 29, 692–693 (2019).
Stockwell, B. R. & Jiang, X. A physiological function for ferroptosis in tumor suppression by the immune system. Cell Metab. 30, 14–15 (2019).
Liao, P. et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40, 365–378 e366 (2022).
Friedmann Angeli, J. P., Xavier da Silva, T. N. & Schilling, B. CD8(+) T cells PUF(A)ing the flames of cancer ferroptotic cell death. Cancer Cell 40, 346–348 (2022).
Gan, B. ACSL4, PUFA, and ferroptosis: new arsenal in anti-tumor immunity. Signal Transduct. Target Ther. 7, 128 (2022).
Kepp, O. & Kroemer, G. Pro-ferroptotic fatty acid metabolism renders cancer cells immunogenic. Trends Cancer 8, 785–787 (2022).
Gocher, A. M., Workman, C. J. & Vignali, D. A. A. Interferon-gamma: teammate or opponent in the tumour microenvironment? Nat. Rev. Immunol. 22, 158–172 (2022).
Wu, L. et al. The establishment of polypeptide PSMA-targeted chimeric antigen receptor-engineered natural killer cells for castration-resistant prostate cancer and the induction of ferroptosis-related cell death. Cancer Commun. 42, 768–783 (2022).
Li, H. et al. HLF regulates ferroptosis, development and chemoresistance of triple-negative breast cancer by activating tumor cell-macrophage crosstalk. J. Hematol. Oncol. 15, 2 (2022).
Bansal, A. et al. Gamma-Glutamyltransferase 1 promotes clear cell renal cell carcinoma initiation and progression. Mol. Cancer Res. 17, 1881–1892 (2019).
Luo, Y. et al. The suppression of cervical cancer ferroptosis by macrophages: The attenuation of ALOX15 in cancer cells by macrophages-derived exosomes. Acta Pharm. Sin. B 13, 2645–2662 (2023).
Bouche, C. & Quail, D. F. Fueling the tumor microenvironment with cancer-associated adipocytes. Cancer Res. 83, 1170–1172 (2023).
Wang, R., Liu, Z., Fan, Z. & Zhan, H. Lipid metabolism reprogramming of CD8(+) T cell and therapeutic implications in cancer. Cancer Lett. 567, 216267 (2023).
Ma, X. et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 33, 1001–1012 e1005 (2021).
Xu, S. et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577 e1567 (2021).
Xiao, L. et al. IL-9/STAT3/fatty acid oxidation-mediated lipid peroxidation contributes to Tc9 cell longevity and enhanced antitumor activity. J. Clin. Invest. 132, e153247 (2022).
Downs-Canner, S. M., Meier, J., Vincent, B. G. & Serody, J. S. B cell function in the tumor microenvironment. Annu Rev. Immunol. 40, 169–193 (2022).
Muri, J., Thut, H., Bornkamm, G. W. & Kopf, M. B1 and marginal zone B Cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 29, 2731-2744 e2734, (2019).
Chen, Q. et al. The role of B-cell ferroptosis in the pathogenesis of systemic lupus erythematosus. Clin. Immunol. 256, 109778 (2023).
Zhu, S. et al. Tumor microenvironment-related dendritic cell deficiency: a target to enhance tumor immunotherapy. Pharm. Res. 159, 104980 (2020).
Cubillos-Ruiz, J. R. et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161, 1527–1538 (2015).
Garris, C. S. & Pittet, M. J. ER stress in dendritic cells promotes. Cancer Cell. 161, 1492–1493 (2015).
Merad, M. & Salmon, H. Cancer: A dendritic-cell brake on antitumour immunity. Nature 523, 294–295 (2015).
Han, L. et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys. Res. Commun. 576, 33–39 (2021).
Rothe, T. et al. 12/15-Lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J. Clin. Invest 125, 1944–1954 (2015).
Zhou, Y., Cheng, L., Liu, L. & Li, X. NK cells are never alone: crosstalk and communication in tumour microenvironments. Mol. Cancer 22, 34 (2023).
Cui, J. X. et al. L-kynurenine induces NK cell loss in gastric cancer microenvironment via promoting ferroptosis. J. Exp. Clin. Cancer Res. 42, 52 (2023).
Tan, S. et al. Exosomal miRNAs in tumor microenvironment. J. Exp. Clin. Cancer Res. 39, 67 (2020).
Kapralov, A. A. et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290 (2020).
Hao, X. et al. Inhibition of APOC1 promotes the transformation of M2 into M1 macrophages via the ferroptosis pathway and enhances anti-PD1 immunotherapy in hepatocellular carcinoma based on single-cell RNA sequencing. Redox Biol. 56, 102463 (2022).
Tang, B. et al. Targeted xCT-mediated ferroptosis and protumoral polarization of macrophages is effective against hcc and enhances the efficacy of the anti-PD-1/L1 response. Adv. Sci. 10, e2203973 (2023).
Gu, Z. et al. Ferroptosis-strengthened metabolic and inflammatory regulation of tumor-associated macrophages provokes potent tumoricidal activities. Nano Lett. 21, 6471–6479 (2021).
Li, L. G. et al. A Dihydroartemisinin-loaded nanoreactor motivates anti-cancer immunotherapy by synergy-induced ferroptosis to activate Cgas/STING for reprogramming of macrophage. Adv. Health. Mater. 12, e2301561 (2023).
Shi, Z. et al. Multifunctional nanomaterials for ferroptotic cancer therapy. Front Chem. 10, 868630 (2022).
Li, L. G. et al. Dihydroartemisinin remodels macrophage into an M1 phenotype via ferroptosis-mediated DNA damage. Front. Pharm. 13, 949835 (2022).
Yan, Y. et al. Metabolic profiles of regulatory T cells and their adaptations to the tumor microenvironment: implications for antitumor immunity. J. Hematol. Oncol. 15, 104 (2022).
Xu, C. et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 35, 109235 (2021).
Li, K. et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target Ther. 6, 362 (2021).
Wu, Y. et al. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol. Cancer 21, 184 (2022).
Du, S., Zeng, F. & Deng, G. Tumor neutrophils ferroptosis: a targetable immunosuppressive mechanism for cancer immunotherapy. Signal Transduct. Target Ther. 8, 77 (2023).
Torti, S. V. & Torti, F. M. Iron and cancer: more ore to be mined. Nat. Rev. Cancer 13, 342–355 (2013).
Manz, D. H. et al. Iron and cancer: recent insights. Ann. N. Y Acad. Sci. 1368, 149–161 (2016).
Tsoi, J. et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 33, 890–904 e895 (2018).
Koeberle, S. C., Kipp, A. P., Stuppner, H. & Koeberle, A. Ferroptosis-modulating small molecules for targeting drug-resistant cancer: Challenges and opportunities in manipulating redox signaling. Med. Res. Rev. 43, 614–682 (2023).
Lei, G. et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 30, 146–162 (2020).
Guo, J. et al. Ferroptosis: A novel anti-tumor action for Cisplatin. Cancer Res. Treat. 50, 445–460 (2018).
Wang, J. et al. RNF2 promotes the progression of colon cancer by regulating ubiquitination and degradation of IRF4. Biochim Biophys. Acta Mol. Cell Res 1869, 119162 (2022).
Yao, X. et al. Simvastatin induced ferroptosis for triple-negative breast cancer therapy. J. Nanobiotechnol. 19, 311 (2021).
Gout, P. W., Buckley, A. R., Simms, C. R. & Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640 (2001).
Chipurupalli, S. et al. Three-dimensional growth sensitizes breast cancer cells to treatment with ferroptosis-promoting drugs. Cell Death Dis. 14, 580 (2023).
Philip, M. & Schietinger, A. CD8(+) T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 22, 209–223 (2022).
Carlino, M. S., Larkin, J. & Long, G. V. Immune checkpoint inhibitors in melanoma. Lancet 398, 1002–1014 (2021).
Kong, R. et al. IFNgamma-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J. Leukoc. Biol. 110, 301–314 (2021).
Jiang, Z. et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J. Clin. Investig. 131, e139434 (2021).
Yang, F. et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 35, 84–100.e108 (2023).
Han, Y. et al. IL-1β-associated NNT acetylation orchestrates iron-sulfur cluster maintenance and cancer immunotherapy resistance. Mol. Cell 83, 1887–1902.e1888 (2023).
Meng, Y. et al. BET inhibitors potentiate melanoma ferroptosis and immunotherapy through AKR1C2 inhibition. Mil. Med. Res. 10, 61 (2023).
Wu, Y., Song, Y., Wang, R. & Wang, T. Molecular mechanisms of tumor resistance to radiotherapy. Mol. Cancer 22, 96 (2023).
Jaffray, D. A. Image-guided radiotherapy: from current concept to future perspectives. Nat. Rev. Clin. Oncol. 9, 688–699 (2012).
Baidoo, K. E., Yong, K. & Brechbiel, M. W. Molecular pathways: targeted alpha-particle radiation therapy. Clin. Cancer Res. 19, 530–537 (2013).
Wan, C. et al. Irradiated tumor cell-derived microparticles mediate tumor eradication via cell killing and immune reprogramming. Sci. Adv. 6, eaay9789 (2020).
Chandra, R. A., Keane, F. K., Voncken, F. E. M. & Thomas, C. R. Jr. Contemporary radiotherapy: present and future. Lancet 398, 171–184 (2021).
Jiang, K. et al. STC2 activates PRMT5 to induce radioresistance through DNA damage repair and ferroptosis pathways in esophageal squamous cell carcinoma. Redox Biol. 60, 102626 (2023).
Chen, Q. et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. 30, 137–151 (2023).
Yang, M. et al. COMMD10 inhibits HIF1alpha/CP loop to enhance ferroptosis and radiosensitivity by disrupting Cu-Fe balance in hepatocellular carcinoma. J. Hepatol. 76, 1138–1150 (2022).
Mou, Y. et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J. Hematol. Oncol. 12, 34 (2019).
Dixon, S. J. & Stockwell, B. R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17 (2014).
Li, Q. et al. Understanding sorafenib-induced ferroptosis and resistance mechanisms: Implications for cancer therapy. Eur. J. Pharm. 955, 175913 (2023).
Xu, X. et al. Increased ATF2 expression predicts poor prognosis and inhibits sorafenib-induced ferroptosis in gastric cancer. Redox Biol. 59, 102564 (2023).
Gao, R. et al. YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol. Med 13, e14351 (2021).
Li, B. et al. CISD2 promotes resistance to sorafenib-induced ferroptosis by regulating autophagy in hepatocellular carcinoma. Front Oncol. 11, 657723 (2021).
Yang, H. et al. The PTBP1‑NCOA4 axis promotes ferroptosis in liver cancer cells. Oncol. Rep. 49, 45 (2023).
Zheng, J. et al. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 12, 698 (2021).
Ni, J., Chen, K., Zhang, J. & Zhang, X. Inhibition of GPX4 or mTOR overcomes resistance to Lapatinib via promoting ferroptosis in NSCLC cells. Biochem Biophys. Res Commun. 567, 154–160 (2021).
Ma, S., Henson, E. S., Chen, Y. & Gibson, S. B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 7, e2307 (2016).
Ma, S. et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One 12, e0182921 (2017).
Nagpal, A. et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Res. 21, 94 (2019).
Ma, H. et al. Neratinib inhibits proliferation and promotes apoptosis of acute myeloid leukemia cells by activating autophagy-dependent ferroptosis. Drug Dev. Res. 83, 1641–1653 (2022).
Park, S. Y. et al. Irreversible HER2 inhibitors overcome resistance to the RSL3 ferroptosis inducer in non-HER2 amplified luminal breast cancer. Cell Death Dis. 14, 532 (2023).
Rottenberg, S., Disler, C. & Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 21, 37–50 (2021).
Han, Y., Wen, P., Li, J. & Kataoka, K. Targeted nanomedicine in cisplatin-based cancer therapeutics. J. Control. Rel. 345, 709–720 (2022).
Zamble, D. B. & Lippard, S. J. Cisplatin and D. N. A. repair in cancer chemotherapy. Trends Biochem Sci. 20, 435–439 (1995).
Roh, J. L. et al. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 381, 96–103 (2016).
Fu, D., Wang, C., Yu, L. & Yu, R. Induction of ferroptosis by ATF3 elevation alleviates cisplatin resistance in gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol. Biol. Lett. 26, 26 (2021).
Han, L., Li, L. & Wu, G. Induction of ferroptosis by carnosic acid-mediated inactivation of Nrf2/HO-1 potentiates cisplatin responsiveness in OSCC cells. Mol. Cell Probes 64, 101821 (2022).
Roh, J. L., Kim, E. H., Jang, H. & Shin, D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 11, 254–262 (2017).
Sato, M. et al. The ferroptosis inducer erastin irreversibly inhibits system x(c)- and synergizes with cisplatin to increase cisplatin’s cytotoxicity in cancer cells. Sci. Rep. 8, 968 (2018).
Wang, Y. et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. 29, 2190–2202 (2022).
Zhou, Z. et al. Cisplatin promotes the efficacy of immune checkpoint inhibitor therapy by inducing ferroptosis and activating neutrophils. Front. Pharmacol. 13, 870178 (2022).
Mini, E. et al. Cellular pharmacology of gemcitabine. Ann. Oncol. 17, v7–12, (2006).
He, H. et al. KIF20A is associated with clinical prognosis and synergistic effect of gemcitabine combined with ferroptosis inducer in lung adenocarcinoma. Front. Pharm. 13, 1007429 (2022).
Qi, R. et al. Cancer-associated fibroblasts suppress ferroptosis and induce gemcitabine resistance in pancreatic cancer cells by secreting exosome-derived ACSL4-targeting miRNAs. Drug Resist. Updat. 68, 100960 (2023).
Combe, B. et al. Efficacy, safety and patient-reported outcomes of combination etanercept and sulfasalazine versus etanercept alone in patients with rheumatoid arthritis: a double-blind randomised 2-year study. Ann. Rheum. Dis. 68, 1146–1152 (2009).
Pardieu, B. et al. Cystine uptake inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia 36, 1585–1595 (2022).
Idei, U. et al. Mechanism of cell death by combined treatment with an xCT Inhibitor and Paclitaxel: An alternative therapeutic strategy for patients with ovarian clear cell carcinoma. Int J. Mol. Sci. 24, 11781 (2023).
Kerkhove, L. et al. Repurposing Sulfasalazine as a radiosensitizer in hypoxic human colorectal cancer. Cancers 15, 2363 (2023).
Ruiz de Galarreta, M. et al. beta-Catenin activation promotes immune escape and resistance to Anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9, 1124–1141 (2019).
Jin, H. et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 595, 730–734 (2021).
Meng, J. et al. Ferroptosis-enhanced immunotherapy with an injectable dextran-chitosan hydrogel for the treatment of malignant ascites in hepatocellular carcinoma. Adv. Sci. (Weinh.) 10, e2300517 (2023).
Mach, F. et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur. Heart J. 41, 111–188 (2020).
Li, Q. et al. Novel function of fluvastatin in attenuating oxidized low-density lipoprotein-induced endothelial cell ferroptosis in a glutathione peroxidase4- and cystine-glutamate antiporter-dependent manner. Exp. Ther. Med. 22, 1275 (2021).
Zhang, Q. et al. Atorvastatin induces mitochondria-dependent ferroptosis via the modulation of Nrf2-xCT/GPx4 Axis. Front. Cell Dev. Biol. 10, 806081 (2022).
Sahebkar, A. et al. Ferroptosis, a new pathogenetic mechanism in cardiometabolic diseases and cancer: Is there a role for statin therapy? Metab.: Clin. Exp. 146, 155659 (2023).
Mao, W. et al. Statin shapes inflamed tumor microenvironment and enhances immune checkpoint blockade in non-small cell lung cancer. JCI insight 7, e161940 (2022).
Klayman, D. L. Qinghaosu (artemisinin): an antimalarial drug from China. Science 228, 1049–1055 (1985).
Li, J. & Zhou, B. Biological actions of artemisinin: insights from medicinal chemistry studies. Molecules 15, 1378–1397 (2010).
Ooko, E. et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine 22, 1045–1054 (2015).
Markowitsch, S. D. et al. Artesunate Inhibits Growth Of Sunitinib-resistant Renal Cell Carcinoma Cells Through Cell Cycle Arrest And Induction Of Ferroptosis. Cancers 12, 3150 (2020).
Li, Z. J. et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta Pharm. Sin. 42, 301–310 (2021).
Chen, Y. et al. Artesunate synergistically promotes sorafenib‑induced apoptosis and ferroptosis in non‑Hodgkin lymphoma cells through inhibition of the STAT3 pathway. Oncol. Rep. 50, 147 (2023).
Crespo-Ortiz, M. P. & Wei, M. Q. Antitumor activity of artemisinin and its derivatives: from a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012, 247597 (2012).
Yuan, B. et al. Dihydroartemisinin Inhibits The Proliferation, Colony Formation And Induces Ferroptosis Of Lung Cancer Cells By Inhibiting PRIM2/SLC7A11 Axis. Oncol Targets Ther. 13, 10829–10840 (2020).
Lai, X. Y., Shi, Y. M. & Zhou, M. M. Dihydroartemisinin enhances gefitinib cytotoxicity against lung adenocarcinoma cells by inducing ROS-dependent apoptosis and ferroptosis. Kaohsiung J. Med. Sci. 39, 699–709 (2023).
Cui, Z. et al. Dihydroartemisinin enhances the inhibitory effect of sorafenib on HepG2 cells by inducing ferroptosis and inhibiting energy metabolism. J. Pharm. Sci. 148, 73–85 (2022).
Du, J. et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. 12, 705 (2021).
Froemming, J. S., Lam, Y. W., Jann, M. W. & Davis, C. M. Pharmacokinetics of haloperidol. Clin. Pharmacokinet. 17, 396–423 (1989).
Bakadlag, R., Jandaghi, P., Hoheisel, J. D. & Riazalhosseini, Y. The potential of dopamine receptor D2 (DRD2) as a therapeutic target for tackling pancreatic cancer. Expert Opin. Ther. Targets 23, 365–367 (2019).
Liu, Z. et al. Synergistic suppression of glioblastoma cell growth by combined application of Temozolomide and Dopamine D2 receptor antagonists. World Neurosurg. 128, e468–e477 (2019).
Shi, L. et al. The DRD2 antagonist Haloperidol mediates autophagy-induced ferroptosis to increase Temozolomide sensitivity by promoting endoplasmic reticulum stress in Glioblastoma. Clin. Cancer Res. 29, 3172–3188 (2023).
Bai, T. et al. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 491, 919–925 (2017).
Adkins, J. C., Peters, D. H. & Faulds, D. Zalcitabine. An update of its pharmacodynamic and pharmacokinetic properties and clinical efficacy in the management of HIV infection. Drugs 53, 1054–1080 (1997).
Ma, X. et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 33, 1001–1012.e1005 (2021).
Zhai, B. et al. Drug delivery systems for elemene, its main active ingredient beta-elemene, and its derivatives in cancer therapy. Int J. Nanomed. 13, 6279–6296 (2018).
Zhao, L. P. et al. beta-Elemene induced ferroptosis via TFEB-mediated GPX4 degradation in EGFR wide-type non-small cell lung cancer. J. Adv. Res. https://doi.org/10.1016/j.jare.2023.08.018, (2023).
Tang, Q. et al. Withaferin A triggers G2/M arrest and intrinsic apoptosis in glioblastoma cells via ATF4-ATF3-CHOP axis. Cell Prolif. 53, e12706 (2020).
Hassannia, B. et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 128, 3341–3355 (2018).
Zhang, Y. et al. Implications of Withaferin A for the metastatic potential and drug resistance in hepatocellular carcinoma cells via Nrf2-mediated EMT and ferroptosis. Toxicol. Mech. Methods 33, 47–55 (2023).
Rao, Z. et al. Iron-based metal-organic framework co-loaded with buthionine sulfoximine and oxaliplatin for enhanced cancer chemo-ferrotherapy via sustainable glutathione elimination. J. Nanobiotechnology 21, 265 (2023).
Lippmann, J., Petri, K., Fulda, S. & Liese, J. Redox modulation and induction of ferroptosis as a new therapeutic strategy in hepatocellular carcinoma. Transl. Oncol. 13, 100785 (2020).
Zeng, L. et al. A MOF-based potent ferroptosis inducer for enhanced radiotherapy of triple negative breast cancer. ACS Nano 17, 13195–13210 (2023).
Yang, C. et al. De novo pyrimidine biosynthetic complexes support cancer cell proliferation and ferroptosis defence. Nat. Cell Biol. 25, 836–847 (2023).
Zhang, R. et al. Curcumenol triggered ferroptosis in lung cancer cells via lncRNA H19/miR-19b-3p/FTH1 axis. Bioact. Mater. 13, 23–36 (2022).
Feng, H. & Stockwell, B. R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 16, e2006203 (2018).
Hassannia, B., Vandenabeele, P. & Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 35, 830–849 (2019).
Zeng, F. et al. Ferroptosis detection: from approaches to applications. Angew. Chem. (Int. ed. Engl.) 62, e202300379 (2023).
Yan, R. et al. The structure of erastin-bound xCT-4F2hc complex reveals molecular mechanisms underlying erastin-induced ferroptosis. Cell Res. 32, 687–690 (2022).
Gan, B. How erastin assassinates cells by ferroptosis revealed. Protein Cell 14, 84–86 (2023).
Cramer, S. L. et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat. Med. 23, 120–127 (2017).
Sun, Y. et al. Fin56-induced ferroptosis is supported by autophagy-mediated GPX4 degradation and functions synergistically with mTOR inhibition to kill bladder cancer cells. Cell Death Dis. 12, 1028 (2021).
Zhang, X., Guo, Y., Li, H. & Han, L. FIN56, a novel ferroptosis inducer, triggers lysosomal membrane permeabilization in a TFEB-dependent manner in glioblastoma. J. Cancer 12, 6610–6619 (2021).
Zhao, L. X. et al. Graphdiyne nanoplatforms for photothermal-ferroptosis combination therapy against glioblastoma. J. Control Rel. 359, 12–25 (2023).
Liu, X. et al. Targeting NRF2 uncovered an intrinsic susceptibility of acute myeloid leukemia cells to ferroptosis. Exp. Hematol. Oncol. 12, 47 (2023).
Song, R. et al. Acidity-activatable dynamic nanoparticles boosting ferroptotic cell death for immunotherapy of cancer. Adv. Mater. 33, e2101155 (2021).
Chen, K. et al. Injectable alginate hydrogel promotes antitumor immunity through glucose oxidase and Fe(3+) amplified RSL3-induced ferroptosis. Carbohydr. Polym. 326, 121643 (2024).
Anstee, Q. M. et al. From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16, 411–428 (2019).
Umemura, A. et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 29, 935–948 (2016).
Yao, L. et al. Cancer-associated fibroblasts impair the cytotoxic function of NK cells in gastric cancer by inducing ferroptosis via iron regulation. Redox Biol. 67, 102923 (2023).
Zhang, S. et al. Chemotherapy impairs ovarian function through excessive ROS-induced ferroptosis. Cell Death Dis. 14, 340 (2023).
Mishima, E. et al. Drugs repurposed as antiferroptosis agents suppress organ damage, including AKI, by functioning as lipid peroxyl radical scavengers. J. Am. Soc. Nephrol. 31, 280–296 (2020).
Fang, X. et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl Acad. Sci. USA 116, 2672–2680 (2019).
Kong, P. et al. Ferroptosis triggered by STAT1- IRF1-ACSL4 pathway was involved in radiation-induced intestinal injury. Redox Biol. 66, 102857 (2023).
Wang, D. et al. LCN2 secreted by tissue-infiltrating neutrophils induces the ferroptosis and wasting of adipose and muscle tissues in lung cancer cachexia. J. Hematol. Oncol. 16, 30 (2023).
Wang, D. et al. Antiferroptotic activity of non-oxidative dopamine. Biochem. Biophys. Res. Commun. 480, 602–607 (2016).
Gaschler, M. M. et al. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem. Biol. 13, 1013–1020 (2018).
Hao, L., Zhong, Y. M., Tan, C. P. & Mao, Z. W. Quantitative tracking of endoplasmic reticulum viscosity during ferroptosis by an iridium complex via TPPLM. Chem. Commun. 57, 5040–5042 (2021).
Dixon, S. J. et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523 (2014).
Hong, S. H. et al. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget 8, 115164–115178 (2017).
Saito, A. et al. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J. Biol. Chem. 286, 4809–4818 (2011).
Su, N. & Kilberg, M. S. C/EBP homology protein (CHOP) interacts with activating transcription factor 4 (ATF4) and negatively regulates the stress-dependent induction of the asparagine synthetase gene. J. Biol. Chem. 283, 35106–35117 (2008).
Lee, A. S. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 14, 263–276 (2014).
Gao, H. et al. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res Commun. 503, 1550–1556 (2018).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Kang, R. & Tang, D. Autophagy and Ferroptosis - What’s the connection? Curr. Pathobiol. Rep. 5, 153–159 (2017).
Torii, S. et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 473, 769–777 (2016).
Wu, Y. et al. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol. Cancer 19, 39 (2020).
Chen, X., Kang, R., Kroemer, G. & Tang, D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 28, 2843–2856 (2021).
Stockwell, B. R. et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Cui, S. et al. Identification of hyperoxidized PRDX3 as a ferroptosis marker reveals ferroptotic damage in chronic liver diseases. Mol. Cell 83, 3931–3939.e3935 (2023).
Zhang, Y. et al. Imidazole Ketone Erastin Induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623–633.e629 (2019).
Chen, X. et al. A small interfering CD147-targeting RNA inhibited the proliferation, invasiveness, and metastatic activity of malignant melanoma. Cancer Res. 66, 11323–11330 (2006).
Zeng, F. et al. Prognostic implications of metabolism related gene signature in cutaneous melanoma. Front. Oncol. 10, 1710 (2020).
Ding, Y., Fei, Y. & Lu, B. Emerging new concepts of degrader technologies. Trends Pharm. Sci. 41, 464–474 (2020).
Luo, T. et al. Intracellular delivery of glutathione peroxidase degrader induces ferroptosis in vivo. Angew. Chem. Int Ed. Engl. 61, e202206277 (2022).
Banik, S. M. et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82103183 to FZ, 82102803, 82272849 to GD), National Natural Science Foundation of Hunan Province (Grant Nos. 2022JJ40767 to FZ) and Natural Science Fund for Outstanding Youths in Hunan Province (2023JJ20093 to GD). We thank Biorender (https://www.biorender.com/) for the assistance for the illustration.
Author information
Authors and Affiliations
Contributions
G.D., X.C., and F.Z. designed the review; Q.Z., Y.M., D.L., L.Y., and J.L., searched for literature and wrote the manuscript; Y.M., D.L., and L.Y., drew the figures; Y.L., and Y.S, helped edit and revise the manuscript. G.D., and F.Z., provided funding support. All authors have read and approved the article and agree with publication in this journal.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, Q., Meng, Y., Li, D. et al. Ferroptosis in cancer: from molecular mechanisms to therapeutic strategies. Sig Transduct Target Ther 9, 55 (2024). https://doi.org/10.1038/s41392-024-01769-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01769-5
- Springer Nature Limited
This article is cited by
-
METTL16-SENP3-LTF axis confers ferroptosis resistance and facilitates tumorigenesis in hepatocellular carcinoma
Journal of Hematology & Oncology (2024)
-
METTL3 silencing inhibits ferroptosis to suppress ovarian fibrosis in PCOS by upregulating m6A modification of GPX4
Journal of Molecular Histology (2024)
-
The Mutual Regulatory Role of Ferroptosis and Immunotherapy in Anti-tumor Therapy
Apoptosis (2024)
-
Ferroptosis crosstalk in anti-tumor immunotherapy: molecular mechanisms, tumor microenvironment, application prospects
Apoptosis (2024)