
Abstract
The transcription factor c-MYC regulates a multiplicity of genes involved in cellular growth, proliferation, metabolism and DNA damage response and its overexpression is a hallmark of many tumours. Since MYC promotes apoptosis under conditions of stress, such as limited availability of nutrients or cytokines, MYC-driven cells are very much dependent on signals that inhibit cell death. Stress signals trigger apoptosis via the pathway regulated by opposing fractions of the BCL-2 protein family and previous genetic studies have shown that the development of B lymphoid tumours in Eµ-Myc mice is critically dependent on expression of pro-survival BCL-2 relatives MCL-1, BCL-W and, to a lesser extent, BCL-XL, but not BCL-2 itself, and that sustained growth of these lymphomas is dependent on MCL-1. Using recently developed mice that lack expression of all three functional pro-survival A1 genes, we show here that the kinetics of lymphoma development in Eµ-Myc mice and the competitive repopulation capacity of Eµ-Myc haemopoietic stem and progenitor cells is unaffected by the absence of A1. However, conditional loss of a single remaining functional A1 gene from transplanted A1-a−/−A1-bfl/flA1-c−/− Eµ-Myc lymphomas slowed their expansion, significantly extending the life of the transplant recipients. Thus, A1 contributes to the survival of malignant Eµ-Myc-driven B lymphoid cells. These results strengthen the case for BFL-1, the human homologue of A1, being a valid target for drug development for MYC-driven tumours.
Similar content being viewed by others

c-MYC (hereafter MYC), a basic helix–loop–helix leucine zipper transcription factor that regulates a multiplicity of genes involved in cell growth, proliferation and metabolism [1,2,3], has been implicated in the aetiology of many, perhaps all, human malignancies [4]. MYC levels are tightly regulated in normal cells but a wide range of mutations can override transcriptional and post-transcriptional controls and many provoke over-expression. High levels of MYC provoke apoptosis when cells experience stress from, for example, limited availability of cytokines or nutrients [5,6,7] and this imposes a brake on proliferation [8]. Consequently, anti-apoptotic mutations release that brake and synergise with MYC to promote malignant transformation [9,10,11,12,13,14].
MYC induces apoptosis via the pathway regulated by the BCL-2 family of proteins. BCL-2 and its closest homologues (BCL-XL, BCL-W, MCL-1 and A1/BFL-1) promote cell survival by neutralising pro-apoptotic relatives: BAX and BAK, and more distant relatives known as BH3-only proteins (for recent reviews see [15,16,17]). In healthy cells, BAX and BAK are primarily in an inactive monomeric state and any activated monomers are restrained by the pro-survival proteins. Stress conditions, such as deprivation of cytokines, oncogene expression or DNA damage, provoke up-regulation of pro-apoptotic BH3-only proteins, which bind tightly to the hydrophobic surface groove of pro-survival proteins via their BH3 domains, preventing them from inhibiting BAX and BAK. Certain BH3-only proteins (BIM, tBID, PUMA) can also bind transiently to the analogous surface groove of BAX and BAK, provoking a dramatic conformational change that prompts their homo-dimerisation [18,19,20]. BAX/BAK homo-dimers then aggregate to form homo-oligomeric pores, through which cytochrome c egresses to initiate sequential activation of caspases, which cleave hundreds of vital proteins, thereby dooming the cell.
Chromosome translocations found in Burkitt’s lymphoma and mouse plasmacytomas link the c-MYC gene to Ig gene loci [21], thereby subjugating MYC expression to strong Ig gene enhancers. Transgenic Eµ-Myc mice developed to model such translocations [22, 23] have greatly advanced our understanding of MYC-driven lymphomagenesis. Overexpression of MYC promotes polyclonal expansion of highly proliferative non-malignant pre-B cells [22,23,24] that are highly susceptible to apoptosis [11] and progression to malignancy depends upon acquisition of additional synergistic somatic changes such as mutation of RAS [25] or of the p19ARF/p53 pathway [12]. Of note, lymphomagenesis in these mice is accelerated by overexpression of BCL-2 and other pro-survival homologues [10, 26, 27] or loss of BH3-only proteins BIM [13, 28], PUMA [14], BMF or BAD [29].
Different cell types have a greater or lesser dependence on individual pro-survival family members, depending on the relative expression of other BCL-2 family members. Tumour cells are particularly dependent, because they express high levels of BH3-only proteins such as BIM due to the stresses and mutations suffered en route to malignancy [30, 31]. Gene knockout studies have shown that the development and expansion of tumours in Eµ-Myc mice is critically dependent on expression of endogenous MCL-1 [32, 33], BCL-W [34] and, to a lesser extent, BCL-XL [32, 35] but not BCL-2 [36].
Understanding of the physiologic importance of pro-survival A1/BFL-1 has lagged due to the fact that the mouse A1 locus contains three very similar functional homologues (A1-a, A1-b and A1-d) and a pseudogene (A1-c) [37]. A1 is expressed in multiple organs in the mouse embryo but apparently only in haemopoietic cells in the adult [38]. Expression in lymphoid and myeloid cells is normally low but is rapidly induced following a variety of stimuli [38]. Mice lacking just the A1-a gene are normal, albeit with some minor defects in their neutrophils and mast cells [39, 40]. Transgenic A1RNAi mice having reduced levels of all functional A1 isoforms have diminished numbers of B cells as well as impaired myelopoiesis and T cell development [41,42,43]. However, recently developed mouse strains lacking all three functional A1 genes are relatively normal, with only minor decreases in γδ T cells, CD4 T cells and conventional dendritic cells [44, 45].
In this study, we have explored the role of A1 in lymphoma development by crossing the A1-a−/− A1-b−/− A1-d−/− mice (hereafter A1−/− mice) [44] with Eµ-Myc transgenic mice [22, 23]. We also investigated whether A1 was important for the expansion of established Eµ-Myc lymphomas by conditional deletion of a single remaining functional A1 gene in transplanted tumour cells.
Results
A1 expression in mouse lymphoid tumours
Prior to commencing this study, mouse haemopoietic tumours of varying genetic provenance were surveyed for expression of A1 protein, including Eµ-Myc lymphomas, Eµ-Myc/Eµ-Bcl-2 progenitor cell tumours, vavP-MYC T cell lymphomas, Eµ-v-Abl plasmacytomas, p53−/− T lymphomas and MLL-AF9 and AML-ETO myeloid leukaemias (Supplementary Figure S1 and data not shown). Many of the Eµ-Myc lymphomas had readily detectable A1 (Supplementary Figure S1a) but levels were low in most other tumour types except for vavP-MYC T cell lymphomas and some Eµ-v-Abl plasmacytomas. We therefore decided to focus on the Eµ-Myc model to investigate the impact of loss of A1 on tumorigenesis.
Loss of endogenous A1 has no impact on tumour development in Eµ-Myc mice
To generate A1−/− Eµ-Myc mice, two independent lines of A1−/− mice (A1–1 and A1–2) [44] were crossed with Eµ-Myc mice [22, 23] (all on a C57BL/6 background) and A1+/− Eµ-Myc offspring were then interbred. Cohorts of A1+/+, A1+/− and A1−/− Eµ-Myc mice were then monitored for tumour development and euthanased at ethical endpoint. No significant differences were detected between the A1–1 and A1–2 cohorts (Supplementary Figure S2a) or between males and females (not shown) and so the results have been pooled in all subsequent Figures.
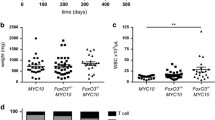
Neither heterozygous nor homozygous loss of A1 had any significant impact on the kinetics of morbidity (Fig. 1a). Phenotypic analysis showed that, as for A1+/+ Eµ-Myc mice, all the tumours in the A1+/− Eµ-Myc and A1−/− Eµ-Myc mice were either B220+sIg−(denoted by pro/pre-B), B220+sIg+ (denoted by B) or a mixture of these phenotypes (denoted by mixed). No significant differences were apparent between genotypes in either the incidence (Fig. 1b) or kinetics (Fig. 1c) of lymphoma type. Furthermore, when sick mice were autopsied, there were no significant differences in the leukaemic burden in the blood and haemopoietic organs (Fig. 1d, Supplementary Figure S2b).

Loss of A1 does not perturb lymphomagenesis in Eµ-Myc mice. a Kaplan–Meier survival curves for A1+/+ Eµ-Myc (n = 27, median survival of 92 days), A1+/− Eµ-Myc (n = 64, median survival of 95 days) and A1−/− Eµ-Myc (n = 49, median survival of 94 days) mice. Data for A1-1 and A1-2 cohorts (Supplementary Figure S2a) have been pooled. The difference in survival between the genotypes was not significant (log−rank test). b Stacked bar graph showing the percentages of pro/pre-B (B220+sIg-), B (B220+sIg+) and mixed pre-B/B cell lymphomas observed for A1+/+ Eµ-Myc (n = 21), A1+/− Eµ-Myc (n = 31) and A1-/- Eµ-Myc (n = 36) mice. c Kaplan–Meier survival curves for pro/pre-B and B lymphoma development in A1+/+ Eµ-Myc (n = 24), A1+/− Eµ-Myc (n = 34) and A1−/− Eµ-Myc (n = 40) mice. Only mice for which immunophenotyping was performed, or that survived until the endpoint, are included in these plots. No significant differences were apparent between genotypes in either the incidence (b) or kinetics (c) of pro/preB and B lymphomas. d Scatter plots of white blood cell counts (WBC) and weights of spleen, lymph nodes (inguinal, axillary and brachial lymph nodes) and thymus of autopsied sick A1+/+ Eµ-Myc, A1+/− Eµ-Myc and A1−/− Eµ-Myc mice that had reached ethical endpoint. Bars represent mean ± SEM. No significant differences were found (Student’s T-test)
Eµ-Myc-driven tumour development is believed to initiate in the expanded pool of pro and pre-B cells in the bone marrow and spleen of young mice. [23] To ascertain whether loss of A1 had perturbed the premalignant phenotype, we analysed haemopoietic tissues of healthy young 4-week-old mice using immunostaining and flow cytometry. Consistent with the unchanged kinetics of tumour development, the A1+/+ Eµ-Myc and A1-/- Eµ-Myc mice had comparable numbers of B220+ sIg− cells in the bone marrow and spleen (Fig. 2, Supplementary Table S1). sIg+ B cells and all other major cell populations were also comparable. There were no notable differences between the A1+/+ and A1-/- non-transgenic mice, as also reported elsewhere. [44]

Loss of A1 does not perturb the premalignant phenotype in Eµ-Myc mice. Analysis of spleens of healthy A1+/+, A1+/−, A1−/−, A1+/+ Eµ-Myc, A1+/− Eµ-Myc and A1−/− Eµ-Myc mice (n = 8–9 per genotype, sex matched) euthanased at 28–30 days of age. The total cellularity and compositional analysis of B, T and myeloid cells are shown. Data for bone marrow, blood, lymph nodes and thymus are presented in Supplementary Table S1. Bars represent mean ± SEM. * P < 0.05. No significant differences in the pre-neoplastic phenotype were found between A1+/+ Eµ-Myc and A1−/− Eµ-Myc mice (Ordinary one-way ANOVA, Tukey’s multiple comparisons test)
Immature B lymphoid cells overexpressing MYC are highly susceptible to apoptosis [11,12,13,14]. To determine whether loss of A1 further increased their vulnerability, pro/pre-B cells (B220+sIg−) were isolated from bone marrow using flow cytometry and cultured in vitro without any added cytokines. Analysis over the 48 h-period showed no increase in susceptibility to apoptosis (Fig. 3a).

Loss of A1 does not enhance apoptosis of pre-leukaemic pro/pre-B cells in vitro. a Viability (%) of bone marrow B220+sIg- cells obtained by FACS sorting from healthy (28–30 day-old) A1+/+ Eµ-Myc and A1−/− Eµ-Myc mice (n = 3–6 per genotype) and cultured for 48 h without added cytokines. Viability was measured at 0, 4, 8, 24 and 48 h by flow cytometry after staining with propidium iodide (PI) and Annexin V. Bars represent mean viability (PI- Annexin V−) ± SEM. No significant differences were found between A1+/+ Eµ-Myc (light grey bars) and A1−/− Eµ-Myc (dark grey bars) cells (multiple Student’s T-tests). b Analysis of A1 expression in bone marrow pro/pre-B (B220+sIg−) and splenic B (B220+sIg+) cells obtained by FACS sorting from healthy young A1+/+, A1−/−, A1+/+ Eµ-Myc, and A1−/− Eµ-Myc mice. Cell lysates (15 µg) from independent mice were run in each lane. Data are representative of two independent experiments. Molecular weight markers are indicated (kD). The immature B cell WEHI-231 line was used as a positive control for A1; treatment with cycloheximide (CHX) resulted in loss of A1 due to its short half-life
A1 expression was analysed in pre-malignant pro/pre-B and B cells from healthy young WT and Eµ-Myc mice by western blot analysis (Fig. 3b). A1 was readily apparent in splenic B cells (B220+sIg+), being higher in the WT than the Eµ-Myc transgenic cells. However, it was not detectable in bone marrow pro/pre-B cells (B220+sIg−) of either genotype. These observations differ from those of Sochalska et al. [46] who, using the same antibody, detected A1 in pre-B as well as B cells and at higher levels in Eµ-Myc than WT populations. The reason for the difference is not clear but could be related to genetic background or other differences (see Discussion). The lack of significant A1 expression in pro/pre-B lymphoid cells in our Eµ-Myc mice would explain why A1+/+ and A1−/− Eµ-Myc pro/pre-B cells did not differ in their sensitivity to apoptosis (Fig. 3a). As previously reported [47], MCL-1 expression was elevated in the pre-leukaemic Eµ-Myc cells, particularly pro/pre-B cells, where MYC levels are higher than those in sIg+ B cells (Fig. 3b). Loss of A1 did not alter the level of MCL-1 or MYC within comparable cell populations.
To investigate whether changes in the expression of other BCL-2 family members or mutation of the p19ARF/p53 pathway might have compensated for the absence of A1 during lymphomagenesis, western blot analysis was performed. A1 protein was readily detected, at varying levels, in pro/pre-B, B and mixed lymphomas taken from lymph nodes of A1+/+ Eµ-Myc mice (Fig. 4a, b and Supplementary Figure S2c). As in Fig. 3b, however, neither of the control A1+/+ Eµ-Myc premalignant pro/pre-B samples (CD19+ cells from bone marrow) expressed A1 at detectable levels. Thus, either A1 levels increase in pro/pre-B cells during malignant transformation or its expression is dependent on the microenvironment.

Expression of BCL-2 protein family members in A1+/+ Eµ-Myc and A1−/− Eµ-Myc lymphomas and premalignant cells. Western blots were performed on a pro/pre-B cell lymphomas from lymph nodes of sick A1+/+ Eµ-Myc (lanes 1–4) and A1−/− Eµ-Myc mice (lanes 5–8) and pre-malignant pro/pre-B cells (MACS-sorted CD19+ bone marrow (BM) samples) from healthy A1+/+ Eµ-Myc and A1−/− Eµ-Myc mice euthanased at 28–30 days of age (lanes 9–12); b B cell lymphomas from lymph nodes of A1+/+ Eµ-Myc (lanes 1–4) and A1−/− Eµ-Myc mice (lanes 5–8). The same premalignant samples and control lysates as in (a) were run to enable comparison between (a) and (b). Lane labels indicate the individual mouse number. WEHI-231 cells served as a positive control for A1 protein, tumour 1/8094 as a positive control for p53 and p19ARF proteins and β-ACTIN as a loading control. Independent blots are separated by a horizontal black line and molecular weight markers (kD) are indicated. Additional lymphomas were analysed (data not shown), in total: 8 pro/pre-B and 8 B A1+/+Eµ-Myc; 7 pro/pre-B and 6 B A1−/−Eµ-Myc
MCL-1 was clearly present in all tumours (16/16) (Fig. 4) but, perhaps counter-intuitively, was lower in the A1-/- than the A1+/+ tumours. BCL-2 was readily detectable in 15/16 tumours, at variable levels, and low in all pre-leukaemic samples. BCL-XL was low in all samples except for two A1+/+ Eµ-Myc B lymphomas. Overall, there was no consistent difference in the expression pattern for A1-positive vs. A1-negative cells, for either the pro-survival proteins or for BH3-only proteins PUMA and BIM.
About 30% of Eµ-Myc tumours carry mutations in the p53 pathway [12, 13]. Consistent with this, p53 and/or high p19ARF was evident in 2 of the 16 lymphomas analysed (Fig. 4), one being A1−/− and the other A1+/+.
Taken together, these observations indicate that absence of endogenous A1 has no impact on the kinetics or phenotype of tumour development in Eµ-Myc mice. Although no consistent compensatory changes were detected, this does not rule out the possibility that individual tumours have compensated for the absence of A1 by modulating the expression of different pro- and anti-apoptosis genes.
Loss of A1 confers no disadvantage during competitive repopulation
We next compared the competitive ability of A1+/+ and A1−/− haemopoietic stem and progenitor cells (HSPCs) from Eµ-Myc mice in a bone marrow reconstitution assay. We used UBC-GFP/Eµ-Myc as competitor bone marrow cells mixed in a 1:1 ratio with either A1+/+ Eµ-Myc or A1−/− Eµ-Myc bone marrow cells. The donor cells, which were all Ly5.2, were transplanted into lethally irradiated Ly5.1 recipients and blood was analysed by flow cytometry 6 weeks later. Fig. 5 shows that loss of A1 did not impair the haemopoietic repopulation capacity of Eµ-Myc HSPCs: those from A1−/− Eµ-Myc mice were as competitive as those from A1+/+ Eµ-Myc mice, as both genotypes (GFP−) constituted ~50% of the Ly5.2+ cells in all cell populations analysed, including pre-B and B cells.

A1−/− Eµ-Myc haemopoietic stem and progenitor cells are as competitive as A1+/+ Eµ-Myc cells in reconstituting haemopoiesis. Bone marrow competitive reconstitution experiments were set up using UBC-GFP/Eµ-Myc cells as the competitor. For analysis these can be distinguished by GFP expression from the A1+/+ Eµ-Myc and A1−/− Eµ-Myc test bone marrow cells, and by Ly5.2 expression from the Ly5.1 recipients. For each experiment UBC-GFP/Eµ-Myc cells were mixed in a 1:1 ratio with either A1+/+ Eµ-Myc or A1−/− Eµ-Myc cells and a total of 3 × 106 cells transplanted into three lethally irradiated Ly5.1 recipients. Six weeks post reconstitution, mice were bled and analysed for GFP, Ly5.2, B220, IgM, IgD, CD4, CD8, Mac1 and Gr1 expression by flow cytometry. The data shown here represents mean ± SEM of 6 independent experiments. Data plotted is % GFP−of total Ly5.2+, so is a comparison of the test bone marrow vs only UBC-GFP/Eµ-Myc (Ly5.1 cells have been excluded)
Impact of loss of A1 in transplanted lymphomas
Finally, to ascertain whether A1 plays a role in supporting expansion of fully malignant MYC-driven cells in vivo, we undertook conditional deletion in transplanted Eµ-Myc tumours, the strategy used to establish the essential role of MCL-1 for the survival of Eµ-Myc lymphoma cells in vivo [32]. To do so, we first generated Eµ-Myc lymphomas carrying floxed A1-b alleles and an inducible Cre gene, by crossing Eµ-Myc mice with A1-a−/− A1-bfl/fl A1-d−/− (hereafter A1fl/fl) mice [44] and Rosa26CreERT2 mice (hereafter CreERT2) [48], which express Cre recombinase that is inactive in the absence of tamoxifen (see Materials and Methods).
When we compared A1 expression levels in premalignant B lymphoid cells from healthy young 4-week-old mice (Supplementary Figure S3a), perhaps not surprisingly, A1 expression was substantially less in the premalignant B cells of A1fl/fl (ie A1-a−/− A1-bfl/fl A1-d−/−) Eµ-MycCreERT2 mice compared to those from A1+/+ Eµ-MycCreERT2 mice and the pro/pre-B cells did not detectably express A1, consistent with the data presented above for premalignant Eµ-Myc pro/pre-B cells (Figs 3b, 4).
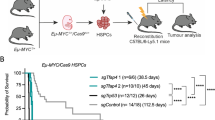
Lymphomas arose with comparable kinetics in each of the Eµ-Myc genotypes: A1+/+ Eµ-Myc, A1+/+ Eµ-MycCreERT2, A1fl/+ Eµ-MycCreERT2 and A1fl/fl Eµ-MycCreERT2 (all Ly5.2+) (Supplementary Figure S3b). Multiple lymphomas of each genotype were transplanted intravenously into non-irradiated C57BL/6-Ly5.1 recipients (3 × 106 cells into each of 6 recipients per tumour). On days 5 and 6, three of the recipients were treated with tamoxifen by oral gavage and three received vehicle, then survival was monitored until ethical endpoint (Fig. 6a, Supplementary Figure S3c). Notably, mice transplanted with lymphomas carrying floxed A1-b alleles that were subsequently treated with tamoxifen (red) had a median survival of 6–7 days longer than those treated with vehicle (A1fl/+ Eµ-MycCreERT2, P = 0.0019 and A1fl/fl Eµ-MycCreERT2, P = 0.0075). The implication is that loss of A1 had impaired the expansion of the tumour cells by enhancing apoptosis.

Conditional deletion of floxed A1-b gene in transplanted Eµ-Myc lymphomas enhances survival of transplanted mice. a Survival curves of mice transplanted with Eµ-Myc-driven lymphomas and then treated with tamoxifen or vehicle. Each individual (Ly5.2+) tumour from female primary mice (Supplementary Figure S3b) was transplanted intravenously into 6 female Ly5.1+ recipients (3 × 106 cells/recipient), three of which received tamoxifen (200 mg/kg body weight) on d5 and d6 post-transplantation and three received vehicle alone. Mice transplanted with A1+/+ Eµ-Myc (n = 7 independent primary tumours) or A1+/+ Eµ-MycCreERT2 tumours (n = 7) showed no significant difference in survival between tamoxifen and vehicle treatment. However, mice transplanted with A1fl/+ Eµ-MycCreERT2 tumours (n = 8) survived significantly longer following tamoxifen treatment (median survival 14 d for vehicle-treated mice vs. 21 d for tamoxifen-treated mice; P = 0.0019, log-rank test), as did mice transplanted with A1fl/fl Eµ-MycCreERT2 tumours (n = 12) (median survival of 17 d for vehicle-treated mice vs. 23 d for tamoxifen-treated mice; P = 0.0075, log-rank test). Data for A1-1 and A1-2 cohorts have been pooled. (see Supplementary Figure S3C for the data divided into the individual A1 lines). b Analysis of lymphomas arising in the lymph nodes of transplant recipients after treatment with tamoxifen or vehicle. DNA was purified from sorted Ly5.2+ lymphoma cells (to exclude any recipient cells, which would not have floxed A1 alleles), after which PCR was performed to confirm the presence of Eµ-Myc, RosaCreERT2, and to determine which A1-b alleles were present (A1-d was also analysed as a control). The number of the primary tumour that was transplanted is indicated above the lanes, and whether the recipient had been treated with tamoxifen or vehicle is indicated as + or − respectively. The survival (in days) of recipients post transplantation is indicated below the blot. In all, 30 tumours were analysed by PCR (11 A1fl/+ Eµ-Myc CreERT2 and 19 A1fl/fl Eµ-Myc CreERT2 lymphomas) and in every case deletion of the floxed A1b alleles was very efficient following tamoxifen treatment
To ascertain whether tamoxifen had indeed induced deletion of the floxed A1-b gene, the tumours that eventually killed the recipients were analysed by PCR. For all tumours analysed (n = 30), A1-b floxed allele(s) were efficiently deleted by tamoxifen activation of CreERT2 (e.g. Fig. 6b).
Discussion
The development of lymphomas in Eµ-Myc mice is dependent on expression of BCL-2 family members MCL-1 [33], BCL-XL [35] and BCL-W[34] but not BCL-2 [36]. This dependence on pro-survival BCL-2 family members is attributed to the increased susceptibility to apoptosis of developing B lymphoid cells in Eµ-Myc mice [11, 12, 49].
Recently, Sochalska et al. [46] reported that knockdown of all A1 genes did not alter the kinetics of Eµ-Myc lymphomagenesis but, finding that pre-B cells with reduced A1 levels were underrepresented in haemopoietic organs and that the tumours that arose had escaped A1 knockdown, suggested a vital role for A1 in the development of Eµ-Myc lymphomas.
We found no evidence of a requirement for A1 during lymphoma development in Eµ-Myc mice. There was no difference in the onset of tumour-induced morbidity in A1 nullizygous vs. WT Eµ-Myc mice (Fig. 1) and neither was there any alteration in the number of premalignant B lymphoid cells (Fig. 2) or in the repopulating capacity of A1-/- Eµ-Myc haemopoietic stem and progenitor cells (Fig. 5). It is not surprising that premalignant cell numbers were not altered by A1 deletion as we were unable to detect A1 expression in pre-B cells from either WT or Eµ-Myc mice (Fig. 3b). In contrast, A1 expression was readily detectable in pro/pre B lymphoma cells of A1+/+ Eµ-Myc mice (Fig. 4), consistent with the selective pressure observed by Sochalska et al. to maintain A1 during lymphomagenesis [46].
Differences found in the aspects of the two studies may reflect major differences in the experimental systems. We used germline deleted A1 knockout mice (A1-a−/− A1-b−/− A1-d−/−) [44] and, during their lengthy stepwise derivation, these mice may well have adapted to lack of A1. In contrast, Sochalska et al. used haemopoietic-specific mosaic expression of transgenic A1 shRNA to constitutively knock down all three functional A1 genes [46]. Furthermore, while the (VV-A1) mice generated by Sochalska et al. had been bred to C57BL/6 mice, residual genetic differences also seem possible, as the line was originally generated and maintained in a C57BL/6 x CBA F1 background [41]. Since A1 expression levels vary with the activation status of B cells [43, 50] differences in the pathogen load within the animal facilities may also have played a role. In this regard we note that the impaired early T-cell differentiation, B cell homoeostasis and granulopoiesis reported for constitutive A1 knockdown [41] were not observed in the A1 knockout mice derived in our animal facility [44].
The importance of individual pro-survival BCL-2 family members varies at different stages of Eµ-Myc lymphomagenesis. While BCL-XL expression helps prevent apoptosis during the development of Eµ-Myc lymphomas [35], it is dispensable for the sustained growth of fully malignant lymphoma cells in transplant recipients [32]. In contrast, MCL-1 is essential for Eµ-Myc lymphomas at both stages [32, 33]. Cognisant of such differences, we also assessed the requirement for A1 during the expansion of established Eµ-Myc lymphoma cells and observed a significant delay in tumour progression of transplanted A1fl/fl Eµ-Myc lymphoma cells following activation of Cre-mediated deletion (P = 0.0075, Fig. 6). In view of the possibility of adaptation to the lower level of A1 in A1-a−/−A1-bfl/flA1-c−/− Eµ-Myc mice (Supplementary Figure S3a), the impact of A1 loss might be even greater should it be possible to simultaneously delete all three functional alleles. Similarly, inhibition of BFL-1, the sole A1 analogue in humans, may have substantial impact.
In summary, taken together with the study by Sochalska et al. [46], our data suggest that, like MCL-1, BFL-1 is a potential target for the treatment of MYC-driven human tumours. A BFL-1-specific BH3 mimetic should be a useful avenue to pursue, since BFL-1 is overexpressed in a variety of haemopoietic and other malignancies: ALL, CLL, AML, DLBCL, melanoma, stomach and colon cancers and breast cancer [51].
Materials and methods
Mice
Experimental protocols involved in the use of mice were conducted according to the guidelines of the Animal Ethics Committee of the Walter and Eliza Hall Institute (WEHI). All mice were on a C57BL/6 background and bred at WEHI. To generate A1−/− Eµ-Myc mice, Eµ-Myc transgenic males [22] were crossed with two strains of A1−/− females (A1-1 and A1-2) [44], and offspring were interbred separately. A1fl/fl Eµ-MycCreERT2 mice were generated by interbreeding Eµ-Myc transgenic males with Rosa26CreERT2 females [48] and A1fl/fl (A1-a−/− A1-bfl/fl A1-c−/−) females (A1-1 and A1-2) followed by interbreeding of offspring. Genotyping was performed as previously described [44]. Tg(UBC-GFP) 30Scha/J female mice [52] were bred with Eµ-Myc transgenic males to produce double transgenic offspring. Cohorts of mice were aged to ethical end point or euthanased at 28–30 days of age for premalignant analysis. Ethical endpoint was determined independently by trained animal technicians; criteria included splenomegaly, lymphadenopathy, hind-limb paralysis, hunched stature, weight loss, laboured breathing.
Tumour analysis
Mice were euthanased according to the guidelines of the Institute’s Animal Ethics Committee. Haemopoietic organs (spleen, inguinal lymph nodes, axillary lymph nodes, brachial lymph nodes, mesenteric lymph node and thymus) were weighed and a peripheral blood sample was collected by either eye bleed or heart bleed. Blood counts and composition were determined using an ADVIA 2120 haematology analyser (Siemens, Erlangen, Germany). Cell suspensions were prepared from lymphomas and cryopreserved. Lymphomas were immunophenotyped by staining with α-B220-APC (clone RA3–6B2), α-IgM−PE (clone 5.1) and α-IgD−FITC (clone 11–26 C) produced in house. Cells were analysed on an LSR II flow cytometer (BD Biosciences) using FlowJo software Version 9.3.2 (TreeStar, Ashland, OR, USA).
Premalignant analysis
Healthy mice were euthanased at 28–30 days of age; their spleen, lymph nodes and thymus were collected and weighed; bone marrow (both femurs) and peripheral blood were also collected. Peripheral blood cell counts were enumerated using an ADVIA 2120 analyser (Siemens). Red blood cells were removed using 0.168 M ammonium chloride. Single cell suspensions were prepared from haemopoietic tissues and cell counts enumerated on a CASY Cell Counter (Scharfe System GmbH, Reutlingen, Germany). Cell composition was determined by immunostaining and flow cytometry (LSR II flow cytometer, BD Biosciences), using FlowJo software and the following fluorochrome-labelled surface marker-specific monoclonal antibodies produced in house: α-CD8-PE (clone YTS169); α-CD4-APC (clone H129.19); α-TCRβ-FITC (clone H57–597); α-B220-PE (clone RA3–6B2); α-IgM-FITC (clone 5.1); α-IgD-FITC (clone 11–26 C); α-IgD-PE; α-CD43-APC (clone S7); α-Mac1-PE (clone M1/70); α-Gr1-APC (clone RB6–8C5); α-Thy1-PE (clone T24/31); α-Ter119-APC.
Pro/pre-B cell survival assay
Bone marrow cells (10 × 106) from 4 week-old mice were stained by incubating with α-B220-PE, α-IgM-FITC and α-IgD-FITC, then washed and resuspended in 4 μg/mL propidium iodide (PI). Viable pro/pre-B cells (B220+IgM-IgD−PI−) were isolated by flow cytometry then cultured at 1 × 106 cells/mL in high-glucose Dulbecco’s Modified Eagle’s medium supplemented with 10% foetal calf serum, 50 μM 2-ME and 100 μM asparagine without additional cytokines to observe spontaneous death. Cell viability was determined at 0, 4, 8, 24, 48 and 72 h by staining with annexin-V-FITC and 4 μg/mL PI followed by analysis on an LSR II flow cytometer.
Western blot analysis
Western blots were performed according to standard procedures using protein lysates prepared from cryopreserved cell pellets using RIPA buffer (300 mM NaCl, 2% IGEPAL CA-630, 1% deoxycholic acid, 0.2% SDS, 100 mM Tris-HCl pH 8.0) containing complete ULTRA protease inhibitors (Roche, Basel, Switzerland). Protein concentration was determined by Bradford assay. Samples (15–20 μg total protein) were run on NuPAGE Bis-Tris gels (Life Technologies) and transferred to nitrocellulose membranes with an iBlot (Life Technologies) according to the manufacturer’s protocol. Membranes were subsequently probed with the following antibodies: A1 (clone 6D6, WEHI mAB lab), BCL-2 (clone 7, BD Biosciences), BCL-XL (clone 44, BD Biosciences), MCL-1 (clone 19C4–15, WEHI mAb lab), PUMA (polyclonal, Abcam), BIM (clone 3C5, WEHI mAb lab), p53 (FL-393, Santa Cruz Biotechnology, Santa Cruz, CA, USA), p19ARF (p19ARF exon 2, Rockland, Gilbertsville, PA, USA), c-MYC (D84C12, Cell Signaling Techology, Danvers, MA, USA) and β-ACTIN (clone AC-74, Sigma-Aldrich). Blots were visualised using LuminataTM Forte western HRP substrate (Merck-Millipore) on a ChemiDoc Touch (Bio-Rad, Hercules, CA, USA) and analysed using Image Lab software (Bio-Rad).
Haemopoietic competitive reconstitution
Bone marrow was collected from 4-week-old female UBC-GFP/Eµ-Myc, A1+/+ Eµ-Myc and A1−/− Eµ-Myc mice and resuspended in phosphate buffered saline to 15 × 106/mL. UBC-GFP/Eµ-Myc cells were mixed at a 1:1 ratio with A1+/+ Eµ-Myc or A1−/− Eµ-Myc cells and 3 × 106 cells were injected into lethally irradiated (2 × 5.5 Gy spaced by 2 h) C57BL/6-Ly5.1 mice. For each competitive bone marrow mixture, 3 recipient mice were used. To prevent infections, transplanted animals were initially provided with water containing neomycin (Sigma). Six weeks later, when their haemopoietic system had re-established, blood was collected from the retro-orbital plexus for ADVIA and FACS analysis.
Conditional A1 deletion in transplanted lymphomas
Lymphomas originating from A1+/+ Eµ-Myc, A1+/+ Eµ-MycCreERT2, A1fl/+ Eµ-MycCreERT2 and A1fl/fl Eµ-MycCreERT2 female mice were cryopreserved as cell suspensions for later use. Tumour cells were thawed and resuspended at 15 × 106 cells/mL in PBS, then 3 × 106 cells (200 µL) injected into the tail veins of 6 female C57BL/6-Ly5.1 recipient mice (unirradiated). On d5 and d6 post-transplantation, 3 out of 6 mice were treated with 200 mg tamoxifen/kg body weight (Sigma-Aldrich) in peanut oil/10% ethanol by oral gavage, while the remaining 3 mice received only vehicle (peanut oil/10% ethanol). Any transplantations deemed unsuccessful (i.e. the vehicle-treated mice did not all become sick at a similar time) were excluded from analysis. When transplanted mice reached ethical endpoint, they were euthanased and their lymphomas cryopreserved as single-cell suspensions for later use. Lymphoma cells were thawed and stained with α-Ly5.1-APC (A20.1) and α-Ly5.2-PE (S-450–15.2). Viable donor-derived (Ly5.2+PI−) tumour cells were purified by flow cytometry and DNA was isolated using a DNeasy Qiagen kit and analysed for A1-b, A1-d, Eµ-Myc and Rosa26CreER genes by PCR and gel electrophoresis.
Statistical analysis
GraphPad Prism (GraphPad Software Inc.) was used to graph and statistically analyse data. For analysis of Kaplan–Meier mouse survival curves, significance was determined using the log−rank (Mantel-Cox) test. Ordinary one-way ANOVA with Tukey’s multiple comparisons test or Student’s T-test was used for statistical analysis; P values <0.05 were considered to be statistically significant.
References
Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–66.
Diolaiti D, McFerrin L, Carroll PA, Eisenman RN. Functional interactions among members of the MAX and MLX transcriptional network during oncogenesis. Biochim Biophys Acta. 2015;1849:484–500.
Tu WB, Helander S, Pilstal R, Hickman KA, Lourenco C, Jurisica I, et al. Myc and its interactors take shape. Biochim Biophys Acta. 2015;1849:469–83.
Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35.
Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–22.
Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28.
Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonnette RP, Green DR. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science. 1992;257:212–4.
Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30.
Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2.
Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–3.
Strasser A, Elefanty AG, Harris AW, Cory S. Progenitor tumours from Em-bcl-2-myc transgenic mice have lymphomyeloid differentiation potential and reveal developmental differences in cell survival. EMBO J. 1996;15:3823–34.
Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999;13:2658–69.
Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. 2004;101:6164–9.
Michalak EM, Jansen ES, Happo L, Cragg MS, Tai L, Smyth GK, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16:684–96.
Cory S, Roberts A, Colman PM, Adams JM. Targeting BCL-2-like proteins to kill cancer cells. Trends Cancer. 2016;2:443–60.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63.
Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Biochem Sci. 2014;39:101–11.
Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–31.
Brouwer JM, Westphal D, Dewson G, Robin AY, Uren RT, Bartolo R, et al. Bak core and latch domains separate during activation, and freed core domains form symmetric homodimers. Mol Cell. 2014;55:938–46.
Dewson G, Ma S, Frederick P, Hockings C, Tan I, Kratina T, et al. Bax dimerizes via a symmetric BH3:groove interface during apoptosis. Cell Death Differ. 2012;19:661–70.
Cory S. Activation of cellular oncogenes in hemopoietic cells by chromosome translocation. Adv Cancer Res. 1986;47:189–234.
Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–8.
Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eµ-myc transgenic mouse: a model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–71.
Langdon WY, Harris AW, Cory S, Adams JM. The c-myc oncogene perturbs B lymphocyte development in Eµ-myc transgenic mice. Cell. 1986;47:11–18.
Alexander WS, Bernard O, Cory S, Adams JM. Lymphomagenesis in Em-myc transgenic mice can involve ras mutations. Oncogene. 1989;4:575–81.
Swanson PJ, Kuslak SL, Fang W, Tze L, Gaffney P, Selby S, et al. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J Immunol. 2004;172:6684–91.
Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, et al. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood. 2010;116:3197–207.
Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW. Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci USA. 2004;101:9333–8.
Frenzel A, Labi V, Chmelewskij W, Ploner C, Geley S, Fiegl H, et al. Suppression of B-cell lymphomagenesis by the BH3-only proteins Bmf and Bad. Blood. 2010;115:995–1005.
Certo M, Moore Vdel G, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65.
Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117:112–21.
Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014;28:58–70.
Grabow S, Delbridge AR, Aubrey BJ, Vandenberg CJ, Strasser A. Loss of a single Mcl-1 allele inhibits MYC-driven lymphomagenesis by sensitizing pro-B cells to apoptosis. Cell Rep. 2016;14:2337–47.
Adams CM, Kim AS, Mitra R, Choi JK, Gong JZ, Eischen CM. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J Clin Invest. 2017;127:635–50.
Kelly PN, Grabow S, Delbridge ARD, Strasser A, Adams JM. Endogenous Bcl-xL is essential for Myc-driven lymphomagenesis in mice. Blood. 2011;118:6380–6.
Kelly PN, Puthalakath H, Adams JM, Strasser A. Endogenous bcl-2 is not required for the development of Eµ-myc-induced B-cell lymphoma. Blood. 2007;109:4907–13.
Hatakeyama S, Hamasaki A, Negishi I, Loh DY, Sendo F, Nakayama K. Multiple gene duplication and expression of mouse bcl-2-related genes, A1. Int Immunol. 1998;10:631–7.
Ottina E, Tischner D, Herold MJ, Villunger A. A1/Bfl-1 in leukocyte development and cell death. Exp Cell Res. 2012;318:1291–303.
Hamasaki A, Sendo F, Nakayama K, Ishida N, Negishi I, Nakayama K-I, et al. Accelerated neutrophil apoptosis in mice lacking A1-a, a subtype of the bcl-2-related A1 gene. J Exp Med. 1998;188:1985–92.
Xiang Z, Ahmed AA, Moller C, Nakayama K, Hatakeyama S, Nilsson G. Essential role of the prosurvival bcl-2 homologue A1 in mast cell survival after allergic activation. J Exp Med. 2001;194:1561–9.
Ottina E, Grespi F, Tischner D, Soratroi C, Geley S, Ploner A, et al. Targeting antiapoptotic A1/Bfl-1 by in vivo RNAi reveals multiple roles in leukocyte development in mice. Blood. 2012;119:6032–42.
Ottina E, Pellegrini M, Villunger A. Guarding effector T-cell survival: all for one, Mcl-1 for all? Cell Death Differ. 2013;20:969–71.
Sochalska M, Ottina E, Tuzlak S, Herzog S, Herold M, Villunger A. Conditional knockdown of BCL2A1 reveals rate-limiting roles in BCR-dependent B-cell survival. Cell Death Differ. 2016;23:628–39.
Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE, et al. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ. 2017;24:534–45.
Tuzlak S, Schenk RL, Vasanthakumar A, Preston SP, Haschka MD, Zotos D, et al. The BCL-2 pro-survival protein A1 is dispensable for T cell homeostasis on viral infection. Cell Death Differ. 2017;24:523–33.
Sochalska M, Schuler F, Weiss JG, Prchal-Murphy M, Sexl V, Villunger A. MYC selects against reduced BCL2A1/A1 protein expression during B cell lymphomagenesis. Oncogene. 2017;36:2066–73.
Mason KD, Vandenberg CJ, Scott CL, Wei AH, Cory S, Huang DC, et al. In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proc Natl Acad Sci USA. 2008;105:17961–6.
Seibler J, Zevnik B, Kuter-Luks B, Andreas S, Kern H, Hennek T, et al. Rapid generation of inducible mouse mutants. Nucl Acid Res. 2003;31:e12.
Jacobsen KA, Prasad VS, Sidman CL, Osmond DG. Apoptosis and macrophage-mediated deletion of precursor B cells in the bone marrow of Em-myc transgenic mice. Blood. 1994;84:2784–94.
Trescol-Biemont MC, Verschelde C, Cottalorda A, Bonnefoy-Berard N. Regulation of A1/Bfl-1 expression in peripheral splenic B cells. Biochimie. 2004;86:287–94.
Vogler M. BCL2A1: the underdog in the BCL2 family. Cell Death Differ. 2012;19:67–74.
Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–22.
Acknowledgements
We thank K Hughes, C D’Alessandro, G Siciliano, J Corbin, J McManus and T Nikolaou for technical assistance; K Campbell, A Delbridge and S Glaser for tumour samples; and the institute’s flow cytometry facility for skilled support. This work was supported by funding from the NHMRC (Australia) program grant 1016701; US Leukaemia and Lymphoma Society Specialized Center for Research Grant 7001–13; and infrastructure support to the institute from the NHMRC Independent Research Institute Infrastructure Support Scheme (IRISS 9000220) and the Victorian State Government Operational Infrastructure Support (OIS).
Author contributions
CJV and SC conceived the studies, planned experiments, analysed the data and wrote the manuscript. MM, NSA, MR and CJV performed the experiments and analysed the data. RLS and MJH provided the mice and intellectual input.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Suzanne Cory and Cassandra J. Vandenberg are Joint senior authors.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mensink, M., Anstee, N.S., Robati, M. et al. Anti-apoptotic A1 is not essential for lymphoma development in Eµ-Myc mice but helps sustain transplanted Eµ-Myc tumour cells. Cell Death Differ 25, 797–808 (2018). https://doi.org/10.1038/s41418-017-0045-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-017-0045-8
- Springer Nature Limited