
Abstract
Pro-apoptotic multi-domain proteins of the BCL2 family such as BAX and BAK are well known for their important role in the induction of mitochondrial outer membrane permeabilization (MOMP), which is the rate-limiting step of the intrinsic pathway of apoptosis. Human or mouse cells lacking both BAX and BAK (due to a double knockout, DKO) are notoriously resistant to MOMP and cell death induction. Here we report the surprising finding that BAX/BAK DKO cells proliferate less than control cells expressing both BAX and BAK (or either BAX or BAK) when they are driven into tetraploidy by transient exposure to the microtubule inhibitor nocodazole. Mechanistically, in contrast to their BAX/BAK-sufficient controls, tetraploid DKO cells activate a senescent program, as indicated by the overexpression of several cyclin-dependent kinase inhibitors and the activation of β-galactosidase. Moreover, DKO cells manifest alterations in ionomycin-mobilizable endoplasmic reticulum (ER) Ca2+ stores and store-operated Ca2+ entry that are affected by tetraploidization. DKO cells manifested reduced expression of endogenous sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (Serca2a) and transfection-enforced reintroduction of Serca2a, or reintroduction of an ER-targeted variant of BAK into DKO cells reestablished the same pattern of Ca2+ fluxes as observed in BAX/BAK-sufficient control cells. Serca2a reexpression and ER-targeted BAK also abolished the tetraploidy-induced senescence of DKO cells, placing ER Ca2+ fluxes downstream of the regulation of senescence by BAX/BAK. In conclusion, it appears that BAX/BAK prevent the induction of a tetraploidization-associated senescence program. Speculatively, this may contribute to the low incidence of cancers in BAX/BAK DKO mice and explain why human cancers rarely lose the expression of both BAX and BAK.
Similar content being viewed by others

Introduction
BCL2 (B-cell lymphoma 2) family is known for their role in regulating the intrinsic (mitochondrial) pathway of apoptosis. According to their domain structure (organized around so-called BCL2 homology [BH] domains) and their functional contribution to mitochondrial outer membrane permeabilization (MOMP), which can be MOMP stimulatory (pro-apoptotic) or MOMP inhibitory (anti-apoptotic), the BCL2 proteins are classified into a multi-domain anti-apoptotic subfamily (BCL2 itself, BCL1L1, BCLW, and MCL1, which all have the full set of BH1 + BH2 + BH3 + BH4 domains), a multi-domain pro-apoptotic subfamily (BCL2-associated X protein (BAX), BAK, and BOK, which all have BH1 + BH2 + BH3 domains but lack BH4), and the so-called BH3-only proteins, which are all pro-apoptotic [1, 2]. Recent years have witnessed an ever-more-profound depth of insights into the dynamic structural alterations induced by the protein–protein interactions of these molecules [3, 4].
Although much work has helped to elucidate the detailed mechanisms through which BAX/BAK/BOK insert into the outer mitochondrial membrane to form higher-order oligomeric structure facilitating MOMP, how BCL2/BCL1L1/BCLW/MCL1 prevent MOMP, and how BH3-only proteins facilitate MOMP (either by the activation of BAX/BAK/BOK or the inhibition of BCL2/BCL1L1/BCLW/MCL1) [5, 6], it has become clear that these proteins have important functions in regulating other cellular functions [7]. Thus, BCL2 proteins have an impact on cellular metabolism (especially at the level of bioenergetics) [8], affect Ca2+ fluxes (especially at the level of the endoplasmic reticulum, ER) [9, 10], influence the advancement of the cell cycle [11], and regulate macroautophagy [12], just to mention a few among the “moonlighting” (non-MOMP-related) functions of the BCL2 family.
Members of the Bcl-2 family have been proposed to be regulators of intracellular Ca2+ signaling in cell survival and cell death by regulating Ca2+ transport systems located in the ER and mitochondria membranes, and at the plasma membrane [13]. It has been widely demonstrated that Ca2+ is involved in cell cycle progression [14,15,16]. Ca2+ controls the cell cycle mainly through the Ca2+/calmodulin complex that regulates the expression of cyclin-dependent protein kinases (CDKs), which, in turn, control the cell cycle. However, Ca2+ fluxes undergo alterations related to checkpoints found throughout the cell cycle [17, 18]. Errors occurring during the cell cycle are detected by these checkpoints leading to cell cycle arrest or even cell death [19]. The cell cycle arrest may be temporary or permanent, depending on whether the cellular machinery can repair the failure or not. Ca2+ signaling also participates in the re-entry of arrested cells (G0) into the cell cycle by the activation of transcription factors such as FOS, JUN, cyclic AMP-response element, and serum response element [16].
BAX and BAK localize at both the mitochondria and ER. Cells deficient for Bax, Bak (double knockout, DKO) display lowered steady-state ER Ca2+ concentrations ([Ca2+]ER) and secondarily decreased mitochondrial Ca2+ uptake [20]. DKO cells show reduced ER Ca2+ levels due to increased Ca2+ leakage and increased Ca2+ permeability due to the hyperphosphorylated state of the IP3R1. Members of the BCL2 family regulate IP3R1 phosphorylation, thereby controlling Ca2+ leakage from the ER. At the ER, BCL2 and IP3R1 interact, and this binding is increased in the absence of BAX and BAK [21]. On the other hand, expression of sarcoplasmic/ER Ca2+ ATPase 2a (Serca2a) restores [Ca2+]ER and mitochondrial Ca2+ uptake in DKO cells reestablishing the mitochondrial pathway of apoptosis [22]. Moreover, DKO cells exclusively expressing BAK in the ER can undergo apoptosis independently of the canonical BAX, BAK-dependent mitochondrial apoptotic pathway [23]. Therefore, there is clear evidence that BAX and BAK somehow control Ca2+ flux. Multiple studies have investigated the functional role of BAX and BAK in Ca2+ control. In healthy cells, BAX and BAK adopt a monomer conformation in the mitochondrial outer membrane. Various stress signals induce a conformational change of these proteins by assembling into oligomeric complexes giving rise to pores of different sizes leading to MOMP during apoptosis [24,25,26]. In addition, ER stress inducers can lead to conformational changes and oligomerization of BAX and BAK on the ER, similar to the mitochondria [27]. BAX inhibitor-1 is a Ca2+ leak channel involved in the ER stress response [28,29,30,31]. It remains to be clarified whether these pores directly account for the passage of Ca2+ from the ER to the cytosol during apoptosis.
Of note, the DKO of BAX and BAK (and, more recently, the triple knockout [TKO] of BAX, BAK, and BOK) have been extensively studied to reveal the essential role of these proteins in the activation of MOMP and hence the intrinsic pathway of apoptosis [26, 32,33,34]. Nonetheless, it turned that BAX/BAK DKO cells do succumb in response to specific cell death stimuli, as exemplified by granzyme B [35], chelethrine [36], N-(3-oxododecanoyl)-homoserine lactone [37], or combined inhibition of glutathione and thioredoxin metabolism [38]. Although it can be argued that these cell death pathways do not rely on a physiological route towards MOMP, it appears that the cytoprotective action of BAX/BAK inactivation is not absolute. In this context, it appears intriguing that the phenotype of BAX/BAK and BAX/BAK/BOK-knockout mice is somehow deceptive. Indeed, although it has been speculated for long that apoptotic (developmental) cell death would play a major role in embryogenesis and fetal development [39,40,41,42,43], such mice are born with a clearly recognizable murine morphology (instead of forming amorphous cell masses) and—even though they contain supernumerary cells in multiple organ systems—rarely develop tumors [44, 45].
Intrigued by these observations, we tested a number of cell death-inducing chemotherapeutic drugs on BAX/BAK DKO cells. In this context, we observed that such cells do not tolerate tetraploidization in the sense that they undergo senescence in response to transient exposure to tetraploidy-inducing microtubule inhibitors. Here we describe this unsuspected phenotype and attempt to explain it in mechanistic terms.
Materials and methods
Materials
Unless otherwise indicated, the media and supplements used in cell culture were purchased from Gibco–Thermo Fisher Scientific (Waltham, MA, USA), the chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA), and plasticware from Corning B.V. Life Sciences (Schiphol-Rijk, The Netherlands).
Antibodies
Rabbit monoclonal antibody (Ab) against Bax (#2772) and rabbit polyclonal Ab against Bak (#3814) were purchased from Cell Signaling Technology (Danvers, MA, USA). Mouse monoclonal Ab against p27 Kip1 (ab193379), rabbit monoclonal Abs against p16INK4 (ab51243), p21Cip1 (ab109199), p57 Kip2 (ab75974), and β-actin–horseradish peroxidase (HRP) (ab49900) were purchased from Abcam (Cambridge, UK). Alexa fluor secondary Abs were purchased from Thermo Fisher. HRP Abs were purchased from Southern Biotechnologies Associates (Birmingham, UK).
Cell lines and culture conditions
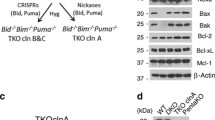
All cell lines were kept in culture at 37 °C under 5% of CO2. Wild type (WT) or genetically modified mouse embryonic fibroblasts (MEFs) for Bax−/−, Bak−/−, Bax/Bak1−/− (DKO), Pmaip1−/−, Bcl2l11−/−, Bbc3−/− [32], Bbc3−/−Pmaip1−/−, Bcl2l11−/−Bbc3−/−, Bcl2l11−/−Bid−/− [46], DKO expressing rabbit Serca2a (DKO-Sercaoe, [22], and DKO expressing Bak at the ER (DKO-BakER), provided by Hetz and colleagues [47], were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4500 mg L−1, 1 mM sodium pyruvate and supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES buffer, 100 U mL−1 penicillin sodium, and 100 μg mL−1 streptomycin sulfate. Immortalized MEFs derived from C57Bl/6 WT or Bax/Bak−/− mice, kindly provided by Strasser and colleagues [40], were cultured in DMEM containing 1000 mg L−1, 1 mM sodium pyruvate, and supplemented with 10% FBS, 50 mM 2-mercaptoethanol, 100 mM asparagine, and 100 U mL−1 penicillin sodium and 100 μg mL−1 streptomycin sulfate. Human colon carcinoma HCT116 cells (WT and DKO) were cultured in McCoy’s 5A (Modified) Medium supplemented with 10 % FBS, 1 mM sodium pyruvate, 10 mM HEPES, and 100 U mL−1 penicillin sodium and 100 μg mL−1 streptomycin sulfate.
Generation of HCT116 DKO cells
HCT116 WT cells were purchased from the American Type Culture Collection (Manassas, VA, USA). HCT116 BAX−/−BAK−/− (DKO) were generated from HCT116 BAX−/− kindly provided by Vogelstein and colleagues [48, 49]. HCT116 BAX−/− were transfected with Human BAK ZFN plasmid from Sigma-Aldrich (St Louis, MO, USA) following the manufacturer’s instructions. Transfected cells were cloned and characterized for BAX and BAK expression by immunoblotting.
RNA isolation and reverse-transcription PCR
Total RNA was extracted and purified using RNeasy mini kit from QIAGEN (Hilden, Germany). Genomic DNA was eliminated by RNase-free DNase I from Invitrogen (Carlsbad, CA, USA). RNA quantification was performed using the NanoDrop2000 (Thermo Fisher Scientific). Reverse transcription (RT) of total RNA (2.5 μg) was performed using SuperScript™ IV VILO™ Master Mix (Invitrogen). Murine Serca2a (mSerca2a) RNA expression was calculated by RT-quantitative real-time PCR (qPCR). cDNA was amplified with specific mSerca2a probe (Mm01201431, Thermo Fisher Scientific) using Taqman Fast Master Mix (Applied Biosystems, Foster City, CA, USA). qPCR was performed on the StepOne Real-time PCR system (Applied Biosystems) according to the manufacturer’s instructions. Relative gene expression was calculated according to the 2−ΔΔCt method, normalizing the level of mSerca2a to peptidylprolyl isomerase A mRNA, as endogenous control. Rabbit Serca2a (rbSerca2a) RNA expression was evaluated by RT semi-qPCR. rbSerca2a primers (Fwd: 5′-GGGCTGTCAACCAGGATA-3′; Rev: 5′-TGCAATGCAAATAAGGGA -3′; product length = 218 bp) were designed using Primer Premier 5 Design Program (Premier Biosoft International, San Francisco, CA, USA). β-Actin was used as internal control to normalized rbSerca2a expression (Fwd 5′-GCACCACACCTTCTACAATG -3′; Rev: 5′-TGCTTGCTGATCCACATCTG -3′; product length = 822). cDNA for rbSerca2a and β-actin were amplified using GoTaq® Hot Start Colorless Master Mix (Promega, Madison, WI, USA). PCR reactions were as follows: initial denaturation at 94 °C for 2 min, followed by 40 amplification cycles at 94 °C for 20 s during denaturation, annealing at 55 °C for 45 s, primer extension at 72 °C for 3 min, and final extension at 72 °C for 7 min. Amplified products were loaded on 2% agarose gels containing 10 μl of SYBR Green (Invitrogen). Images were acquired with the G:Box chemiluminescent system (GeneSys software) from Syngene (Cambridge, UK) and analyzed using the Image J software (National Institutes of Health, USA).
Cytofluorometry
Cell death
Cells were seeded in 12-well plates. Next day, cells were treated with the following pharmacological agents: cis-diaminodicloroplatino(II) (10, 20, and 40 μM), oxaliplatin (10, 25, and 50 μM), carboplatin (1, 100, and 200 μM), nocodazole (Noco) (50, 100, and 200 nM), paclitaxel (150, 300, and 600 nM), rotenone (1, 5, and 10 μM), RO3280 (5, 25, and 50 μM), AZD1152 (0.5, 5, and 50 μM), or M2I-1 (25, 50, and 100 μM). After 48 h of treatment, apoptosis was measured in unfixed cells co-stained with 40 nM 3,3’dihexiloxalocarbocyanine iodide (DiOC6(3)) (Life Technologies, Carlsbad, CA, Estados Unidos) to quantify mitochondrial transmembrane potential (ΔΨm) plus 1 μg/mL propidium iodide (PI) (Life Technologies) to identify plasma membrane breakdown.
Senescence
Cells were seeded in 12-well plates. Next day, cells were treated with Noco 100 nM or cytochalasin D (CytD) 1.2 μM. After 48 h of treatment or 48 h of treatment + 4 days in drug-free culture medium, cells were labeled with the β-galactosidase substrate 5-Dodecanoylaminofluorescein Di-β-d-Galactopyranoside (C12FDG) (Thermo Fisher Scientific) plus 1 μg/mL PI and 10 μM Hoechst 33342 (Life Technologies).
Cytofluorometric determinations were performed by means of an Attune® LSRII Fortessa flow cytometer (BD Biosciences) (San Jose, CA, USA). Data analyses were carried out by using the FlowJo software, upon gating on the events characterized by normal forward scatter and side scatter values.
Fluorescence imaging of cytosolic Ca2+
Cells were plated onto 12 mm ∅ coverslips previously coated with 0.01 mg/ml poly-l-lysine. Next day, cells were treated with Noco 100 nM. After 48 h of treatment, cells were loaded with 4 µM Fura2/AM (Life Technologies) for 1 h in external saline solution (ESS) containing (in mM): 145 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 Hepes/Na+ (pH 7.42). Cytosolic Ca2+ concentration ([Ca2+]cyt) was monitored as reported previously [50] by fluorescence imaging of cells using an OrcaER Hamamatsu digital camera (Hamamatsu Photonics, Hamamatsu, Japan) connected to an inverted Zeiss Axiovert microscope (Zeiss, Oberkochen, Germany), while being continuously perfused with ESS at 37 °C. Cells were epi-illuminated alternately at 340 and 380 nm using bandpass filters. Light emitted above 520 nm at both excitation lights was filtered by a dichroic mirror and collected every 5–10 s with a ×40, 1.4 numerical aperture, oil objective. To evaluate Ca2+ store content, cells were perfused with Ca2+-depleted ESS containing 0.5 mM of EGTA and 100 nM of the Ca2+ ionophore ionomycin. Intracellular free [Ca2+]cyt, rise in these conditions, reflects an estimation of the Ca2+ store content as previously reported [50]. For monitoring store-operated Ca2+ entry (SOCE), cells were treated with 10 μM cyclopiazonic acid for 10 min or 1 µM thapsigargin for 10 min in Ca2+-depleted ESS containing 0.5 mM of EGTA before the calcium imaging experiments, to deplete Ca2+ stores and to enable SOCE activation. Then, cells were perfused with 1 mM Ca2+-containing ESS to monitor [Ca2+]cyt increase that is an estimation of SOCE.
Clonogenicity assay
Cells were seeded in 175 cm2 flasks. After 24 h, cells were treated, or not, with Noco 100 nM or CytD 1.2 μM. After 48 h, cells were washed and kept in drug-free culture medium for 4 days more, followed by cloning cells. Prior to cloning, cells were labeled with 10 μM Hoechst 33342 for 1 h at 37 °C under 5% of CO2. Cells characterized by 2n or >4n DNA content were sorted on a FACs Influx cell sorter (BD Bioscience). Hoechst signal was read off the 405 nm laser, using the 460/50 nm detector. A control tube (cells without treatment) was used to position the gates for 2n and >4n. Then, 2n or >4n populations were sorted into 96-well plates (100 cells/well) and cultured for 2 weeks in drug-free culture medium. Then, wells showing cell proliferation were quantified.
Clonogenic assay
Cells were seeded in 6-well plates at 1000, 2000, 5000, 10,000, 50,000, or 100,000 cells per well. After 24 h, cells were treated with Noco 100 nM for 48 h. Then, cells were washed and cultured in drug-free medium for 1 week (plates with 10,000, 50,000, and 100,000 cells per well) or 2 weeks (plates with 1000, 2000, and 5000 cells per well). After 1 or 2 weeks, supernatant was removed and 500 μl of Crystal Violet (Sigma) was added. Crystal violet was discarded after 10 min and wells were washed with deionized water. Colony area was quantified through Image J “ColonyArea” Plugin [51].
Cell proliferation assay
Cells were prepared similarly to the clonogenicity assay. Cells characterized by >4n DNA content were sorted (300 cells/well in 96-well plates). Cell concentration was evaluated at 4, 6, 9, 11, and 14 days after sorting by flow cytometry (Attune® LSRII Fortessa).
Immunoblotting
Cells were seeded into 75 or 175 cm2 flasks for control and treated conditions, respectively. After 24 h, cells were treated with Noco 100 nM. After 48 h, control cells were collected and treated cells washed and kept in drug-free culture medium for 4 days more. Then, only attached cells were collected, washed with phosphate-buffered saline, their pellets were lysed, and proteins quantified following standard procedures. At least 20 μg of protein were loaded on Bis-Tris 4–12% pre-cast gels (Thermo Fisher) and transferred to nitrocellulose or polyvinyldifluoride membranes (Millipore, Bedford, USA). Membranes were incubated for 1 h in Tris-buffered saline (TBS)-Tween 20 (0.05%) supplemented with 5% non-fat powdered milk or bovine serum albumin to block nonspecific binding sites. Primary Abs were incubated overnight at 4 °C, detected with the appropriate HRP-labeled secondary Abs, and revealed with Amersham ECL+ (GE Healthcare, Little Chalfont, UK). β-Actin was used as protein loading control.
Statistical analysis
The experiments were repeated at least three times, which in our experience is optimal for obtaining significant results, with similar results. The specific n value for each experiment is specified in the corresponding figure. Outliers values were identified with the extreme studentized deviate method Grubb’s test by Graphpad. Given the variability in the size of the replicates, instead of using normal distribution test and equal variance test, we use two-sided (mixed) linear models. All statistical analysis from data without outliers were done within R environment [52]. We used the standard linear model inference (“lm” function) or the mixed model inference (nlme package) [53] and p-values estimations, according to the statistical question set in every figure legend. See Supplementary Table ST1 for details.
Results
Loss of clonogenicity of tetraploid Bax/Bak DKO cells
As compared to normal WT controls, Bax/Bak DKO MEFs (Supplementary Fig. S1A) are highly resistant against a variety of anticancer drugs, including the DNA-damaging platinum-based chemotherapeutics cisplatin, oxaliplatin, and carboplatin, as determined by cell staining with the mitochondrial transmembrane (ΔΨm)-sensitive fluorochrome DiOC6(3) and the vital dye PI to identify dying (DiOC6(3)low PI−) and dead (DiOC6(3)low PI+) cells (Fig. 1A–D). In contrast, Bax/Bak DKO are as susceptible as WT cells to killing by a variety of microtubular inhibitors such as Noco, paclitaxel, and high-dose rotenone (Fig. 1A, E–G), as well as by several agents that target the spindle checkpoint such as RO3380 (an inhibitor of polo-like kinase-1), AZD1152 (an inhibitor of aurora kinase B), and M2I-1 (an inhibitor of mitotic arrest deficient 2) (Fig. 1A, H–J). Similar results were obtained with BAX/BAK DKO human colon cancer HCT116 cells (Supplementary Fig. S1B), which are more resistant than WT cells in response to mitoxantrone, cisplatin, and oxaliplatin, similarly susceptible to CytD and only partially resistant to Noco, paclitaxel, docetaxel, and vinblastine (Supplementary Fig. S1C). Altogether, these results indicate that deletion of Bax/Bak (or BAX/BAK) confers a variable degree of resistance to anticancer drugs, in line with previously published studies [32, 54].

Wild type (WT) and DKO MEFs were cultured for 48 h in the absence (control, Co) or presence of 10, 20, and 40 μM cis-diaminodicloroplatino(II) (CDDP) (A, B); 10, 25, and 50 μM oxaliplatin (OXA) (C); 1, 100, and 200 μM carboplatin (CARB) (D); 50, 100, and 200 nM nocodazole (Noco) (A, E); 150, 300, and 600 nM paclitaxel (PCT) (F); 1, 5, and 10 μM rotenone (ROT) (G); 5, 25, and 50 μM RO3280 (RO) (H); 0.5, 5, and 50 μM AZD1152 (I); or 25, 50, and 100 μM M2I-1 (A, J). Then, cells were stained with the vital dye propidium iodide (PI) and the mitochondrial membrane potential (ΔΨm)–sensitive dye DiOC6(3) to measure apoptotic cell death (A–J). A Illustrates representative dot plots of WT and DKO MEFs in the absence of any treatment (Co) or upon incubation with μM CDDP, 100 nM Noco, or 100 nM M2I-1. Numbers indicate the percentage of cells in each quadrant. In B–J, black columns represent the percentage of dead (PIhigh) cells and white columns represent the percentage of dying cells (DiOC6(3)lowPIlow). Error bars indicate SEM. Total columns (DiOC6(3)lowPIlow + PIhigh) were compared by R software using standard linear model inference, “lm” function. #p < 0.05, ##p < 0.01, ###p < 0.001 treatment vs. control. *p < 0.05, **p < 0.01 treatment effect, DKO (“D”, in red) vs. WT (“W”, in blue).
Intrigued by the fact that Bax/Bak (or BAX/BAK) deficiency confers relatively poor protection against cell cycle-perturbing agents (such as microtubular and spindle checkpoint inhibitors), we investigated the effects of Noco, which is a reversible microtubular inhibitor that can be washed out so that cells resume proliferation [55, 56]. WT and Bax/Bak MEFs responded similarly by an accumulation of tetraploid cells to transient exposure to Noco (Fig. 2A, B). Surprisingly, purified tetraploid Bax/Bak MEFs were unable to resume proliferation and hence were poorly clonogenic (<5% cells), whereas ~50% of WT MEFs were able to do so and formed macroscopic clones upon cytofluorometric sorting and culture in 96-well plates (Fig. 2A, C). We performed an extensive comparison of different MEF genotypes, finding that the single knockout of BAX had a minor effect, but that of BAK alone compromised clonogenic potential to about half of that of WT cells. However, the strongest phenotype was seen for the Bax/Bak DKO MEFs, whereas other DKOs (for instance of Bbc3 and Pmaip1, Bcl2l11, and Bbc3, Bcl2l11, and Bid) had no such effects (Fig. 2C). Of note, DKO cells expressed close-to-normal levels (with variations up to 40%) of other genes from the BCL2 family (Supplementary Fig. S2).

Mouse embryonic fibroblasts (MEFs) (A–D), human colon carcinoma HCT116 cells (E), or immortalized MEFs derived from C57Bl/6 mice (MEF C57Bl/6) (F) were seeded on day 0, cultured from day 1 to day 3 (48 h) in drug-free medium or treated with 100 nM Nocodazole (Noco) (A–D and F) or 1.2 μM cytochalasin D (CytD) (A, E), then washed and kept in a drug-free medium for 4 more days, and finally sorted on day 7 (100 cells per well). Diploid (2n) cells were derived from cells grown in a drug-free medium (A–C, E, F) or from cells treated with CytD (E), whereas polyploid (>4n) cells were derived from cells treated with Noco (A–D and F) or CytD (E). The cell cycle and the DNA content gates were established by cell staining with 10 μM Hoechst 33342 for 1 h (B). Clonogenicity of MEF (C), HCT116 (E), or MEF C57Bl/6 (F) cells deficient for one or two pro-apoptotic proteins was quantified at day 21 as percentage of wells in which there was cell proliferation. Alternatively, >4n wild type (WT) and Bax−/−Bak−/− (DKO) MEFs sorted on day 7 were subjected for cell proliferation assay at different time points between days 7 and 21 (D). Bars with blue contours represent WT cells and bars with red contours represent Bax/Bak DKO cells. White columns represent clonogenicity of 2n cells and black columns represent clonogenicity of >4n cells. Error bars indicate SEM. Data were compared by R software using standard linear model inference, “lm” function. >4n Samples were compared to 2n samples for each cell line (##p < 0.01, ###p < 0.001 vs. 2n). Clonogenicity of >4n cells was compared between all MEFs deficient for different pro-apoptotic proteins vs. DKO cells (p-values in red, C); or between DKO (“D”) and WT (“W”) cells (E, F) (*p < 0.05, **p < 0.01). Cell proliferation of >4n DKO (red) vs. >4n WT (blue) MEFs (D) was compared (**p < 0.01).
Hence, the loss of clonogenicity of tetraploid cells is a feature of Bax/Bak DKO MEFs, which fail to resume proliferation after Noco washout (Fig. 2D). Tetraploid BAX/BAK DKO HCT116 cells also lost much of their clonogenic potential after CytD treatment as compared to tetraploid WT cells (Fig. 2E), confirming the data obtained with MEF. As an additional control, we took advantage of another source of WT and Bax/Bak DKO MEFs (which were derived from Bax/Bak KO mice), replicating the defective clonogenicity of tetraploid Bax/Bak DKO cells.
Senescence of tetraploid Bax/Bak DKO cells
Next, we attempted to understand the reasons why Bax/Bak DKO cells lose their clonogenic survival upon tetraploidization. Noco undistinguishably induced ER stress in WT and Bax/Bak (or BAX/BAK) DKO cells, as indicated by the phosphorylation of eukaryotic initiation factor 2α and the nuclear translocation of the ER stress-linked transcription factors ATF6 and XBP1. These ER stress markers were determined by multicolor immunofluorescence staining and image cytometry, comparing WT and Bax/Bak DKO MEFs (Supplementary Fig. S3) or WT and BAX/BAK DKO HCT116 cells (Supplementary Fig. S4). Similarly, WT and Bax/Bak DKO MEFs produced similarly elevated levels of reactive oxygen species (ROS) in response to Noco, as measured with the ROS sensor Oxidative Stress Detection Reagent (Supplementary Fig. S5A) and contained similar levels of the autophagy markers LC3-II and SQSTM1 (also known as p62). Although an increase in LC3 lipidation is observed, autophagic flux is blocked after Noco treatmen, as, in neither line, lipidation is accompanied by effective degradation of SQSTM1 (Supplementary Fig. S5B). Finally, WT and Bax/Bak DKO MEFs manifested a similar increase in Noco-induced autophagosomes detectable by transmission electron microcopy (Supplementary Fig. S5C).
Next, we performed triple staining with the vital dye PI, the cell-permeable chromatin stain Hoechst 33342, and the β-galactose substrate C12FDG, a senescence marker [57]. Noco treatment led to a similar minor loss of viability among WT and Bax/Bak DKO MEFs. However, the percentage of C12FDGhigh cells was higher among viable Bax/Bak DKO MEFs than WT controls (Fig. 3A, B). These C12FDGhigh cells were especially elevated among hyperploid (>4n) cells (Fig. 3C), indicating that tetraploid Bax/Bak DKO cells are particularly prone to senescence (Fig. 3D, E). This result was replicated in HCT116 cells subjected to experimental tetraploidization, indicating that the lack of BAX/BAK predisposes to senescence (Fig. 3F, G). Moreover, Noco-induced higher levels of CDK inhibitor 2A protein p16INK4a and CDK inhibitor 1A protein p21Cip1 in Bax/Bak DKO compared to WT MEFs, but not CDK inhibitor 1C protein p57Kip2, as determined by immunoblotting (Fig. 3H–K). In conclusion, it appears that the loss of Bax/Bak (or BAX/BAK) causes tetraploid cells to undergo a higher degree of senescence, explaining their loss of clonogenic potential.

Mouse embryonic fibroblasts (MEFs) (A–E, H–K) or human colon carcinoma HCT116 cells (F, G) were seeded, treated or not with 100 nM Nocodazole (Noco) or 1.2 μM cytochalasin D (CytD) for 48 h, then washed and kept in a drug-free medium for 4 days. Then, cells were stained with the vital dye propidium iodide (PI), the β-galactosidase substrate, C12FDG, and the fluorescent stain for DNA, Hoechst 33342. Live cells (A) were subjected to DNA content analysis (B). C12FDG was analyzed both in the total population (D, F) and according to ploidy (2n or >4n) (C, E, G). Alternatively, p16INK4a (H, I), p21Cip1 (H, J), and p57Kip2 (H, K) in MEFs were determined by immunoblotting. Bars with blue contours represent WT (“W”) cells and bars with red contours represent Bax/Bak DKO (“D”) cells. White columns represent control or 2n cells and black columns represent treated or >4n cells. Error bars indicate SEM. Data were compared by R software using standard linear model inference, “lm” function. ##p < 0.01, ###p < 0.001 treatment vs. control. ++p < 0.01, +++p < 0.001 > 4n vs. 2n. *p < 0.05, **p < 0.01, **p < 0.001 for treatment effect, DKO vs. WT.
Aberrant Ca2+ signaling in tetraploid Bax/Bak DKO cells
BCL2 family proteins including BAX and BAK have been suspected to affect calcium (Ca2+) signaling, as determined by cell-free experiment and measurement of Ca2+ fluxes in stressed cells [13, 20, 22]. Bax/Bak DKO MEFs manifested a reduced ionomycin-induced release of Ca2+ from the ER into the cytosol at the basal level compared to WT MEFs, as determined with the ratiometric probe Fura-2AM and fluorescence microscopy (Fig. 4A, B). Of note, tetraploidization did not affect ER Ca2+ stores in WT cells, but did increase such stores in Bax/Bak DKO MEFs (Fig. 4A, B). Moreover, DKO cells manifested a higher SOCE than WT controls and this increase was reduced by Noco (Fig. 4A, C). Interestingly, DKO cells manifested an underexpression of endogenous Serca2a (official name Atp2a2) and overexpression of inositol 1,4,5-Trisphosphate Receptor Type 2 and 3 (Itpr2 and Itpr3), which might explain the effect on ER Ca2+ stores. Moreover, DKO cells overexpress stromal interaction molecule 1, which is involved in SOCE (Supplementary Fig. S6). Considering that restoration of [Ca2+]ER in DKO cells with reintroduction of Serca2a and Bak into the ER has previously been described [22, 23], we wondered whether they would also play a role in restoring Ca2+ signaling in tetraploid Bax/Bak DKO cells. We took advantage of DKO cells transfected with rbSerca2a or an ER-targeted version of Bak (BakER) (Fig. 5A–D) and compared Ca2+ fluxes in untreated vs. Noco-treated WT, DKO cells, and DKO cells expressing rbSerca2a or BakER. Of note, the modulation of Ca2+ fluxes by the DKO (a decrease of ionomycin-induced cytosolic Ca2+ in untreated cells and an increase of store-operated Ca2+ in Noco-treated cells) was lost upon introduction of rbSerca2a or BakER into these DKO cells (Fig. 5E–G).

WT and DKO mouse embryonic fibroblasts (MEFs) were seeded and treated or not with 100 nM Nocodazole (Noco) for 48 h. Then, cells were MEFs loaded with 4 µM Fura2/AM and subjected to fluorescence imaging of cytosolic Ca2+ (A). Ca2+ store content (A, B) was evaluated by perfusing cells with 100 nM ionomycin (Iono) for 1 min in Ca2+-free medium. Store-operated Ca2+ entry (SOCE) (C) was evaluated by perfusing cells with 10 μM cyclopiazonic acid for 10 min or 1 µM thapsigargin for 10 min in Ca2+-depleted medium. Then, cells were perfused with 1 mM Ca2+-containing medium. B, C On the left are shown [Ca2+]cyt traces of control (Co) cells (purple) and treated (Noco) cells (orange), and on the right the quantification. Bars with blue contours represent WT cells and bars with red contours represent Bax/Bak DKO cells. White columns represent control cells and black columns represent treated cells. Error bars indicate SEM. Data were compared by R software using standard linear model inference, “lm” function. ##p < 0.01 treatment vs. control. *p < 0.05, for treatment effect, DKO vs. WT. Scale bar, 10 μm.

WT, DKO, and DKO mouse embryonic fibroblasts (MEFs) stably transfected (A) with rabbit SERCA2a (DKO with Serca2a-overexpressed, DKO-Sercaoe) (B, C), or an ER-targeted version of Bak (DKO-BakER) (B, D) were seeded and treated or not with 100 nM Nocodazole (Noco) for 48 h. Then, cells were MEFs were loaded with 4 µM Fura2/AM and subjected to fluorescence imaging of cytosolic Ca2+ (E). Ca2+ store content (F) was evaluated by perfusing cells with 100 nM ionomycin (Iono) for 1 min. Store-operated Ca2+ entry (SOCE) (G) was evaluated by perfusing cells with 10 μM cyclopiazonic acid for 10 min or 1 µM thapsigargin for 10 min in Ca2+-depleted medium. Then, cells were perfused with 1 mM Ca2+-containing medium. Bars with blue contours represent WT cells and bars with red contours represent Bax/Bak DKO cells. White columns represent control cells and black columns represent treated cells. Error bars indicate SEM. Data were compared by R software using standard linear model inference, “lm” function. $p < 0.05, DKO vs. WT in B or vs. DKO-Serca2aoe in C. #p < 0.05, ###p < 0.001 treatment vs. control. *p < 0.05, for treatment effect, DKO vs. WT. &&&p < 0.001 for treatment effect, DKO vs. DKO-Sercaoe. σσσp < 0.001 for treatment effect, DKO vs. DKO-BAKER. Scale bar, 10 μm.
Reversion of tetraploid Bax/Bak DKO senescence by ER-targeted Bak
The aforementioned results suggest that the Bax/Bak DKO and tetraploidization in Bax/Bak DKO cells affect Ca2+ fluxes, yet provide no prove that such alterations account for the senescence of tetraploid Bax/Bak DKO cells. We therefore measured markers of senescence in untreated vs. Noco-exposed WT, DKO MEFs, and DKO cells expressing rbSerca2a or BakER. Importantly, introduction of BakER reduced the Noco-induced expression of the senescence markers p21Cip1 and cyclin-dependent kinase inhibitor 1B p27Kip1 to WT levels (Fig. 6). Therefore, the exclusive presence of Bak in the ER plays a key role in senescence induced by tetraploidizing agents.

WT, DKO, and DKO with Serca2a-overexpressed (DKO-Sercaoe), or DKO expressing an ER-targeted version of Bak (DKO-BakER) mouse embryonic fibroblasts (MEFs) were seeded, treated, or not with 100 nM Nocodazole (Noco) for 48 h, then washed and kept in a drug-free medium for 4 days. Attached cells were collected and p16INK4a (A), p21Cip1 (B), p27Kip1 (C), and p57Kip2 (D) were determined by immunoblotting. Inserts show representative experiments. Densitometry data shown as mean value ± SEM are depicted. Bars with blue contours represent WT cells and bars with red contours represent Bax/Bak DKO cells. Data were compared by R software using standard linear model inference, “lm” function. #p < 0.05, ##p < 0.01, ###p < 0.001 treatment vs. control. *p < 0.05, **p < 0.01, ***p < 0.001, for treatment effect, DKO, or DKO-Sercaoe vs. WT. σσp < 0.01, σσσp < 0.001, for treatment effect, DKO vs. DKO-BAKER.
At the functional level, both BakER and SERCA2a were able to restore the clonogenic potential of tetraploid Bax/Bak DKO cells, as determined by two distinct experimental approaches. In the first protocol, WT, DKO MEFs, and DKO MEFs expressing rbSerca2a or BakER were transiently cultured in Noco, washed, re-cultured without Noco, stained with Hoechst 33324, subjected to cytofluorometric purification of tetraploid cells, and seeded in 96-well plates (100 cells per well). Using this assay, we found that tetraploid DKO cells expressing rbSerca2a or BakER recovered a normal clonogenic potential comparable to WT MEFs (Fig. 7A, B). As an alternative approach that does not involve Hoechst 33324 staining, cells were treated for 48 h with Noco, washed, allowed to rest for 4 days, and cultured in six-well plates for 2–3 weeks, followed by visualization of clones by crystal violet staining (Fig. 7C). Of note, only Bax/Bak DKO cells manifested a reduction in clonogenic potential as compared to WT controls. This phenotype was completely lost upon introduction of BakER into the cells and was attenuated by SERCA2a as well (Fig. 7D, E).

WT, DKO, and DKO with Serca2a-overexpressed (DKO-Sercaoe), or DKO expressing an ER-targeted version of Bak (DKO-BakER) mouse embryonic fibroblasts (MEFs) were seeded, treated or not with 100 nM Nocodazole (Noco) for 48 h, then washed and kept in a drug-free medium for 4 days, and finally sorted (100 cells per well) on day 7. Diploid (2n) cells were derived from cells grown in a drug-free medium, whereas polyploid (>4n) cells were derived from cells treated with Noco (A). Clonogenicity of different MEFs was quantified at day 21 as percentage of wells in which there was cell proliferation (B). Alternatively, WT, DKO, DKO-Sercaoe, or DKO-BakER MEFs were seeded, treated with 100 nM Nocodazole (Noco) for 48 h, then washed and seeded again in a six-well plate at different cell concentrations. After 14 or 21 days, colonies were stained with crystal violet (C). Pictures were taken and colony area analyzed (D, E). Pictures show a representative experiment. Scale bar = 1 cm. Data shown as mean value ± SEM. White bars represent control cells and black or gray bars represent treated cells. Bars with blue contours represent WT cells, bars with red contours represent Bax/Bak DKO cells, light gray bars represent treated DKO-Sercaoe cells, and dark gray bars represent DKO-BakER cells. Data in B were compared by R software using standard linear model inference, “lm” function. Data in D and E were compared by R software using linear mixed-effects model, “lme” function. ###p < 0.001 treatment vs. control. p-Values in black, *p < 0.05, ***p < 0.001, for treatment effect, DKO, DKO-Sercaoe, or DKO-BakER vs. WT. p-Values in red, &&&p < 0.001 for treatment effect, DKO vs. DKO-Sercaoe. p-Values in red, σσσp < 0.001 for treatment effect, DKO vs. DKO-BAKER.
We conclude that Ca2+ fluxes at the level of ER are indeed important to explain the propensity of Bax/Bak DKO cells to undergo senescence upon their tetraploidization.
Discussion
The DKO of BAX and BAK, as well as the more recent TKO of BAX + BAK + BOK strongly inhibits the mitochondrial pathway of apoptosis. [33, 58]. Accordingly, MEFs from such DKO or TKO mice are largely resistant against a series of pro-apoptotic insults and similar results have been obtained in human DKO cells such as the HCT116 colon cancer cells used in this study. Here we report the unexpected finding that BAX and BAK are required for the expansion of tetraploid cells generated as a result of failed mitosis, supporting the idea that BAX and BAK are not only pro-apoptotic proteins but that they may also be required for assuring the fitness of cells in specific circumstances such as tetraploidization. Previous reports have demonstrated “moonlighting” functions for BCL2 family members that also regulate cell death-unrelated functions including autophagy, cellular senescence, inflammation, Ca2+ fluxes, bioenergetics, and redox homeostasis [59]. However, most of these “moonlighting” functions have been attributed to anti-apoptotic BCL2 family members and, to the best of our knowledge, this is the first report on a function of BAX/BAK that favors cellular fitness.
Although the exact pathway explaining how BAX/BAK is required for the proliferation of tetraploid cells remains to be elucidated, our present study highlights two elements in this pathway. First, the presence of BAX/BAK is required for tetraploid cells to suppress a permanent cell cycle arrest referred to as senescence. Following the exposure of cells to the reversible spindle poison Noco (which causes tetraploidization due to “mitotic slippage”) [55, 56], as well as its removal, BAX/BAK-deficient cells usually activate a senescence program, as indicated by the upregulation of several CDK inhibitors and the activation of senescence-associated β-galactosidase. It is well accepted that upregulation of p21 is not sufficient to maintain long-term senescent cell arrest. However, upregulation of p16 is able to maintain the senescent state over time [60]. This would explain why DKO cells cannot resume proliferation, whereas WT can do so. Second, compared to WT, the absence of BAX/BAK is tied to alterations in Ca2+ signaling with reduced ER Ca2+ stores and increased SOCE, yet restored by expressing SERCA2A or an ER-targeted version of BAK in the cells. Both these changes are lost after induction of tetraploidy in BAX/BAK DKO cells. ER-targeted BAK also restore the tetraploidy-associated senescence found in BAX/BAK DKO cells to BAX/BAK-sufficient control cell levels. Collectively, these findings suggest that BAX/BAK act on the ER to affect local Ca2+ fluxes, e.g., by downregulation of SERCA2A and upregulation of IP3R2 and IP3R3 in DKO cells, which then indirectly affect SOCE and senescence. Several models have shown that Ca2+ is involved in senescence. For example, IP3Rs [61, 62] and l-type Ca2+ channels appear to play a role in senescence in vascular smooth muscle cells [63] and β-cells from pancreatic islets [64]. Moreover, so-called BH3 mimetics, which are small molecules that activate MOMP (and also mediate other effects including the induction of autophagy) [65, 66] have been shown to eliminate senescent cells from aging tissues and consequently to mediate “senolysis” [67,68,69]. Molecular connections have been established between some members of the BCL2 family and senescence. However, the precise molecular links between ER Ca2+ storage, BAX/BAK, and senescence remain to be elucidated.
It is fascinating to consider that, depending on the exact genetic background, BAX/BAK-knockout mice are usually born at the expected frequency and then are variably affected by perinatal mortality. Such mice develop close-to-normally, with relatively minor phenotypes, but do not develop malignant disease as a major phenotype [39]. Genetic studies investigating the ploidy of cancer cells have led to the conclusion that at least 40% of solid cancers are the result of an often transient tetraploidization followed by the loss of excessive chromosomes to reestablish a close-to-diploid (but often aneuploid) karyotype [70]. This polyploidization/deploidization cycle may be considered as a major mechanism of genomic instability that favors oncogenesis and tumor progression [70], and is also under the control of the immune system (which tends to eliminate tetraploid or higher-order polyploid cells) [71, 72]. Although formal proof for this conjecture is elusive, it is tempting to speculate that BAX/BAK deficiency is not a major driver of oncogenesis, because it compromises the proliferative potential of tetraploid cells, hence obliterating one major path towards carcinogenesis. Following the same mental construction, the rarity of complete loss of BAX and BAK from cancer cells may be explained. Indeed, tumor cells have to navigate between Scylla and Charybdis, to avoid their death (which often occurs through apoptosis) and their senescence (which is another effective tumor suppressor mechanism). From the point of view of the cancer cell, the loss of BAX/BAK may be desirable for avoiding apoptosis but disastrous for preventing senescence induced by ploidy changes and perhaps other yet-to-be elucidated stressors.
Irrespective of the aforementioned gaps in our knowledge, uncertainties on mechanisms, and speculative issues, it appears clear that the maintenance of BAX/BAK expression may be advantageous for cells in specific circumstances, as illustrated here for tetraploidization.
Data availability
Data are available in ArrayExpress under the ID “E-MTAB-10997” and through the corresponding author on request.
References
Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008;4:600–6.
Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59.
Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80.
Konig SM, Rissler V, Terkelsen T, Lambrughi M, Papaleo E. Alterations of the interactome of Bcl-2 proteins in breast cancer at the transcriptional, mutational and structural level. PLoS Comput Biol. 2019;15:e1007485.
Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25:46–55.
Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5:a008714.
Gross A, Katz SG. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017;24:1348–58.
Gimenez-Cassina A, Danial NN. Regulation of mitochondrial nutrient and energy metabolism by BCL-2 family proteins. Trends Endocrinol Metab. 2015;26:165–75.
Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21.
Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008;70:73–91.
Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006;13:1351–9.
Marquez RT, Xu L. Bcl-2:Beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res. 2012;2:214–21.
Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene 2016;35:5079–92.
Poenie M, Alderton J, Tsien RY, Steinhardt RA. Changes of free calcium levels with stages of the cell division cycle. Nature 1985;315:147–9.
Arredouani A, Yu F, Sun L, Machaca K. Regulation of store-operated Ca2+ entry during the cell cycle. J Cell Sci. 2010;123:2155–62.
Berridge MJ. Calcium signalling and cell proliferation. Bioessays 1995;17:491–500.
Lu KP, Means AR. Regulation of the cell cycle by calcium and calmodulin. Endocr Rev. 1993;14:40–58.
Chen YW, Chen YF, Chen YT, Chiu WT, Shen MR. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci Rep. 2016;6:22142.
Humeau J, Bravo-San Pedro JM, Vitale I, Nunez L, Villalobos C, Kroemer G, et al. Calcium signaling and cell cycle: progression or death. Cell Calcium. 2018;70:3–15.
Oakes SA, Opferman JT, Pozzan T, Korsmeyer SJ, Scorrano L. Regulation of endoplasmic reticulum Ca2+ dynamics by proapoptotic BCL-2 family members. Biochem Pharm. 2003;66:1335–40.
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:105–10.
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, et al. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003;300:135–9.
Klee M, Pallauf K, Alcala S, Fleischer A, Pimentel-Muinos FX. Mitochondrial apoptosis induced by BH3-only molecules in the exclusive presence of endoplasmic reticular Bak. EMBO J. 2009;28:1757–68.
Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta. 2011;1813:521–31.
Uren RT, Iyer S, Kluck RM. Pore formation by dimeric Bak and Bax: an unusual pore? Philos Trans R Soc Lond B Biol Sci. 2017;372:20160218.
Cosentino K, Garcia-Saez AJ. Bax and Bak pores: are we closing the circle? Trends Cell Biol. 2017;27:266–75.
Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, et al. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69.
Lisak D, Schacht T, Gawlitza A, Albrecht P, Aktas O, Koop B, et al. BAX inhibitor-1 is a Ca(2+) channel critically important for immune cell function and survival. Cell Death Differ. 2016;23:358–68.
Henke N, Lisak DA, Schneider L, Habicht J, Pergande M, Methner A. The ancient cell death suppressor BAX inhibitor-1. Cell Calcium. 2011;50:251–60.
Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, et al. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016;35:389–401.
Grosse L, Wurm CA, Bruser C, Neumann D, Jans DC, Jakobs S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016;35:402–13.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727–30.
Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–32.
Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, et al. BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 2016;165:421–33.
Thomas DA, Scorrano L, Putcha GV, Korsmeyer SJ, Ley TJ. Granzyme B can cause mitochondrial depolarization and cell death in the absence of BID, BAX, and BAK. Proc Natl Acad Sci USA. 2001;98:14985–90.
Wan KF, Chan SL, Sukumaran SK, Lee MC, Yu VC. Chelerythrine induces apoptosis through a Bax/Bak-independent mitochondrial mechanism. J Biol Chem. 2008;283:8423–33.
Schwarzer C, Fu Z, Morita T, Whitt AG, Neely AM, Li C, et al. Paraoxonase 2 serves a proapopotic function in mouse and human cells in response to the Pseudomonas aeruginosa quorum-sensing molecule N-(3-Oxododecanoyl)-homoserine lactone. J Biol Chem. 2015;290:7247–58.
Sobhakumari A, Love-Homan L, Fletcher EV, Martin SM, Parsons AD, Spitz DR, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE. 2012;7:e48175.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–99.
Ke FFS, Vanyai HK, Cowan AD, Delbridge ARD, Whitehead L, Grabow S, et al. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell 2018;173:1217–30. e17.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell 2011;147:742–58.
Opferman JT, Kothari A. Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 2018;25:37–45.
Giam M, Huang DC, Bouillet P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008;27:S128–36. Suppl 1
Ke F, Bouillet P, Kaufmann T, Strasser A, Kerr J, Voss AK. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 2013;4:e650.
Voss AK, Strasser A. The essentials of developmental apoptosis. F1000Res. 2020;9:F1000 Faculty Rev-148.
Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 2010;330:1390–3.
Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006;312:572–6.
Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 1999;401:616–20.
Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science 2000;290:989–92.
Calvo-Rodriguez M, Hernando-Perez E, Nunez L, Villalobos C. Amyloid beta oligomers increase ER-mitochondria Ca(2+) cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca(2+) remodeling. Front Cell Neurosci. 2019;13:22.
Guzman C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE. 2014;9:e92444.
Team RC. R: A language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2020).
Pinheiro J, Bates D, DebRoy S, Sarkar D, The R Core Team. nlme: Linear and Nonlinear Mixed Effects Models. R package version 3.1-151. 2021. https://CRAN.R-project.org/package=nlme.
Olejniczak SH, Hernandez-Ilizaliturri FJ, Clements JL, Czuczman MS. Acquired resistance to rituximab is associated with chemotherapy resistance resulting from decreased Bax and Bak expression. Clin Cancer Res. 2008;14:1550–60.
Hayne C, Tzivion G, Luo Z. Raf-1/MEK/MAPK pathway is necessary for the G2/M transition induced by nocodazole. J Biol Chem. 2000;275:31876–82.
Zieve GW. Nocodazole and cytochalasin D induce tetraploidy in mammalian cells. Am J Physiol. 1984;246:C154–6.
Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–806.
Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science 2004;305:626–9.
Chong SJF, Marchi S, Petroni G, Kroemer G, Galluzzi L, Pervaiz S. Noncanonical cell fate regulation by Bcl-2 proteins. Trends Cell Biol. 2020;30:537–55.
Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19:2109–17.
Borodkina AV, Shatrova AN, Deryabin PI, Griukova AA, Abushik PA, Antonov SM, et al. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging (Albany NY). 2016;8:3400–18.
Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun. 2014;5:3792.
Kim SG, Sung JY, Kim JR, Choi HC. Nifedipine-induced AMPK activation alleviates senescence by increasing autophagy and suppressing of Ca(2+) levels in vascular smooth muscle cells. Mech Ageing Dev. 2020;190:111314.
Sun L, Wang X, Gu T, Hu B, Luo J, Qin Y, et al. Nicotine triggers islet beta cell senescence to facilitate the progression of type 2 diabetes. Toxicology 2020;441:152502.
Villalobos-Ortiz M, Ryan J, Mashaka TN, Opferman JT, Letai A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 2020;27:999–1007.
Malik SA, Shen S, Marino G, BenYounes A, Maiuri MC, Kroemer G. BH3 mimetics reveal the network properties of autophagy-regulatory signaling cascades. Autophagy 2011;7:914–6.
Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016;7:11190.
Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. 2019;29:1045–60.e10.
Ritschka B, Knauer-Meyer T, Goncalves DS, Mas A, Plassat JL, Durik M, et al. The senotherapeutic drug ABT-737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev. 2020;34:489–94.
Vitale I, Galluzzi L, Senovilla L, Criollo A, Jemaa M, Castedo M, et al. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2011;18:1403–13.
Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science 2012;337:1678–84.
Aranda F, Chaba K, Bloy N, Garcia P, Bordenave C, Martins I, et al. Immune effectors responsible for the elimination of hyperploid cancer cells. Oncoimmunology 2018;7:e1463947.
Acknowledgements
We are indebted to Andreas Strasser and Francine Ke (The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia), for the gift of immortalized MEFs derived from C57Bl/6 WT or Bax/Bak−/− mice. We thank the genomics platform of the Institut Gustave Roussy (Taxe d’apprentissage TA2017, P28_JUHU) for performing whole-genome expression arrays.
Funding
LS is supported by Beatriz Galindo senior program of the Spanish Ministry of Universities; Strategic Program “Instituto de Biología y Genética Molecular (IBGM), Junta de Castilla y León” (Ref. CCVC8485). GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)–Projets blancs; AMMICa US23/CNRS UMS3655; Association “Ruban Rose”; Cancéropôle Ile-de-France; Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onco-Pheno-Screen; European Joint Programme on Rare Diseases (EJPRD); Gustave Roussy Odyssea, the European Union Horizon 2020 Projects Oncobiome and Crimson; Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LabEx Immuno-Oncology (ANR-18-IDEX-0001); the Leducq Foundation; a Cancer Research ASPIRE Award from the Mark Foundation; Programme de cooperation ECO-Sud (C17S02); the RHU Torino Lumière; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001. CV and LN are supported by research grants #RTI2018-099298-B-100 from Ministerio de Ciencia e Innovación, Spain, #CA9751 from Junta de Castilla y León, and #CCVC8485 Programa Estratégico IBGM, Junta de Castilla y León, Spain.
Author information
Authors and Affiliations
Contributions
JD and LGG acquired, analyzed, and interpretated the data. GS designed and interpreted bioinformatics and statistical analyses. OM contributed to the design, acquisition, analysis, and interpretation of data. IM contributed to the cytometry-based experiments. LN contributed to the calcium imaging experiments. JMB-SP and JH generated HCT116 Bax−/−Bak−/− (DKO) cells and contributed to the initial experiments. CB optimized and acquired the initial experiments. JP contributed to data acquisition. HF-T contributed to clonogenic capacity experiments. SS and GP contributed to the electron microscopy experiments. CH provided WT and all genetically modified mouse embryonic fibroblasts (MEFs), reviewed, and contributed to the study supervision. LS designed the experiments and prepared the figures. CV, GK, and LS conceived the study, interpreted data, supervised the whole set of experiments, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
GK has been holding research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, PharmaMar, Samsara, Sanofi, Sotio, Vascage, and Vasculox/Tioma. GK is on the Board of Directors of the Bristol Myers Squibb Foundation France. GK is a scientific co-founder of everImmune, Samsara Therapeutics, and Therafast Bio. GK is the inventor of patents covering therapeutic targeting of aging, cancer, cystic fibrosis, and metabolic disorders.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Gerry Melino
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, J., Gutiérrez, L.G., Stoll, G. et al. Paradoxical implication of BAX/BAK in the persistence of tetraploid cells. Cell Death Dis 12, 1039 (2021). https://doi.org/10.1038/s41419-021-04321-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-04321-3
- Springer Nature Limited