
Abstract
Sestrin2 (SESN2), a highly conserved stress-responsive protein, can be triggered by various noxious stimuli, such as hypoxia, DNA damage, oxidative stress, endoplasmic reticulum (ER) stress, and inflammation. Multiple transcription factors regulate SESN2 expression, including hypoxia-inducible factor 1 (HIF-1), p53, nuclear factor E2-related factor 2 (Nrf2), activating transcription factor 4 (ATF4), ATF6, etc. Upon induction, SESN2 generally leads to activation of adenosine monophosphate-activated protein kinase (AMPK) and inhibition of mechanistic target of rapamycin complex 1 (mTORC1). To maintain cellular homeostasis, SESN2 and its downstream molecules directly scavenge reactive oxygen species or indirectly influence the expression patterns of key genes associated with redox, macroautophagy, mitophagy, ER stress, apoptosis, protein synthesis, and inflammation. In liver diseases including acute liver injury, fatty liver diseases, hepatic fibrosis, and hepatocellular carcinoma (HCC), SESN2 is abnormally expressed and correlated with disease progression. In NAFLD, SESN2 helps with postponing disease progression through balancing glycolipid metabolism and macroautophagy (lipophagy), and rectifying oxidative damage and ER stress. During hepatic fibrosis, SESN2 represses HSCs activation and intrahepatic inflammation, hindering the occurrence and progress of fibrogenesis. However, the role of SESN2 in HCC is controversial due to its paradoxical pro-autophagic and anti-apoptotic effects. In conclusion, this review summarizes the biological functions of SESN2 in hypoxia, genotoxic stress, oxidative stress, ER stress, and inflammation, and specifically emphasizes the pathophysiological significance of SESN2 in liver diseases, aiming to providing a comprehensive understanding for SESN2 as a potential therapeutic target in liver diseases.
Similar content being viewed by others


FACTS
-
SESN2 is a stress-responsive protein with a distinct molecular structure regulating mTORC1.
-
SESN2 is involved in multiple pathophysiological events, such as hypoxia, genotoxic stress, oxidative stress, endoplasmic reticulum stress, inflammation, autophagy, and cell death.
-
SESN2 exerts potent hepatoprotective effects against acute and chronic liver injuries.
OPEN QUESTIONS
-
Does SESN2 have the potential as a viable therapeutic target for liver diseases?
-
The roles of SESN2 in liver cancers should be further explored.
Introduction
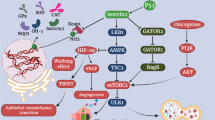
Sestrins (SESNs) belong to an evolutionarily conserved stress-responsive protein family existed in most vertebrates. The SESNs family comprises three members, SESN1, SESN2, and SESN3, among which SESN2 has been the most profoundly investigated [1]. SESN2, also nominated as Hi95, was originally identified inducible by prolonged hypoxia, DNA damage, and oxidative stress [2, 3]. Structurally, SESN2 is composed of two globin-like α-helix-only subdomains, N-terminal domain (NTD; residues 66–220) and C-terminal domain (CTD; residues 339–480), connected by a helix-loop-helix linker (residues 221–338). The NTD contains a homology region (residues 109–139) that corresponds to the helix-turn-helix oxidoreductase motif of an alkyl hydroperoxide reductase AhpD in Mycobacterium tuberculosis. The catalytic cysteine residue (Cys125) and the residues (Tyr127 and His132) mediating the proton delay system of AhpD are well-conserved within the NTD, enabling SESN2 to resemble AhpD in directly scavenging reactive oxygen species (ROS). The CTD contains a helix-loop structure but no helix-turn-helix motif or catalytic residues involved in AhpD oxidoreductase activity, implying that the CTD may not have the antioxidative activity. Aspartic acid residues Asp406 and Asp407 in the CTD may interact with GTPase-activating protein activity towards Rags 2 (GATOR2), liberating GATOR1 from GATOR2-mediated restriction [4]. GATOR1 binds to and inactivates Rag A/B, restricting lysosomal translocation and activation of mechanistic target of rapamycin complex 1 (mTORC1) [5]. Serine residue Ser190 in the NTD is also necessary for GATOR2 binding, suggesting that SESN2 may make multiple contacts with GATOR2 through both NTD and CTD [6]. There is a leucine-binding pocket at the intersection of helices C2, C3, and C7 in the CTD, which enables SESN2 to directly bind with leucine and act as a leucine sensor for transmitting leucine signal to activate mTORC1 [6,7,8,9]. The leucine pocket is in close proximity to the GATOR2 binding sites, which provides a possible mechanism for how leucine binding may cause SESN2 dissociation from GATOR2 that the changes in the leucine binding state alters the position of domains and affects the availability of GATOR2 binding sites [6]. Alternatively, leucine suppresses SESN2 phosphorylation at Thr232, Ser249, and Ser279, forcing SESN2 to dissociate from and activate GATOR2, leading to mTORC1 activation [10]. The unique molecular structure (Fig. 1) endows SESN2 with manifold roles in different biological processes.

SESN2 is shown as a ribbon diagram with NTD, Linker, and CTD labeled as pink, blue, and yellow, respectively. The disordered regions (1–65, 222–232, 241–255, 272–280, 296–309) are labeled as dash lines. The locations of key residues (C125, Y127, H132, S190, T232, S249, S279, D406, and D407) are marked in red.
As a classical stress-responsive protein, SESN2 can be induced in response to diverse stress conditions, such as hypoxia, genotoxic stress, and oxidative stress. In recent years, SESN2 has also been observed to be altered and impacted on endoplasmic reticulum (ER) stress and inflammation. Induced SESN2 may serve as a cellular defender against multiple detrimental stimuli and contribute to the recovery of organ homeostasis from diseases, especially liver diseases. In the liver, SESN2 displays additional functions including regulating glycolipid metabolism, HSCs activation, autophagy, cell survival and death.
In this review, we summarize the biological functions of SESN2 in distinct pathophysiological processes and particularly describe its association with liver diseases, aiming to promote profound understanding for the medical significance of SESN2.
Diversified functions and regulations of SESN2 under stresses
SESN2 in hypoxia
Hypoxia is a complex pathophysiological process occurring under diverse conditions. SESN2 was initially discovered as a prolonged hypoxia-induced molecule independent of p53 [2]. Later studies showed that SESN2 is transcriptionally activated by hypoxia-inducible factor-1 (HIF-1), a primary adaptive responsor to hypoxia [11, 12]. Induced SESN2 contributes to cellular self-protection by mitigating hypoxia-caused oxidative damage and cell death [2, 11, 13]. Another benefit of SESN2 to hypoxic injury is that SESN2 can deprive the induction of HIF-1 alpha subunit (HIF-1α) on vascular endothelial growth factor (VEGF) expression and brain-blood-barrier permeability, where may involve a mechanism that SESN2 facilitates HIF-1α degradation via enhancing the catalytic activity of prolyl hydroxylase (PH), an essential enzyme hydroxylating HIF-1α for ubiquitination [14]. Hence, SESN2 may be a preferable therapeutic target for hypoxia-related diseases.
SESN2 in genotoxic stress
DNA damage can be caused by various endogenous or exogenous insults, including oncogenic mutations, oxidative stress, metabolic stress, etc. [15]. P53 is an important guardian of genome that can be activated under genotoxic and oxidative stress to promote genomic repair through induction of specific target genes. Upon DNA damage, SESN2 is induced in a p53-dependent manner [2]. SESN2 mediates p53-initiated mTORC1 inhibition, eliciting a metabolic checkpoint in response to genotoxic stress and executing the genomic guardianship [3, 16]. Serine/threonine kinase 3 (AKT3) signal is also involved in inducing SESN2 and then performing DNA repair [17]. This shows that SESN2 is a critical gatekeeper of genome.
SESN2 in oxidative stress
Oxidative stress is triggered by redox imbalance after hyperactive oxidative system and/or hypoactive antioxidative defense. In response to oxidative stress, SESN2 can be transcriptionally activated by multiple transcription factors including nuclear factor E2-related factor 2 (Nrf2), CCAAT enhancer binding protein β (C/EBPβ), HIF-1, p53, activator protein-1 (AP-1), forkhead box protein O3 (FoxO3), nuclear factor-kappa B (NF-κB), and activating transcription factor 4 (ATF4) [18,19,20,21,22,23,24,25].
SESN2 architects cellular defense against redox imbalance generated by stimuli such as hydrogen peroxide [21], angiotensin II [26], methylglyoxal [27], etc., mainly via three patterns. Firstly, with the alkyl hydroperoxide reductase-like structure, SESN2 can directly function as an oxidoreductase to scavenge free radicals. However, the catalytically-crucial residue Cys125 within the NTD is surrounded by hydrophobic molecular surfaces, rendering SESN2 preferentially affinitive towards hydrophobic alkyl hydroperoxides rather than hydrogen peroxide. Also, its physiological substrates and reducing partners need clarification [4]. Secondly, SESN2 has physical-biological interactions with multiple redox regulators, among which the interaction between SESN2 and Nrf2-antioxidant-response element (ARE) antioxidant system has been the best studied [28,29,30]. Nrf2 is a core transcriptional regulator of antioxidant systems based on Mafs-mediated heterodimerization and ARE binding machinery. SESN2 but not SESN1 or SESN3 is exclusively induced at both transcriptional and translational levels by Nrf2 agonists and deprived when Nrf2 deletion. In silico and laboratory analyses displayed an ARE sequence (-550 to -539 bp) in the 5’ upstream region of SESN2 gene promoter for Nrf2 binding [31]. Alternatively, induced SESN2 contributes to Nrf2 expression and nuclear translocation, amplifying the transcription of ARE-targeted antioxidant genes, including heme-oxygenase-1 (HO-1) and NAD(P)H: quinone reductase 1 (NQO1) [28].
Thirdly, SESN2 can motivate macroautophagy (hereafter autophagy) machinery to clear defective proteins and organelles and recover redox balance, which is mainly achieved by inhibiting mTORC1, a pivotal checkpoint for autophagy, via both adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms [32,33,34,35]. For AMPK-dependent mechanism, SESN2 increases the transcription of AMPKα1, AMPKβ1, and AMPKγ1 subunits, facilitates the formation of AMPKα1β1γ1 heterotrimer, and evokes AMPK activity via liver kinase B1 (LKB1)-catalyzed AMPKα1 phosphorylation at Thr127 [36]. AMPK phosphorylates tuberous sclerosis 2 (TSC2), the GTPase-activating protein (GAP) of the Ras homolog enriched in brain (Rheb). TSC2 facilitates the hydrolysis of Rheb-bound GTP and converts it to inactivated GDP-bound form, hindering Rheb interaction with the catalytic domain of mTOR and mTORC1 phosphorylation [3]. AMPK also phosphorylates the critical mTORC1 binding subunit regulatory associated protein of mTOR (Raptor) at two highly conserved serines, Ser722 and Ser792, and induces their direct binding to 14-3-3 protein, restricting the kinase activity of mTORC1 towards its downstream substrates [37]. mTORC1 when its catalytic activity is blocked unfreezes Unc-51-like protein kinase 1 (ULK1), which then forms an active complex via autophosphorylation and phosphorylation of autophagy-related protein 13 (Atg13), focal adhesion kinase interacting protein of 200 kD (FIP200), and Atg101, and activates autophagy [38]. ULK1 can also be phosphorylated by AMPK at multiple active residues and directly initiates autophagy [39]. In a second way independent of AMPK, SESN2 physically interacts with GATOR2 and releases GATOR1 acting as a GAP for Rag A/B, limiting mTORC1 translocation to lysosomal surface where to be activated by Rheb and provoking autophagy [40]. Physiological and pharmacological induction of SESN2 can contribute to autophagy marker light chain 3 (LC3)-II expression and autophagosome formation, suppressing mitochondrial dysfunction and oxidative stress [21, 41]. A possible mechanism for SESN2-regulated autophagy to ease oxidative stress may be that SESN2 physically associates with ULK1 and autophagic cargo receptor p62/sequestosome-1 (SQSTM1) to form a complex, facilitating p62/SQSTM1 phosphorylation at Ser403 and autophagic degradation of p62/SQSTM1 and its substrates [42], such as Kelch-like ECH-associated protein 1 (Keap1), a Nrf2 suppressor that can exclusively bind to the evolutionarily conserved N-terminal Neh2 regulatory domain of Nrf2 and facilitate its ubiquitylation and degradation in cytoplasm with the collaboration of Cullin3 and ring-box 1 (RBX1) [43, 44]. The autophagic degradation of Keap1 can promote the expression of Nrf2 downstream genes, including sulfiredoxin (Srx), glutathione-S-transferase (GST), and NQO1 [45]. More specifically, SESN2 can activate mitophagy, a mitochondrion-selective autophagic machinery, to remove damaged mitochondria for restoring redox homeostasis. Parkin is the predominant E3 ubiquitin ligase that can be recruited to mitochondria and phosphorylated by PTEN-induced kinase 1 (PINK1) upon mitochondrial damage. Then, Parkin integrates with ubiquitin and ubiquitylates substrates on mitochondrial outer membrane for recognition by autophagic cargo receptors and mitophagy formation. SESN2 amplifies PINK1/Parkin-mediated mitophagy by two main manners. On one hand, SESN2 interacts with ULK1 to phosphorylate Beclin1 at Ser14, promoting Beclin1 to bind to and phosphorylate Parkin and helping Parkin translocate to mitochondria [46]. Moreover, SESN2 can directly interact with Parkin, reinforcing the mitochondrial accumulation of Parkin [47]. On the other hand, SESN2 can facilitate the perinuclear-clustering of mitochondria by mediating p62/SQSTM1 aggregation and its binding to lysine 63 (K63)-linked ubiquitin on mitochondrial surface [48, 49]. There is another possible mode for SESN2 regulation on mitophagy that SESN2 can directly interact with mitochondrial alpha-subunit of F1-ATP synthase (ATP5A) through the CTD, attracting LC3-coated autolysosomes to locate ROS-damaged mitochondria for degradation [50]. There also forms a loop between ULK1 and SESN2 that SESN2 can be phosphorylated by ULK1 at Ser73 and Ser254, which is required for mitochondrial fusion with autophagosomes [50]. The multicomponent redox regulatory network centering on SESN2 is shown in Fig. 2.

SESN2 is up-regulated during oxidative stress induced by hydrogen peroxide, methylglyoxal, angiotensin II, etc., which is facilitated by transcription factors including C/EBPβ, HIF-1, p53, Nrf2, AP-1, FoxO3, NF-κB, and ATF4. (i) SESN2 restrains mTORC1 activity through directly binding to GATOR2. GATOR1 is released from GATOR1/2 complex and inactivates Rag A/B via promoting GTP hydrolysis, which prevents mTORC1 binding and recruitment to lysosome. ULK1 is dissociated, then auto-phosphorylates, phosphorylates the complex components including Atg13, FIP200, and Atg101, and promotes autophagy. SESN2 also indirectly inactivates mTORC1 in an AMPK-dependent way. SESN2 promotes AMPK activation and Raptor phosphorylation to inhibit mTORC1 activation. Alternatively, AMPK activation leads to TSC2 phosphorylation and inhibits GTP binding to Rheb, which halts mTORC1 activation. (ii) SESN2-ULK1 interaction promotes the autophagic degradation of Keap1 in a p62/SQSTM1-dependent manner, and accelerates Nrf2 nuclear translocation, which contributes to the formation of Nrf2/Mafs/ARE complex and the expression of downstream antioxidant genes including SESN2. (iii) SESN2 interacts with ULK1 and phosphorylates Beclin1, which anchors Parkin before its location to mitochondria, reinforces PINK1-Parkin interaction, and initiates mitophagy. SESN2 also interacts with Parkin, facilitating its mitochondrial translocation and mitophagy. Alternatively, SESN2 colocalizes with ATP5A on the outer mitochondrial membrane where ATP5A attaches LC3 directly to trigger mitophagy.
SESN2 in ER stress
Prolonged and unresolved ER stress are closely related to homeostasis disequilibrium and cell death. ER stress can activate three unfolded protein response (UPR) branches to orchestrate the recovery of ER function, including protein kinase RNA-like ER kinase (PERK)-eukaryotic translation initiation factor 2α (eIF2α)-ATF4 branch, inositol-requiring enzyme 1 alpha (IRE1α)-X-box binding protein 1 (XBP1) branch, and ATF6 branch [51, 52]. PERK-eIF2α branch can reduce protein synthesis. Paradoxically, preferential translation of ATF4 binds to C/EBP homologous protein (CHOP), a pro-apoptotic transcript, aggravates protein synthesis, ATP depletion, oxidative stress, and cell death [53, 54]. IRE1α-XBP1 and ATF6 branches can up-regulate the transcription of XBP1, glucose regulated protein 78 (GRP78), and GRP94, accelerating misfolded protein degradation and accurate protein folding [55, 56]. All three branches contribute to ER stress-mediated SESN2 induction, but the pathways involved vary among different stress inducers. ATF4 mediates both thapsigargin (Tg)- and brefeldin A (BFA)-induced SESN2 transactivation possibly by binding the site (-221 to -228 bp) within the upstream region of SESN2 transcription start site [57, 58]. XBP1 also transmits the activation signal of Tg to SESN2, but the regulatory pattern requires further verification [57]. ATF6 mainly governs tunicamycin (Tm)-triggered SESN2 enhancement, which can be implemented by being affinitive to UPR-element-like element 1 (UPRE-LE1) (-549 to -544 bp) and UPRE-LE6 (-235 to -230 bp) in the proximal half region of SESN2 gene promoter [59].
Inducible SESN2 functions as a feedback modulator for ER stress and manipulates cell fates. SESN2 contributes to cell survival under Tg and BFA treatment, which is especially obvious at the early stimulation phase, indicating that progressive auto-activated SESN2 can mitigate mild ER stress [57]. Several studies have reached a consensus that SESN2 links ER stress to AMPK-mTORC1 signaling [60,61,62,63,64,65]. SESN2 deficiency augments ER stress-related signaling including PERK and IRE1α, which is associated with AMPK inactivation [61]. SESN2 knockdown also maintains PERK-eIF2α-CHOP signal transduction, leading to mTOR activation [66]. SESN2-mediated mTORC1 inhibition recovers ER homeostasis by two ways. One is that lack of mTOR-catalyzed phosphorylation of downstream molecules [ribosomal protein S6 kinase (S6K) and eIF4E-binding protein (4E-BP)] interrupts protein synthesis [60, 65]. The other one is that mTORC1 suppression initiates autophagy to eliminate misfolded proteins [63, 66, 67]. Cell death as a consequence of unresolved ER stress is also impressed by SESN2. SESN2 expression lessens ER stress-related apoptosis of dendritic cells (DCs), endothelial cells, and trophoblast cells [68,69,70]. SESN2 deficiency augments PERK-ATF4-CHOP signaling to induce NACHT, LRR, and PYD domains-containing protein 3 (NLRP3)/apoptosis-associated speck-like protein containing CARD (ASC)/Caspase-1-dependent cell pyroptosis [71, 72]. SESN2 can also defend against ER stress-associated non-canonical necroptotic death [73]. Furthermore, SESN2 can convalesce intracellular redox homeostasis to recover ER quality and function by enhancing the transcriptional activity of Nrf2 [74]. The modes of SESN2 to recover ER homeostasis is summarized in Fig. 3.

ER stress agonists, thapsigargin, brefeldin A, and tunicamycin, can excite ER stress via inducing GRP78 dissociation and liberation of IRE1α, PERK, and ATF6. IRE1α phosphorylates and forms a dimer, cleaving XBP1 mRNA into an active form XBP1s. ATF6 when liberated translocates into Golgi and is lysed to ATF6p50 by S1P/S2P. Both XBP1 and ATF6p50 as transcription factors can up-regulate the transcription of downstream genes including XBP1, GRP78, GRP94, and SESN2, which promotes protein folding and misfolded protein degradation, and relieves ER stress. Induced SESN2 can arrest protein synthesis and enhance autophagy via AMPK-mTORC1 pathway and reduces oxidative damage via Nrf2. PERK phosphorylates and dimerizes, phosphorylating downstream elF2α and subsequently promoting the transcriptional activity of ATF4. ATF4 can initiate the transcription of CHOP, enhancing protein synthesis, apoptosis, and pyroptosis. ATF4 can also induce SESN2 expression.
SESN2 in inflammation
Inflammation generally involves both immune cells and non-immune cells. SESN2 is expressed in diverse immune cells, especially in macrophages and monocytes that are essential for innate immune response [75]. In bone marrow-derived mononuclear macrophages (BMDMs), NO and hypoxia transcriptionally activate SESN2 in a HIF-1α-dependent manner to resist cellular oxidative damage [20]. HIF-1α also mediates globular adiponectin (gAcrp)-induced SESN2 expression and anti-inflammatory response, which is under the regulation of extracellular regulated protein kinase (ERK)/phosphoinositide 3-kinase (PI3K) signaling [76]. Lipopolysaccharide (LPS) and other toll-like receptor (TLR) ligands (e.g. polyI:C and peptidoglycan) activate PI3K and p38 MAPK signals by conjugating with TLR, prompting AP-1 and Nrf2 induction on the transcription and translation of SESN2 [22, 77]. Inducible nitric oxide synthase (iNOS)-mediated NO production is also implicated in LPS-stimulated SESN2 elevation [48]. SESN2 can restrain p38 MAPK and c-Jun N-terminal kinase (JNK) phosphorylation to inhibit the DNA-binding activity of AP-1 and the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, limiting release of pro-inflammatory cytokines [e.g. tumor necrosis factor alpha (TNF-α), IL-6, and IL-1β], ROS production, and cell death [77]. Oxidized low-density lipoprotein (OxLDL) elicits SESN2 expression by JNK/c-Jun signaling pathway to decay ROS generation and apoptosis in macrophages [78]. Furthermore, in a model of myocardial infarction, SESN2 is up-regulated in both pro-inflammatory M1 and anti-inflammatory M2 type cardiac macrophages, and SESN2 suppresses inflammatory response of M1 macrophages via inhibiting mTORC1 signaling and enhances M2 type macrophage polarization [79]. In cochlear tissues, SESN2 loss occurs with age and accelerates age-related sensory cell degeneration, which is correlated with overproduction of pro-inflammatory cytokines including TNF, chemokine (c-c motif) ligand 2 (CCL2/MCP-1), CCL3, CCL4, and IL-1β in cochlear macrophages [80]. In a model of acute cerebral ischemic stroke, ectopic SESN2 expression promotes the shifting of brain-resident macrophage/microglia from M1 to M2 phenotype and alleviates neuroinflammation by inhibiting mTOR pathway and restoring autophagic flux [81].
In monocytes, human acute monocytic leukemia cell line THP-1 cells for example, SESN2 is induced by LPS dose- and time-dependently, establishing a compensatory mechanism under p38 MAPK and PI3K activation by augmenting AMPK phosphorylation, decreasing NF-κB phosphorylation, and reducing secretion of pro-inflammatory cytokines (TNF-α, CCL2/MCP-1, and IL-6) [22, 61]. A later study supplemented that both high glucose and OxLDL can mediate monocyte polarization, which is characterized by increased M1 markers like iNOS, IL-6, TNF-α, etc. and decreased M2 markers like TGF-β, IL-10, etc., and monocyte adhesion to endothelial cells via SESN2-AMPK-mTOR nexus [82].
NLRP3 is predominantly expressed in immune cells from the myeloid lineage, such as macrophages, monocytes, and DCs [83]. In Sesn2-knockout BMDMs, mitophagy is deficient but NLRP3 inflammasome is hyperactivated when primed with LPS and ATP. Mechanistically, SESN2 enhances p62/SQSTM1 aggregation to K63-ubiquitinated mitochondria. Synchronously, SESN2 facilitates ULK1-mediated initiation of autophagic machinery and launches the degradation of primed mitochondria, which diminishes mitochondrial ROS and cytosolic oxidized mitochondrial DNA generation, and suppresses prolonged NLRP3 inflammasome activation and inflammatory cytokine release (IL-1β and IL-18) [48, 84]. In an acute lung injury model, SESN2 suppresses NLRP3 activation and pyroptosis in alveolar macrophages via promoting PINK/Parkin-mediated mitophagy [49]. In sepsis models, SESN2 levels in blood monocytes negatively correlate with serum IL-1β and IL-18 levels and disease progression [48]. SESN2 also reduces gasdermin D (GSDMD)-dependent pyroptosis of splenic DCs in the context of sepsis via inhibiting PERK-ATF4-CHOP signaling-triggered NLRP3/ASC pyroptosome formation and Caspase-1 activation [71]. Collectively, SESN2 has pleiotropic functions in immune cells and exert robust anti-inflammatory activities (Fig. 4).

SESN2 is expressed in immune cells including macrophages, monocytes, and dendritic cells, and architects cellular responses to inflammatory insults. SESN2 when induced by stimuli can exert robust anti-inflammatory function, which is associated with scavenging ROS, promoting autophagy, reducing inflammasome, and diminishing cell death.
Dynamics and control of SESN2 in liver diseases and implications
Acute liver injury (ALI)
ALI is pathologically featured by extensive hepatocyte death and hypohepatia, which is generally caused by virus infection, drug abuse, hepatectomy, etc. Several studies collaboratively confirmed that preventative SESN2 overexpression can suppress galactosamine/LPS or acetaminophen-induced acute hepatocyte apoptosis and serum cytokine elevation, which is attributed to the antioxidative property of SESN2 [22, 77, 85]. Pharmacological induction of SESN2 by oleanolic acid also prevents hepatic ischemia reperfusion injury [86]. These findings uncover the potential of SESN2 regulation in the prevention of liver-related conditions.
Fatty liver diseases
Non-alcoholic fatty liver disease (NAFLD)
NAFLD is one of the most common chronic liver diseases with a soaring worldwide prevalence, which generally starts from simple steatosis and progresses to steatohepatitis, hepatic cirrhosis, and/or hepatobiliary malignancies such as hepatocellular carcinoma (HCC) [87].
Glucose and lipid metabolism
Excessive fat deposition in hepatocytes, also mentioned as hepatocyte steatosis, is the most remarkable pathological manifestation of NAFLD, where obesity and insulin resistance are major risk factors [88]. SESN2 is the only isoform among three family members that is inducible by saturated fatty acids in hepatoma cell line HepG2 cells. However, under chronic NAFLD conditions, SESN2 gene expression in both human and murine livers is decreased [89]. Sesn2 knockout aggravates glucose intolerance, insulin resistance, hepatocyte apoptosis, macrophage infiltration, and hepatic stellate cells (HSCs) activation in wild-type C57BL/6 J mice fed with high-fat diet (HFD) or in Lepob/ob mice, which involves AMPK inhibition and mTORC1-S6K activation; Ectopic SESN2 reconstitution can rescue SESN2-deficient mice from HFD-caused liver damage [60, 90]. Kowalsky et al. further elaborated that adenovirus-mediated systematic SESN2 overexpression decreases the expression of gluconeogenic and lipogenic genes in the liver, including acetyl-CoA carboxylase alpha (ACACA), ACACB, and fatty acid synthase (FASN), resulting in lower basal and insulin-reduced blood glucose levels, and hepatic lipid accumulation under HFD condition. Molecularly, they found that SESN2 induces AKT activation, an essential signal molecule responsible for glucose and lipid regulation, via mTORC2 but not mTORC1 or AMPK. SESN2 indirectly binds to mTORC2 relying on SESN2-GATOR2-mTORC2 interaction via WD repeat domain 24 (WDR24) and WDR59, which then facilitates AKT S473 phosphorylation. SESN2 also directly binds to the pleckstrin homology domain of AKT and induces AKT translocation to the plasma membrane. PI3K is also involved in the activation of AKT by SESN2 [91]. The regulatory action of SESN2 on hepatic lipogenesis is also relevant to liver X receptor alpha (LXRα), a transcription factor controlling de novo fatty acid synthesis. SESN2 induced by resveratrol represses LXRα-retinoid X receptor alpha (RXRα) DNA-binding activity and restricts the expression of lipogenic gene sterol regulatory element-binding protein-1c (SREBP-1c) and its target genes, including ACC, FASN, and stearoyl-CoA desaturase-1 (SCD1) [92, 93].
Accumulative evidence has shown that autophagy machine is impaired during the onset of NAFLD [94]. SESN2-deficient murine livers under HFD exposure show more and larger lipid droplets and lesser colocalization of lipid droplets and autophagic vesicles under transmission electron microscope, which implies deficient autophagic degradation of lipid droplets (as known as lipophagy) [90]. Notably, SESN2 induced by carbon monoxide restores autophagy in mouse hepatocytes AML12 cells and murine livers under methionine/choline deficiency (MCD) conditions via AMPK-mTORC1 axis, reducing triglyceride accumulation and hepatocyte damage [95]. These suggest that autophagy maintenance or enhancement may be a strategy for clearing excessive lipid droplets and ameliorating steatosis.
Oxidative stress and ER stress
Oxidative stress is another hallmark of NAFLD. Excessive free radicals can attack unsaturated fatty acids on biological membranes, inducing lipid peroxidation, destroying membrane structure, and leading to cell damage [96]. SESN2 is an important endogenous defender with prominent antioxidant capacity. Bae et al. found that SESN2 enhances p62/SQSTM1-mediated autophagic degradation of Keap1 and facilitates Nrf2 release and activation, thereby alleviating oxidative liver damage [45]. Han et al. further verified that pharmacological induction of SESN2 by liraglutide promotes the transduction of Nrf2/HO-1 pathway and initiates the translation of downstream targets, including catalase (CAT), NQO1, and glutamate cysteine ligase modifier subunit (GCLM), in livers of HFD mice, which contributes to the recovery of redox balance [97].
ER stress is also involved in the pathological mechanism of NAFLD. HFD evokes hepatic ER stress, characterized by increased expression of GRP78, ATF6, IRE1α, and eIF2α [60, 98]. Park et al. found that SESN2 is up-regulated by HFD via PERK-eIF2α-c/EBPβ signaling, which then regulates AMPK-mTORC1 axis to impede protein synthesis, relieve ER stress, and balance liver metabolism during obesity [60]. Jegal et al. linked SESN2 with oxidative stress and ER stress and confirmed the role of Nrf2-induced SESN2 in relieving Tm-induced ER stress-related liver injury [99].
Alcoholic fatty liver disease (AFLD)
AFLD, which is caused by chronic alcohol binge, is another major branch of fatty liver diseases. The disease spectrum of AFLD is similar to NAFLD and AFLD shares a large set of common pathological manifestations with NAFLD, such as hepatosteatosis, oxidative stress, ER stress, autophagy malfunction, etc. This suggests that the cytoprotective function and mechanism of SESN2 in AFLD may be analogous to that in NAFLD but still awaits further verification. More recently, Zhou et al. discovered that SESN2 is declined in murine livers after chronic alcohol exposure for 4 weeks and in human hepatocyte HL-7702 cells after 24-h alcohol stimulation. Pharmacological induction of SESN2 by pterostilbene significantly improves AFLD, featured by decreased serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) activity and reduced intrahepatic CD45+ leukocyte and F4/80+ macrophage/Kupffer cell infiltration. Mechanistically, pharmacologically-forced SESN2 expression promotes autophagic machinery and selective degradation of cellular communication network factor 1 (CCN1) via p62/SQSTM1, then mitigating hepatocyte senescence and senescence-associated secretory phenotype under alcohol exposure [100].
Together, SESN2 confers hepatocyte protection during fatty liver diseases, which is probably associated with regulation on glycolipid metabolism, oxidative stress, ER stress, autophagy, senescence, etc. (Table 1). Pharmacological induction and genetical reconstitution of SESN2 may be medically favorable for the improvement of fatty liver diseases.
Hepatic fibrosis and cirrhosis
Hepatic fibrosis and cirrhosis are common advanced stages after chronic liver injuries and inflammatory response, including fatty liver diseases, viral hepatitis, etc. Fibrogenesis is featured by excessive extracellular matrix accumulation, which destroys intrahepatic structure, disrupts biological exchange of substances between hepatocytes and hepatic sinusoids, and is accompanied by extensive hepatocyte death, HSCs activation, and hypohepatia [101].
HSCs activation
SESN2 was firstly found to confer protection against hepatic fibrogenesis in obese mice and then in carbon tetrachloride (CCl4)- and bile duct ligation (BDL)-insulted fibrotic mice [60, 102, 103]. HSCs activation is the key event driving hepatic fibrogenesis. In primary HSCs isolated from murine livers that exposed to single dose of CCl4, SESN2 expression is markedly elevated. Concertedly, SESN2 is transcriptionally up-regulated in primary murine HSCs during in vitro auto-activation or in immortalized human HSCs line LX-2 cells stimulated by transforming growth factor-β (TGF-β) for 0–12 h [103]. Intriguingly, TGF-β induction for 48 h or long-term CCl4 damage for 8 weeks results in SESN2 reduction in rat HSC-T6 cells or murine livers [102]. Clinical liver specimens from advanced cirrhotic patients also show decreased hepatic SESN2 expression [103]. These findings suggested that SESN2 expression varies in different trends during early and advanced fibrotic responses and this may be valuable to the therapeutic discovery, but the molecular basis for this dynamic alternation needs further exploration. HSCs-specific delivery of SESN2 reduces α-SMA-labeled activated HSCs and collagen deposition, thereby ameliorating prolonged CCl4- or BDL-induced hepatic fibrosis in mice. Mechanistically, there has a possible interaction between SESN2 and TGF-β, a signal molecule that can activate adjacent quiescent HSCs, transform HSCs into myofibroblasts, and promote fibrosis development. TGF-β induction causes Smad3 phosphorylation and augments the binding of p-Smad3 to a putative Smad-binding element within SESN2 gene promoter (-964 to -956 bp). In addition to Smad-dependent pathway, TGF-β-induced p38 MAPK activation and ROS production are also involved in SESN2 induction. SESN2 inhibits Smad3 phosphorylation but enhances Smad7 expression [102, 103]. Smad7 is a negative regulator of TGF-β/Smad pathway as Smad7 can bind to TGF-β receptor I (TGFβRI) and prevent the phosphorylation of Smad2 and Smad3, or recruit E3 ubiquitin ligase Smad ubiquitination regulatory factors (Smurfs) to Smad2 and TGFβRI and ubiquitinate and degrade the two proteins [104].
Inflammation
Liver inflammation is a key driver of HSCs activation and fibrogenesis. Hu et al. observed that lentiviral SESN2 overexpression abrogates CCl4-induced elevation of pro-inflammatory cytokines including TNF-α, IL-1β, and CCL2/MCP-1 in murine livers [102]. Yang et al. also found that recombinant adenovirus expressing SESN2 reduces CD45+ leukocytes in murine fibrotic livers caused by CCl4 or BDL [103]. Recently, Zhou et al. delineated that pharmacological induction of SESN2 decreases the number of CD45+ leukocytes and F4/80+ macrophages/Kupffer cells in murine livers under long-term alcohol exposure [100]. These findings imply that the anti-inflammatory action of SESN2 may be based on its modulation of inflammatory cell infiltration and activation in the liver.
In summary, current studies have preliminarily revealed the implication of SESN2 in ameliorating hepatic fibrosis (Table 2). However, fibrogenesis involves multiple types of hepatic cells including HSCs, hepatocytes, Kupffer cells, liver sinusoidal endothelial cell, etc., thus, whether and how SESN2 in other types of hepatic cells influences the process of hepatic fibrosis remain inconclusive.
Liver cancer
Liver cancer has the sixth highest incidence and the fourth highest mortality rate among cancers worldwide, which can be derived from chronic liver diseases, etc. HCC accounts for approximately 90% of liver cancer cases [105]. Chen et al. and Qi et al. reported that SESN2 expression is dramatically lower in HCC tissues than that in adjacent non-cancerous tissues, which is highly correlated with lymph node metastasis, tumor progression, and poor prognosis in HCC patients [106, 107]. However, Dai et al. disputed that SESN2 abundance is higher in HCC tissues than that in corresponding adjacent non-cancerous liver tissues. Coherently, SESN2 levels are higher in HCC cell lines, including Bel-7404, SNU-368, HLE, HLF, and Hep3B cells, comparing with normal human hepatocytes HL-7702 cells [108]. The contradictory findings between the studies may be owing to the discrepancy and insufficiency of HCC samples or the comorbidities in HCC patients, which needs more comprehensive explorations.
Autophagy
SESN2 has been well-documented in initiating autophagy, however, the role of autophagy machinery in HCC is paradoxical, so is SESN2. Wang et al. found that fangchinoline induces autophagic death of hepatoma cell lines HepG2 and PLC/PRF/5 cells via activating p53/SESN2/AMPK signaling [109]. Qi et al. confirmed that SESN2/AMPK/mTOR1 signaling induced by muscone triggers autophagy-dependent apoptosis of HepG2 cells [107]. The researches highlight the anti-oncogenic effect of SESN2 and autophagy. However, autophagy is defined as a double-edged sword as it can be beneficial to cancer cells by preventing oxidative stress, DNA damage, and inflammation, or starvation [110]. Jegal et al. found that SESN2-dependent autophagy induced by eupatilin protects HepG2 cells from arachidonic acid and iron-induced oxidative stress and promotes cell survival [41]. The heterophany of SESN2-dependent autophagy in HCC may be associated with the difference in metabolic environments of HCC cells or drug administration that may activate unrevealed signaling cascades.
Cell survival and death
Induction of apoptotic cell death can restrain the proliferation, invasion, and migration of HCC cells, which may be a viable anti-cancer strategy [111, 112]. Several studies have confirmed that SESN2 can promote apoptosis of multiple types of cancer cells, including human head and neck cancer cells, lung adenocarcinoma cells, and colon cancer cells [113,114,115]. However, intriguingly, up-regulated SESN2 in HepG2 cells halts cell apoptosis and exacerbates primary resistance to sorafenib, which is attributed to activation of pro-survival AKT and AMPK signaling pathways [108]. Kumar et al. found that SESN2 forms a complex with JNK and FoxO1 and promotes FoxO1 nuclear translocation, elevating the transcriptional level of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). PGC-1α can promote glutamine metabolism, increase mitochondrial biogenesis, and decrease the expression of pro-apoptotic genes, such as p53 up-regulated modulator of apoptosis (PUMA) and B-cell lymphoma-2-associated X protein (Bax), facilitating the survival of HepG2 cells under glucose starvation conditions [116]. The function of SESN2 in different cancer cells may depend on the species or cellular metabolic conditions.
Altogether, SESN2 has been preliminarily shown to regulate autophagy and cell status in HCC (Table 3), but its concrete effects are far from clear. In vivo studies are needed to further determinate the association between SESN2 and HCC pathology, including tumor size and number, tumor stage, survival rate, prognosis, etc., and testify the therapeutic implication of SESN2 modulation.
Conclusion and future directions
In this review, we summarize that SESN2 regulates multiple cellular events including glycolipid metabolism, oxidative stress, ER stress, HSCs activation, inflammation, autophagy, cell survival and death and integrate the multicomponent network involved in SESN2 action. From preclinical studies, we also conclude that SESN2 is involved in the development and progression of acute and chronic liver diseases and serve as an endogenous hepatoprotective molecule.
Evidence has shown that SESN2 is of great clinical significance in a variety of diseases. Circulating SESN2 levels have been identified viable in indicating disease severity or prognosis, including cardiovascular diseases [117,118,119], respiratory diseases [120, 121], neurodegenerative diseases [122, 123], metabolic diseases [124, 125], cancers [106, 126], etc. Since SESN2 expression is dynamically changed during liver pathology, we hypothesize that SESN2 has a good potential as a clinical biomarker and prognostic indicator for liver diseases. In addition, the findings from preclinical studies uncover the favorable outcome of SESN2 regulation in the intervention of liver damage with no obvious side effects, which suggests that SESN2 may be a promising therapeutic target for liver diseases. Future work focusing on identifying compounds that induce or activate SESN2 may drive the development of hepatoprotective strategies. Moreover, direct targeting at SESN2 using genetical techniques like viral vector delivery system may be tested in future clinical trials.
References
Haidurov A, Budanov AV. Sestrin family—the stem controlling healthy ageing. Mech Ageing Dev. 2020;192:111379.
Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–31.
Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–60.
Kim H, An S, Ro SH, Teixeira F, Park GJ, Kim C, et al. Janus-faced Sestrin2 controls ROS and mTOR signalling through two separate functional domains. Nat Commun. 2015;6:10025.
Kim JS, Ro SH, Kim M, Park HW, Semple IA, Park H, et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep. 2015;5:9502.
Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science. 2016;351:53–8.
Lynch CJ, Fox HL, Vary TC, Jefferson LS, Kimball SR. Regulation of amino acid-sensitive TOR signaling by leucine analogues in adipocytes. J Cell Biochem. 2000;77:234–51.
Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, et al. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9:1–8.
Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351:43–8.
Kimball SR, Gordon BS, Moyer JE, Dennis MD, Jefferson LS. Leucine induced dephosphorylation of Sestrin2 promotes mTORC1 activation. Cell Signal. 2016;28:896–906.
Olson N, Hristova M, Heintz NH, Lounsbury KM, van der Vliet A. Activation of hypoxia-inducible factor-1 protects airway epithelium against oxidant-induced barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2011;301:L993–L1002.
Shi X, Doycheva DM, Xu L, Tang J, Yan M, Zhang JH. Sestrin2 induced by hypoxia inducible factor1 alpha protects the blood-brain barrier via inhibiting VEGF after severe hypoxic-ischemic injury in neonatal rats. Neurobiol Dis. 2016;95:111–21.
Liu Y, Li M, Sun M, Zhang Y, Li X, Sun W, et al. Sestrin2 is an endogenous antioxidant that improves contractile function in the heart during exposure to ischemia and reperfusion stress. Free Radic Biol Med. 2021;165:385–94.
Seo K, Seo S, Ki SH, Shin SM. Sestrin2 inhibits hypoxia-inducible factor-1alpha accumulation via AMPK-mediated prolyl hydroxylase regulation. Free Radic Biol Med. 2016;101:511–23.
Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. 2014;20:967–77.
Hay N. p53 strikes mTORC1 by employing sestrins. Cell Metab. 2008;8:184–5.
Zhao B, Shah P, Qiang L, He TC, Budanov A, He YY. Distinct role of Sesn2 in response to UVB-induced DNA damage and UVA-induced oxidative stress in melanocytes. Photochem Photobiol. 2017;93:375–81.
Yang JH, Shin BY, Han JY, Kim MG, Wi JE, Kim YW, et al. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol Appl Pharm. 2014;274:293–301.
Papadia S, Soriano FX, Leveille F, Martel MA, Dakin KA, Hansen HH, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci. 2008;11:476–87.
Essler S, Dehne N, Brune B. Role of sestrin2 in peroxide signaling in macrophages. FEBS Lett. 2009;583:3531–5.
Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Ren Physiol. 2013;305:F495–509.
Kim MG, Yang JH, Kim KM, Jang CH, Jung JY, Cho IJ, et al. Regulation of Toll-like receptor-mediated Sestrin2 induction by AP-1, Nrf2, and the ubiquitin-proteasome system in macrophages. Toxicol Sci. 2015;144:425–35.
Hanus J, Zhang H, Chen DH, Zhou Q, Jin P, Liu Q, et al. Gossypol acetic acid prevents oxidative stress-induced retinal pigment epithelial necrosis by regulating the FoxO3/Sestrin2 pathway. Mol Cell Biol. 2015;35:1952–63.
Wu CL, Chen SD, Yin JH, Hwang CS, Yang DI. Nuclear factor-kappaB-dependent sestrin2 induction mediates the antioxidant effects of BDNF against mitochondrial inhibition in rat cortical neurons. Mol Neurobiol. 2016;53:4126–42.
Liu J, Amar F, Corona C, So RWL, Andrews SJ, Nagy PL, et al. Brain-derived neurotrophic factor elevates activating transcription factor 4 (ATF4) in neurons and promotes ATF4-dependent induction of Sesn2. Front Mol Neurosci. 2018;11:62.
Yi L, Li F, Yong Y, Jianting D, Liting Z, Xuansheng H, et al. Upregulation of sestrin-2 expression protects against endothelial toxicity of angiotensin II. Cell Biol Toxicol. 2014;30:147–56.
Seo K, Seo S, Han JY, Ki SH, Shin SM. Resveratrol attenuates methylglyoxal-induced mitochondrial dysfunction and apoptosis by Sestrin2 induction. Toxicol Appl Pharm. 2014;280:314–22.
Fan Y, Xing Y, Xiong L, Wang J. Sestrin2 overexpression alleviates hydrogen peroxide-induced apoptosis and oxidative stress in retinal ganglion cells by enhancing Nrf2 activation via Keap1 downregulation. Chem Biol Interact. 2020;324:109086.
Chen M, Xi Y, Chen K, Xiao P, Li S, Sun X, et al. Upregulation Sestrin2 protects against hydrogen peroxide-induced oxidative damage bovine mammary epithelial cells via a Keap1-Nrf2/ARE pathway. J Cell Physiol. 2021;236:392–404.
Li Y, Wu J, Yu S, Zhu J, Zhou Y, Wang P, et al. Sestrin2 promotes angiogenesis to alleviate brain injury by activating Nrf2 through regulating the interaction between p62 and Keap1 following photothrombotic stroke in rats. Brain Res. 2020;1745:146948.
Shin BY, Jin SH, Cho IJ, Ki SH. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free Radic Biol Med. 2012;53:834–41.
Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–53.
Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–7.
Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, et al. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle. 2009;8:1571–6.
Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–80.
Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One. 2012;7:e32035.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26.
Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2:pe51.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456–61.
Kovaleva IE, Tokarchuk AV, Zheltukhin AO, Dalina AA, Safronov GG, Evstafieva AG, et al. Mitochondrial localization of SESN2. PLoS One. 2020;15:e0226862.
Jegal KH, Ko HL, Park SM, Byun SH, Kang KW, Cho IJ, et al. Eupatilin induces Sestrin2-dependent autophagy to prevent oxidative stress. Apoptosis 2016;21:642–56.
Ro SH, Semple IA, Park H, Park H, Park HW, Kim M, et al. Sestrin2 promotes Unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014;281:3816–27.
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86.
Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–51.
Bae SH, Sung SH, Oh SY, Lim JM, Lee SK, Park YN, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013;17:73–84.
Kumar A, Shaha C. SESN2 facilitates mitophagy by helping Parkin translocation through ULK1 mediated Beclin1 phosphorylation. Sci Rep. 2018;8:615.
Wang P, Wang L, Lu J, Hu Y, Wang Q, Li Z, et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J Mol Cell Cardiol. 2019;133:125–37.
Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, et al. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 2016;12:1272–91.
Wu D, Zhang H, Wu Q, Li F, Wang Y, Liu S, et al. Sestrin 2 protects against LPS-induced acute lung injury by inducing mitophagy in alveolar macrophages. Life Sci. 2021;267:118941.
Kim H, Jeon BT, Kim IM, Bennett SJ, Lorch CM, Viana MP, et al. Sestrin2 phosphorylation by ULK1 induces autophagic degradation of mitochondria damaged by copper-induced oxidative stress. Int J Mol Sci. 2020;21:6130.
Cho IJ, Oh DH, Yoo J, Hwang YC, Ahn KJ, Chung HY, et al. Allopurinol ameliorates high fructose diet induced hepatic steatosis in diabetic rats through modulation of lipid metabolism, inflammation, and ER stress pathway. Sci Rep. 2021;11:9894.
Sahin E, Bagci R, Bektur Aykanat NE, Kacar S, Sahinturk V. Silymarin attenuated nonalcoholic fatty liver disease through the regulation of endoplasmic reticulum stress proteins GRP78 and XBP-1 in mice. J Food Biochem. 2020;44:e13194.
Rashid HO, Kim HK, Junjappa R, Kim HR, Chae HJ. Endoplasmic reticulum stress in the regulation of liver diseases: Involvement of Regulated IRE1alpha and beta-dependent decay and miRNA. J Gastroenterol Hepatol. 2017;32:981–91.
Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–90.
Zhou X, Fouda S, Li D, Zhang K, Ye JM. Involvement of the autophagy-ER stress axis in high fat/carbohydrate diet-induced nonalcoholic fatty liver disease. Nutrients 2020;12:2626.
Kim SY, Kyaw YY, Cheong J. Functional interaction of endoplasmic reticulum stress and hepatitis B virus in the pathogenesis of liver diseases. World J Gastroenterol. 2017;23:7657–65.
Saveljeva S, Cleary P, Mnich K, Ayo A, Pakos-Zebrucka K, Patterson JB, et al. Endoplasmic reticulum stress-mediated induction of SESTRIN 2 potentiates cell survival. Oncotarget 2016;7:12254–66.
Garaeva AA, Kovaleva IE, Chumakov PM, Evstafieva AG. Mitochondrial dysfunction induces SESN2 gene expression through activating Transcription Factor 4. Cell Cycle. 2016;15:64–71.
Jegal KH, Park SM, Cho SS, Byun SH, Ku SK, Kim SC, et al. Activating transcription factor 6-dependent sestrin 2 induction ameliorates ER stress-mediated liver injury. Biochim Biophys Acta Mol Cell Res. 2017;1864:1295–307.
Park HW, Park H, Ro SH, Jang I, Semple IA, Kim DN, et al. Hepatoprotective role of Sestrin2 against chronic ER stress. Nat Commun. 2014;5:4233.
Hwang HJ, Jung TW, Choi JH, Lee HJ, Chung HS, Seo JA, et al. Knockdown of sestrin2 increases pro-inflammatory reactions and ER stress in the endothelium via an AMPK dependent mechanism. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1436–44.
Jia Y, Zheng Z, Yang Y, Zou M, Li J, Wang L, et al. MiR-4756 promotes albumin-induced renal tubular epithelial cell epithelial-to-mesenchymal transition and endoplasmic reticulum stress via targeting Sestrin2. J Cell Physiol. 2019;234:2905–15.
Li Y, Zhang J, Zhou K, Xie L, Xiang G, Fang M, et al. Elevating sestrin2 attenuates endoplasmic reticulum stress and improves functional recovery through autophagy activation after spinal cord injury. Cell Biol Toxicol. 2021;37:401–19.
Yang X, Xue P, Yuan M, Xu X, Wang C, Li W, et al. SESN2 protects against denervated muscle atrophy through unfolded protein response and mitophagy. Cell Death Dis. 2021;12:805.
Yang Y, Guo G, Zhou W, Ge Y, Fan Z, Liu Q, et al. Sestrin2 protects against bavachin induced ER stress through AMPK/mTORC1 signaling pathway in HepG2 cells. J Pharm Sci. 2021;145:175–86.
Tang Z, Wei X, Li T, Wang W, Wu H, Dong H, et al. Sestrin2-mediated autophagy contributes to drug resistance via endoplasmic reticulum stress in human osteosarcoma. Front Cell Dev Biol. 2021;9:722960.
Oh HJ, Lee S, Park PH. ER stress contributes to autophagy induction by adiponectin in macrophages: Implication in cell survival and suppression of inflammatory response. Cytokine. 2020;127:154959.
Wang LX, Zhu XM, Luo YN, Wu Y, Dong N, Tong YL, et al. Sestrin2 protects dendritic cells against endoplasmic reticulum stress-related apoptosis induced by high mobility group box-1 protein. Cell Death Dis. 2020;11:125.
Fatima MT, Hasan M, Abdelsalam SS, Sivaraman SK, El-Gamal H, Zahid MA, et al. Sestrin2 suppression aggravates oxidative stress and apoptosis in endothelial cells subjected to pharmacologically induced endoplasmic reticulum stress. Eur J Pharm. 2021;907:174247.
Lee S, Shin J, Hong Y, Shin SM, Shin HW, Shin J, et al. Sestrin2 alleviates palmitate-induced endoplasmic reticulum stress, apoptosis, and defective invasion of human trophoblast cells. Am J Reprod Immunol. 2020;83:e13222.
Wang LX, Ren C, Yao RQ, Luo YN, Yin Y, Wu Y, et al. Sestrin2 protects against lethal sepsis by suppressing the pyroptosis of dendritic cells. Cell Mol Life Sci. 2021;78:8209–27.
Han D, Kim H, Kim S, Le QA, Han SY, Bae J, et al. Sestrin2 protects against cholestatic liver injury by inhibiting endoplasmic reticulum stress and NLRP3 inflammasome-mediated pyroptosis. Exp Mol Med. 2022;54:239–51.
Ding B, Parmigiani A, Divakaruni AS, Archer K, Murphy AN, Budanov AV. Sestrin2 is induced by glucose starvation via the unfolded protein response and protects cells from non-canonical necroptotic cell death. Sci Rep. 2016;6:22538.
Park HJ, Yang SG, Koo DB. SESN2/NRF2 signaling activates as a direct downstream regulator of the PERK pathway against endoplasmic reticulum stress to improve the in vitro maturation of porcine oocytes. Free Radic Biol Med. 2022;178:413–27.
Xiao T, Zhang L, Huang Y, Shi Y, Wang J, Ji Q, et al. Sestrin2 increases in aortas and plasma from aortic dissection patients and alleviates angiotensin II-induced smooth muscle cell apoptosis via the Nrf2 pathway. Life Sci. 2019;218:132–8.
Lee S, Pham DV, Park PH. Sestrin2 induction contributes to anti-inflammatory responses and cell survival by globular adiponectin in macrophages. Arch Pharm Res. 2022;45:38–50.
Yang JH, Kim KM, Kim MG, Seo KH, Han JY, Ka SO, et al. Role of sestrin2 in the regulation of proinflammatory signaling in macrophages. Free Radic Biol Med. 2015;78:156–67.
Hu HJ, Shi ZY, Lin XL, Chen SM, Wang QY, Tang SY. Upregulation of Sestrin2 expression protects against macrophage apoptosis induced by oxidized low-density lipoprotein. DNA Cell Biol. 2015;34:296–302.
Yang K, Xu C, Zhang Y, He S, Li D. Sestrin2 suppresses classically activated macrophages-mediated inflammatory response in myocardial infarction through inhibition of mTORC1 signaling. Front Immunol. 2017;8:728.
Zhang C, Sun W, Li J, Xiong B, Frye MD, Ding D, et al. Loss of sestrin 2 potentiates the early onset of age-related sensory cell degeneration in the cochlea. Neuroscience. 2017;361:179–91.
He T, Li W, Song Y, Li Z, Tang Y, Zhang Z, et al. Sestrin2 regulates microglia polarization through mTOR-mediated autophagic flux to attenuate inflammation during experimental brain ischemia. J Neuroinflammation. 2020;17:329.
Sundararajan S, Jayachandran I, Balasubramanyam M, Mohan V, Venkatesan B, Manickam N. Sestrin2 regulates monocyte activation through AMPK-mTOR nexus under high-glucose and dyslipidemic conditions. J Cell Biochem. 2018;120:8201–8213.
Shi Y, Yang Y, Hu Y, Wu W, Yang Q, Zheng M, et al. Acute-on-chronic liver failure precipitated by hepatic injury is distinct from that precipitated by extrahepatic insults. Hepatology 2015;62:232–42.
Wu KKL, Long K, Lin H, Siu PMF, Hoo RLC, Ye D, et al. The APPL1-Rab5 axis restricts NLRP3 inflammasome activation through early endosomal-dependent mitophagy in macrophages. Nat Commun. 2021;12:6637.
Kim SJ, Kim KM, Yang JH, Cho SS, Kim JY, Park SJ, et al. Sestrin2 protects against acetaminophen-induced liver injury. Chem Biol Interact. 2017;269:50–8.
Hao BB, Pan XX, Fan Y, Lu L, Qian XF, Wang XH, et al. Oleanolic acid attenuates liver ischemia reperfusion injury by HO-1/Sesn2 signaling pathway. Hepatobiliary Pancreat Dis Int. 2016;15:519–24.
Li M, Xu C, Shi J, Ding J, Wan X, Chen D, et al. Fatty acids promote fatty liver disease via the dysregulation of 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Gut 2018;67:2169–80.
Farzanegi P, Dana A, Ebrahimpoor Z, Asadi M, Azarbayjani MA. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur J Sport Sci. 2019;19:994–1003.
Fang Z, Kim HG, Huang M, Chowdhury K, Li MO, Liangpunsakul S, et al. Sestrin proteins protect against lipotoxicity-induced oxidative stress in the liver via suppression of C-Jun N-terminal kinases. Cell Mol Gastroenterol Hepatol. 2021;12:921–42.
Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012;16:311–21.
Kowalsky AH, Namkoong S, Mettetal E, Park HW, Kazyken D, Fingar DC, et al. The GATOR2-mTORC2 axis mediates Sestrin2-induced AKT Ser/Thr kinase activation. J Biol Chem. 2020;295:1769–80.
Jin SH, Yang JH, Shin BY, Seo K, Shin SM, Cho IJ, et al. Resveratrol inhibits LXRalpha-dependent hepatic lipogenesis through novel antioxidant Sestrin2 gene induction. Toxicol Appl Pharm. 2013;271:95–105.
Heckmann BL, Zhang X, Saarinen AM, Schoiswohl G, Kershaw EE, Zechner R, et al. Liver X receptor alpha mediates hepatic triglyceride accumulation through upregulation of G0/G1 Switch Gene 2 expression. JCI Insight. 2017;2:e88735.
Wu P, Zhao J, Guo Y, Yu Y, Wu X, Xiao H. Ursodeoxycholic acid alleviates nonalcoholic fatty liver disease by inhibiting apoptosis and improving autophagy via activating AMPK. Biochem Biophys Res Commun. 2020;529:834–8.
Kim HJ, Joe Y, Kim SK, Park SU, Park J, Chen Y, et al. Carbon monoxide protects against hepatic steatosis in mice by inducing sestrin-2 via the PERK-eIF2alpha-ATF4 pathway. Free Radic Biol Med. 2017;110:81–91.
Arroyave-Ospina JC, Wu Z, Geng Y, Moshage H. Role of oxidative stress in the pathogenesis of non-alcoholic fatty liver disease: implications for prevention and therapy. Antioxid (Basel). 2021;10:174.
Han X, Ding C, Zhang G, Pan R, Liu Y, Huang N, et al. Liraglutide ameliorates obesity-related nonalcoholic fatty liver disease by regulating Sestrin2-mediated Nrf2/HO-1 pathway. Biochem Biophys Res Commun. 2020;525:895–901.
Zhang J, Yang P, Wang H, Huang Q, Chen T, Li N, et al. N-3 PUFAs inhibited hepatic ER stress induced by feeding of a high-saturated fat diet accompanied by the expression LOX-1. J Nutr Biochem. 2021;88:108481.
Jegal KH, Kim EO, Kim JK, Park SM, Jung DH, Lee GH, et al. Luteolin prevents liver from tunicamycin-induced endoplasmic reticulum stress via nuclear factor erythroid 2-related factor 2-dependent sestrin 2 induction. Toxicol Appl Pharm. 2020;399:115036.
Jiang Y, Zhou Y, Xu W, Wang X, Jin H, Bao X, et al. Induction of Sestrin2 by pterostilbene suppresses ethanol-triggered hepatocyte senescence by degrading CCN1 via p62-dependent selective autophagy. Cell Biol Toxicol. 2021; https://doi.org/10.1007/s10565-021-09635-8.
Ezhilarasan D, Sokal E, Najimi M. Hepatic fibrosis: It is time to go with hepatic stellate cell-specific therapeutic targets. Hepatobiliary Pancreat Dis Int. 2018;17:192–7.
Hu YB, Ye XT, Zhou QQ, Fu RQ. Sestrin 2 attenuates rat hepatic stellate cell (HSC) activation and liver fibrosis via an mTOR/AMPK-dependent mechanism. Cell Physiol Biochem. 2018;51:2111–22.
Yang JH, Kim KM, Cho SS, Shin SM, Ka SO, Na CS, et al. Inhibitory effect of Sestrin 2 on hepatic stellate cell activation and liver fibrosis. Antioxid Redox Signal. 2019;31:243–59.
Hu HH, Chen DQ, Wang YN, Feng YL, Cao G, Vaziri ND, et al. New insights into TGF-beta/Smad signaling in tissue fibrosis. Chem Biol Interact. 2018;292:76–83.
Altundag O. Recent advances in systemic therapy for hepatocellular carcinoma. Exp Clin Transplant. 2022; https://doi.org/10.6002/ect.2021.0478.
Chen S, Yan W, Lang W, Yu J, Xu L, Xu X, et al. SESN2 correlates with advantageous prognosis in hepatocellular carcinoma. Diagn Pathol. 2017;12:13.
Qi W, Li Z, Yang C, Jiangshan Dai J, Zhang Q, Wang D, et al. Inhibitory mechanism of muscone in liver cancer involves the induction of apoptosis and autophagy. Oncol Rep. 2020;43:839–50.
Dai J, Huang Q, Niu K, Wang B, Li Y, Dai C, et al. Sestrin 2 confers primary resistance to sorafenib by simultaneously activating AKT and AMPK in hepatocellular carcinoma. Cancer Med. 2018;7:5691–703.
Wang N, Pan W, Zhu M, Zhang M, Hao X, Liang G, et al. Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. Br J Pharm. 2011;164:731–42.
Yun CW, Jeon J, Go G, Lee JH, Lee SH. The dual role of autophagy in cancer development and a therapeutic strategy for cancer by targeting autophagy. Int J Mol Sci. 2020;22:179.
Yu L, Chen W, Tang Q, Ji KY. Micheliolide inhibits liver cancer cell growth via inducing apoptosis and perturbing actin cytoskeleton. Cancer Manag Res. 2019;11:9203–12.
Yao J, Fu J, Liu Y, Qu W, Wang G, Yan Z. LncRNA CASC9 promotes proliferation, migration and inhibits apoptosis of hepatocellular carcinoma cells by down-regulating miR-424-5p. Ann Hepatol. 2021;23:100297.
Won DH, Chung SH, Shin JA, Hong KO, Yang IH, Yun JW, et al. Induction of sestrin 2 is associated with fisetin-mediated apoptosis in human head and neck cancer cell lines. J Clin Biochem Nutr. 2019;64:97–105.
Ding BX, Parmigiani A, Yang C, Budanov AV. Sestrin2 facilitates death receptor-induced apoptosis in lung adenocarcinoma cells through regulation of XIAP degradation. Cell Cycle. 2015;14:3231–41.
Kim GT, Lee SH, Kim JI, Kim YM. Quercetin regulates the sestrin 2-AMPK-p38 MAPK signaling pathway and induces apoptosis by increasing the generation of intracellular ROS in a p53-independent manner. Int J Mol Med. 2014;33:863–9.
Kumar A, Giri S, Shaha C. Sestrin2 facilitates glutamine-dependent transcription of PGC-1alpha and survival of liver cancer cells under glucose limitation. FEBS J. 2018;285:1326–45.
Kishimoto Y, Aoyama M, Saita E, Ikegami Y, Ohmori R, Kondo K, et al. Association between plasma Sestrin2 levels and the presence and severity of coronary artery disease. Dis Markers. 2020;2020:7439574.
Fang C, Yang Z, Shi L, Zeng T, Shi Y, Liu L, et al. Circulating sestrin levels are increased in hypertension patients. Dis Markers. 2020;2020:3787295.
Kishimoto Y, Saita E, Ohmori R, Kondo K, Momiyama Y. Plasma sestrin2 concentrations and carotid atherosclerosis. Clin Chim Acta. 2020;504:56–9.
Jiang R, Wang Q, Zhai H, Du X, Sun S, Wang H. Explorating the involvement of plasma Sestrin2 in obstructive sleep apnea. Can Respir J. 2019;2019:2047674.
Zhang DW, Wei YY, Ji S, Fei GH. Correlation between sestrin2 expression and airway remodeling in COPD. BMC Pulm Med. 2020;20:297.
Rai N, Kumar R, Desai GR, Venugopalan G, Shekhar S, Chatterjee P, et al. Relative alterations in blood-based levels of sestrin in Alzheimer’s disease and mild cognitive impairment patients. J Alzheimers Dis. 2016;54:1147–55.
Rai N, Upadhyay AD, Goyal V, Dwivedi S, Dey AB, Dey S. Sestrin2 as serum protein marker and potential therapeutic target for Parkinson’s disease. J Gerontol A Biol Sci Med Sci. 2020;75:690–5.
Mao EW, Cheng XB, Li WC, Kan CX, Huang N, Wang HS, et al. Association between serum Sestrin2 level and diabetic peripheral neuropathy in type 2 diabetic patients. World J Clin Cases. 2021;9:11156–64.
Nourbakhsh M, Sharifi R, Ghorbanhosseini SS, Javad A, Ahmadpour F, Razzaghy Azar M, et al. Evaluation of plasma TRB3 and Sestrin 2 levels in obese and normal-weight children. Child Obes. 2017;13:409–14.
Chen KB, Xuan Y, Shi WJ, Chi F, Xing R, Zeng YC. Sestrin2 expression is a favorable prognostic factor in patients with non-small cell lung cancer. Am J Transl Res. 2016;8:1903–9.
Acknowledgements
The authors thank Prof. Yong Ling (School of Pharmacy and Jiangsu Province Key Laboratory for Inflammation and Molecular Drug Target, Nantong University, Nantong, Jiangsu, China) for providing Molecular Operating Environment (MOE) 2020 software support in protein structure presentation.
Funding
This work was supported by National Natural Science Foundation of China (81803606), Natural Science Research Project of Jiangsu Higher Education Institutions (22KJB360015), Innovation and Entrepreneurship Training Program for Undergraduates of Nantong University (2022233), Scientific Research Project of Jiangsu Commission of Health (Z2021007), and The Sixth 226 High Level Talent Training Project of Nantong City.
Author information
Authors and Affiliations
Contributions
CL and WX conceptualized the project. CL, WX, YJ, and XB conducted literature retrieval. CL, WX, and YJ wrote the original manuscript and drew the figures and tables. CL and WX reviewed and revised the manuscript. CL, WX, and XB provided acquisition. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Francesca Pentimalli
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, C., Jiang, Y., Xu, W. et al. Sestrin2: multifaceted functions, molecular basis, and its implications in liver diseases. Cell Death Dis 14, 160 (2023). https://doi.org/10.1038/s41419-023-05669-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-05669-4
- Springer Nature Limited
This article is cited by
-
Glutamate is effective in decreasing opacity formed in galactose-induced cataract model
Scientific Reports (2024)
-
N-Acetylcysteine Alleviates Necrotizing Enterocolitis by Depressing SESN2 Expression to Inhibit Ferroptosis in Intestinal Epithelial Cells
Inflammation (2024)
-
ACE2 Rescues Sepsis-Associated Encephalopathy by Reducing Inflammation, Oxidative Stress, and Neuronal Apoptosis via the Nrf2/Sestrin2 Signaling Pathway
Molecular Neurobiology (2024)