
Abstract
Pancreatic cancer is an aggressive cancer with a poor prognosis. Metabolic abnormalities are one of the hallmarks of pancreatic cancer, and pancreatic cancer cells can adapt to biosynthesis, energy intake, and redox needs through metabolic reprogramming to tolerate nutrient deficiency and hypoxic microenvironments. Pancreatic cancer cells can use glucose, amino acids, and lipids as energy to maintain malignant growth. Moreover, they also metabolically interact with cells in the tumour microenvironment to change cell fate, promote tumour progression, and even affect immune responses. Importantly, metabolic changes at the body level deserve more attention. Basic research and clinical trials based on targeted metabolic therapy or in combination with other treatments are in full swing. A more comprehensive and in-depth understanding of the metabolic regulation of pancreatic cancer cells will not only enrich the understanding of the mechanisms of disease progression but also provide inspiration for new diagnostic and therapeutic approaches.
Similar content being viewed by others


Key facts
-
Metabolic reprogramming is one of the key hallmarks in pancreatic cancer.
-
Targeting key enzymes in metabolic pathways can affect the progression of pancreatic cancer.
-
The complex tumor microenvironment of pancreatic cancer creates metabolic heterogeneity.
-
Clinical trials exploring metabolic-based treatments for pancreatic cancer have been initiated.
-
Advanced technologies (including preclinical models and detection technologies) continue to promote progress in the field.
Key questions
-
Influencing redox homeostasis brings hope for cancer treatment.
-
Polyamine metabolism and microbial metabolic targets still need further exploration.
-
Interactions between metabolism and epigenetics and metabolic cell death are blue oceans.
-
Systemic metabolism can also serve as a potential pathogenic mechanism and therapeutic target.
-
Breakthroughs are urgently needed to overcome the low specificity and side effects of metabolic therapy.
Introduction
Tumour cells have many unique characteristics, and many of these characteristics, are directly controlled by cell metabolism or amenable to regulation by specific metabolites [1]. Since the Warburg effect was identified a century ago [2], a variety of metabolic reprogramming events have been revealed in tumour cells [3]. The Warburg effect refers to persistent glycolysis even when oxygen is abundant, which provides ATP but is primarily a precursors for the synthesis of various biological macromolecules [4]. With the rapid development of new technologies, it has been gradually discovered that… tumour metabolism involves an overall change at multiple levels of the metabolic network and plays an important role in promoting the occurrence and development of tumours [5, 6].
Metabolic reprogramming [7], which refers to the adaptive changes in the balance of anabolism and catabolism that occur in tumour cells during the malignant development process to meet the large demand for materials and energy, has gradually become recognized as one of the crucial hallmarks of cancer [8]. This reprogramming allows tumour cells to gain advantages and survive in the nutrient-starved tumour microenvironment (TME) caused by rapid proliferation [9].
In contrast to the booming research field, only a few metabolism-based cancer drugs have been successfully developed and are still in the nascent stage due to their low specificity and undesirable side effects [10]. These strategies targeting the intrinsic metabolism of cancer cells usually do not consider the complex crosstalk of noncancerous stromal cells and immune cells, still awaiting further research [11,12,13].
Pancreatic cancer (PC), is an aggressive cancer with a poor prognosis [14]. The overall five-year survival rate is less than 13%, and it is expected that PC will rank second in malignant tumour-related deaths in the United States by 2030 [15]. Exploring the metabolic regulatory network and underlying mechanisms, formulating comprehensive treatment strategies, and promoting precise treatment based on individual patients are expected to improve the overall prognosis.
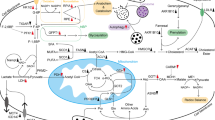
This review summarizes the main themes that are currently under investigation in the context of tumour metabolism and provides an overview of the current status of basic research and clinical trials targeting key enzymes and signaling pathways in PC metabolism (Fig. 1).

Changes in key enzymes and transporters lead to metabolic reprogramming of glucose, amino acids, lipids, etc., and meet the large demand for materials and energy during the malignant proliferation of tumor cells. Enzymes and transporters shown in red indicate significant overexpression; enzymes and transporters shown in green indicate significant downexpression; enzymes shown in black indicate no significant changes or unknown; metabolites shown in orange indicate that they were involved in redox balance; solid arrows imply shifts or bioconversions; dashed lines indicate that the reaction is not direct; italics indicate metabolic pathways or processes. 3-PG 3-bisphosphoglycerate, ACC acetyl-CoA carboxylase, ACAT acetyl-CoA acetyltransferase, ACLY ATP citrate lyase, ACSS acyl-CoA synthetase short-chain family, AHCY adenosylhomocysteinase, ASNS asparagine synthetase, ASL argininosuccinate lythase, ASS argininosuccinate synthase, AK aspartic kinase, AMD aAdenosylmethionine decarboxylase, BCAA branched-chain amino acids, BCKA branched alpha-ketoacids, BCKDH branched alphaketoate dehydrogenase, CE cholesterol ester, CS citrate synthetase, DAG diacylglycerol, DHAP: dihydroxyacetone phosphate, ELOVL FA elongase, FA fatty acid, FABP fatty acid-binding protein, FADS FA desaturase, FASN fatty acid synthase, FATP: fatty acid transport protein, Fructose-6P fructose 6-phosphate, F- 1:6BP fructose 1:6-bisphosphate, F-2:6BP fructose 2:6-bisphosphate, GA3P glyceraldehyde 3-phosphate, GLUT1 glucose transporter 1, GSH glutathione synthesis, GLS glutaminase, GFPT1 glutamine:fructose 6-phosphate amidotransferase 1, Glucose-6P glucose 6- phosphate, GOT1 cytoplasmic aspartate Transaminase, GOT2 mitochondrial aspartate transaminase, HK1/2 hexokinase 1/2, HMGCR: 3-hydroxy-3-methylglutaryl coenzyme A reductase, HMG-CoA 3-hydroxy-3-methylglutaryl coenzyme A, ME1 malic enzyme, LDLR low-density lipoprotein receptor, LDH lactate dehydrogenase, MAG monoacylglycerol, MCT monocarboxylate transporter, MDH1 malate dehydrogenase 1, MTA 5:-methylthioadenosine, MTAP 5-methylthioadenosine, MS methionine synthase, MUFA monounsaturated fatty acid, NAD nicotinamide adenine dinucleotide, NADPH nicotinamide adenine dinucleotide phosphate, NOS nitric oxide synthase, OCT ornithine transcarbamylase, OPLH o-phospho-l-homoserine, PDH pyruvate dehydrogenase, PFK1: phosphofructokinase 1, PRODH1 proline oxidase, PUFA polyunsaturated fatty acid, R-5-P ibose-5-phosphate, RPE ribulose-5-phosphate epimerase, RPIA ribose-5-phoshate isomerase, SAH S-homocysteine; SAM S-adenosine methionine, SCD stearoyl-CoA desaturase, SFA saturated fatty acid, SM squalene monooxygenase, TAG triacylglycerol, TCA tricarboxylic acid.
Metabolic reprogramming of tumour cells
Nucleotide metabolism
The enhanced synthesis and use of nucleotide triphosphates (NTPs) is a universal metabolic dependence across different cancer types [16]. Oncogenic drivers increase nucleotide biosynthetic capacity, which is a prerequisite for cancer initiation and progression [17]. The clinical utility of nucleotide synthesis inhibitors, the first antineoplastic agents discovered, has been demonstrated in many cancers [18]. As they have been extensively studied and due to space limitations, we summarize the key enzymes and inhibitors in nucleotide metabolism in Text Box S1.
In addition to the de novo and salvage pathways, pancreatic ductal adenocarcinoma (PDAC) cells can also acquire nucleotides through autophagy, specifically through the degradation of cellular nucleic acids in autophagosomes, especially in hypoxic and nutrient-poor regions of the TME [19]. Recent studies have also shown that nucleotide metabolism is not only a therapeutic target for chemotherapy but also has the potential to become a sensitization target for immunotherapy, which we will describe later [20].
Glycolysis and OXPHOS
Otto Warburg first discovered in the late 1920s that increased aerobic glycolysis in tumour cells [21], also known as the Warburg effect, allows tumour cells to survive under the harsh conditions of cancer progression [22]. Glycolysis is the primary way PC cells generate energy to sustain malignant proliferation, including under hypoxic conditions [23, 24]. Work in mouse models also demonstrated that glycolysis is the major metabolic effector of oncogenic KRAS [25].
First, the expression of glucose transporter 1 (GLUT1) is upregulated, increasing glucose uptake to enhance subsequent aerobic glycolysis [26]. LIMS1 enhances GLUT1 expression and membrane translocation, which promotes cancer cell survival under oxygen-glucose deprivation [27]. Key enzymes involved in glycolysis play a role in PC. Hexokinase 1/2 (HK1/2) converts glucose into glucose-6-phosphate (G-6-P), the rate-limiting first step in glycolysis. The binding of HK2 to mitochondria increases the glycolytic capacity and promotes PDAC immortalization [28]. Recent research has shown that the endogenous cellular metabolic checkpoint STING can limit aerobic glycolysis by targeting HK2 [29]. Phosphofructokinase (PFK) catalyses the second critical step in glycolysis [30]. The production of the PFKFB-4 and PFKFB-3 isoenzymes is induced by hypoxia in PC [31]. Pyruvate kinase (PK) catalyses the conversion of phosphoenolpyruvate to pyruvate, which is the final rate-limiting step of glycolysis and can transfer metabolites to branch pathways [32]. Some studies have shown that methionine can oxidatively activate PKM2 to promote PC metastasis [33], while other studies have shown that PKM2 expression is not required for carcinogenesis or progression in PDAC [34]. Under conditions of glucose deficiency, fructose can be taken up and metabolized by PDAC cells to promote adaptive survival [35].
The expression of several enzymes involved in glycolysis, such as aldolase [36], glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [37], phosphoglycerate kinase (PGK) [38], phosphoglycerate mutase (PGAM) [39], and alpha enolase (ENO-1) [40], is upregulated in PC. Lactate dehydrogenase (LDH) is overexpressed in PDAC [41], and LDH-mediated catalysis of pyruvate to lactate is significantly increased [42]; thus, LDH may be an effective therapeutic target [43].
In contrast to glycolysis, phosphorylated pyruvate dehydrogenase kinase 1 (PDHK1) inhibits oxidative phosphorylation (OXPHOS) in PDAC [26]. Nonetheless, some studies have shown that OXPHOS is not always suppressed; rather, it can be reactivated under certain conditions [44]. The oncogene KRAS and the loss of LKB1 may drive the upregulation of OXPHOS in cancer [44]. The transcription factor ISL2 can regulate the expression of metabolic genes and affect OXPHOS [45]. As tumours proliferate rapidly, cells in the tumour core become hypoxic, and OXPHOS decreases. Several studies have demonstrated that OXPHOS inhibitors can serve as promising therapeutic agents for treating PDAC [46]. The molecular mechanism underlying the relationship between OXPHOS and PDAC progression still requires in-depth study with bright prospects (Text Box S2).
Amino acid metabolism
Amino acid metabolism, which can affect cancer cell status and systemic metabolism through energy metabolism and signal transduction, is deregulated in PDAC [47].
Glutamine is the most abundant nonessential amino acid in the human body [48]. Cancer cells can convert glutamine into lactic acid, participate in a glucose-free metabolic pathway, take up glutamine, and promote the synthesis of nonessential amino acids and nucleotides through carbon or nitrogen metabolism [49]. Generally, glutamine can be converted in the mitochondria to supplement the tricarboxylic acid (TCA) cycle, or it can be completely oxidized to produce ATP. Many studies have confirmed that glutamine plays a very important role in the migration and invasion of PDAC cells [50,51,52] and relies on noncanonical metabolic pathways. Glutamine-derived aspartate is converted into oxaloacetate by aspartate transaminase GOT1; subsequently, oxaloacetate is converted into malate and then pyruvate, which can potentially maintain redox homeostasis [53]. Loss of SIRT5 promotes tumorigenesis by increasing noncanonical glutamine utilization through GOT1 and is expected to serve as a suppressor in PDAC [54]. GOT1 inhibition-mediated impairment of redox balance synergizes with radiotherapy mouse models [55]. The adaptation of PDAC cells to nutrient deprivation is reversible, and glutamine synthetase (GS) is expected to become a therapeutic target for patients with PDAC [56]. Alanine, serine, and cysteine transporter 2 (ASCT2) is responsible for glutamine transport [57]. Recent studies have shown that CD9 promotes the plasma membrane localization of the glutamine transporter ASCT2, thereby enhancing glutamine uptake [58]. In preclinical models, PI3K-C2γ deficiency can overactivate the mTORC1 pathway and reprogram glutamine metabolism, increasing invasiveness [59]. Targeting glutamine metabolism may provide therapeutic avenues for treating PDAC [60], and glutamine mimicry inhibits tumour progression through asparagine metabolism [61].
In addition, many studies have found that other amino acids, such as serine, tryptophan, methionine, and branched-chain amino acids (BCAAs) such as leucine, play a role in different stages of tumors and are expected to develop new intervention strategies. We summarize them in Text Box S3.
Recently, polyamine metabolism has been redefined as an anticancer strategy and may be involved in antitumour immune responses [62, 63]. The RAS-RAF-MEK-ERK signalling pathway has been shown to control multiple aspects of polyamine metabolism in preclinical models [64]. Arginine deprivation can inhibit the migration, invasion and EMT of PDAC cells [65]. Arginine is the precursor of polyamines, and Lee et al. reported that PDAC cells can synthesize polyamines from glutamine, revealing a new pathway [66]. PDAC can synthesize ornithine from glutamine and support polyamine synthesis through ornithine aminotransferase (OAT), thereby promoting tumour growth [67]. Cancer cells require elevated levels of polyamines to sustain proliferation [67]; microbiota- and diet-related polyamine metabolism will be described later.
Lipid metabolism
Lipids not only provide energy but are also widely distributed in organelles and serve as second messengers to transduce signals within cells [68]. Therefore, lipid metabolism is increasingly recognized as an important pathway for cancer cells [69, 70]. Lipid metabolism reprogramming can be observed even outside histological tumour boundaries [71], and lipidomic analysis of serum holds promise for detecting PDAC [72].
Exogenous fatty acid (FA) uptake confers metabolic flexibility to cancer cells [73]. Cancer cells can absorb fatty acids through fatty acid transporter proteins (FATPs) [74], fatty acid translocase (CD36) [75], and fatty acid binding proteins (FABPs) [76]. CD36 is thought to be involved in the initiation of metastasis [77] and the promotion of fatty acid β-oxidation to support proliferation [78]. Excess exogenous FAs are stored in lipid droplets (LDs), which sequester FAs in the form of triacylglycerides (TAGs) and sterol esters and can be used for energy production or phospholipid synthesis [79]. Increased LD abundance is related to tumour aggressiveness, and reduced LD abundance can reduce the invasive ability of KRAS-mutant PDAC [80]. β-oxidation of stored lipids can produce acetyl-CoA, which is subsequently shuttled through the TCA cycle, providing a valuable source of ATP and NADPH under conditions of metabolic stress [81].
The de novo synthesis of lipids is a metabolic source for tumour cell growth, even in the presence of exogenous lipids [70]. Acetyl-CoA is the main substrate for lipid synthesis. Cancer cells can upregulate acetyl-CoA synthetase 2 (ACSS2) expression to generate acetyl-CoA from acetate [82] or upregulate ATP-citrate lyase (ACLY) expression to convert citrate to acetyl-CoA [83]. Glucose undergoes pyruvate oxidation through the TCA cycle, while glutamine promotes citric acid production through reductive carboxylation [84].
The regulation of de novo lipogenesis mainly occurs at the transcription level, and sterol regulatory element binding proteins (SREBPs) can regulate genes related to fatty acid and cholesterol synthesis and uptake [85]. Liver X receptors (LXRs) [86], whose inhibitory ligands can hinder cell proliferation in various forms of cancer, regulate FA and cholesterol metabolism [87]. Acetyl-CoA carboxylase (ACC) [88], fatty acid synthase (FASN) [89], and stearoyl-CoA desaturase (SCD1) [90] are rate-limiting enzymes for FA synthesis and are significantly highly expressed in PDAC. ACC1 is genetically regulated by SREBP at the transcription level [91]. In recent years, FASN has received increasing attention as a potential target for cancer therapy, and its high expression is associated with poor survival and gemcitabine resistance [92]. Under hypoxic conditions, the flux from glucose to acetyl-CoA is reduced, and saturated fatty acids are regulated by SCD1, reducing the conversion to monounsaturated fatty acids [93]. SCD1 expression is associated with poor prognosis in PDAC patients and can protect cancer cells from ferroptosis, indicating its antitumour potential [94].
Cholesterol plays a key role in maintaining membrane integrity and fluidity and regulating cell signalling events [95]. Abnormal cholesterol metabolism can support PDAC growth [96]. The uptake of cholesterol depends on the LDLR-mediated endocytosis pathway. Low-density lipoprotein (LDL) particles bind to the LDL receptor (LDLR) and are eventually internalized and then reach the lysosome to release free cholesterol [97]. Decreased total cholesterol and LDL levels may be associated with the development of PDAC [98]. Some studies have also shown that LDLR expression is positively correlated with poor prognosis and recurrence [99]. Cholesterol is synthesized through the mevalonate pathway and is regulated by several key enzymes, such as acetyl-CoA acetyltransferase (ACAT) [100], 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) [101], squalene monooxygenase (SM) [102], and sterol-O-acyltransferase (SOAT) [103], which may provide new ideas for the treatment of PDAC [104]. In contrast, the level of high-density lipoprotein (HDL)-cholesterol, which is associated with cholesterol efflux, is significantly inversely related to the risk of cancer [105]. In preclinical models, disruption of cholesterol biosynthesis promotes a shift to a basal phenotype, conferring a poor prognosis [106]. Targeting SOAT1 can affect p53 mutant PDAC organoids that are sensitive to cholesterol metabolism and impair tumor progression [103].
Fatty acid oxidation (FAO), also known as β-oxidation, is increased in many cancer cells because cancer cells can use fatty acid (FA) catabolism to proliferate when ATP is depleted [107, 108]. FAO can participate in tumorigenesis [109] and tumour metastasis [110] and may be related to cachexia [111]. Among different fatty acids, polyunsaturated fatty acids (PUFAs) are easily oxidized, with lipid peroxidation leading to various types of cell death, while saturated fatty acids (SFAs) and monounsaturated fatty acids (MUFAs), on the contrary, are thought to promote cancer growth [112, 113].
Finally, the sophisticated mechanisms and functions of phospholipid metabolism [114] and sphingolipid metabolism [115] have also been increasingly revealed. Increased glycosphingolipid biosynthesis can localize KRAS to the plasma membrane [116]. MUFAs and their linked ether phospholipids play a key role in maintaining reactive oxygen species (ROS) homeostasis [117].
Redox homeostasis
Cellular redox homeostasis is an important process for cell survival, and ROS can cause damage to cellular components [118]. In addition, cancer cells have an increased demand for nicotinamide adenine dinucleotide (NAD+), which leads to a restorative stress state [119]. Several transcription factors are activated by ROS, regulate the redox status of cells and are implicated in carcinogenesis [120]. New technologies for measuring and manipulating reducing stress offer promise for cancer treatment [121].
As mentioned previously, the glutamine metabolic pathway in PDAC cells is often referred to as the noncanonical pathway. Glutamine deprivation [122] and the inhibition of GOT1 [123] or GOT2 [124, 125] increase intracellular ROS. CRISPR/Cas9 ablation of Glutamate–Ammonia Ligase (GLUL) in PDAC mouse models reduced tumor growth [51]. Cancer cells rely on cysteine-derived metabolites such as glutathione (GSH) and CoA to alleviate ROS [118, 126]. GSH is the most abundant antioxidant in cells mainly to scavenge free radicals and maintain cellular redox homeostasis [127]. Cysteine is taken up by SLC7A11 (also called xCT) [128]. The expression of SLC7A11 and cystine uptake can be mediated by mitochondrial calcium uniporter (MCU), potentially inhibiting PDAC metastasis [129].
Ferroptosis is caused by the excessive accumulation of lipid ROS [130]. The Fenton reaction produced by labile iron in cancer cells promotes the peroxidation of membrane-bound lipids containing polyunsaturated fatty acids [131]. In preclinical models, cysteine depletion and promotion of ferroptosis inhibited PDAC growth [132]. NRF2 can activate downstream suppressor genes through transcription, the trans-sulfur pathway can convert methionine into cysteine, and the mTOR pathway can increase GPX4 protein synthesis, all of which can improve ferroptosis resistance [133]. Cysteine depletion can induce ferroptosis [132]. ROS-enhanced phototherapy via the Nrf2-mediated stress-defence pathway and ferroptosis can inhibit PDAC [134]. Increasing the labile iron in cancer cells can induce ferroptosis, while iron accumulation can also promote tumour progression [135, 136]. Recent groundbreaking studies expand our understanding of the mechanisms of 7-Dehydrocholesterol (7-DHC) in ferroptosis through cholesterol metabolism [137, 138]. The unique ferroptosis response mediated by PHLDA2 is likely independent of ACSL4 and does not require common ferroptosis inducers [139].
Several other metabolic targets that modulate ROS toxicity are also being studied preclinically [140, 141]. Inhibiting nicotinamide phosphoribosyl transferase (NAMPT) to block NAD+ synthesis can inhibit tumour growth [142] and has been carried out in clinical trials [143, 144]. New concepts and attempts related to NAD+ metabolism are being made, but its double-edged sword effect reflects the complexity of regulating redox homeostasis in cancer [145, 146].
Other metabolic alterations
There are also several metabolism-related targets or pathways that have not been covered in previous chapters. KRAS mutations are among the earliest events in pancreatic carcinogenesis and can drive common metabolic programs and promote tumour progression [147]. In the normal pancreas, acinar cells use amino acids to synthesize digestive enzymes, and ductal cells are mainly responsible for the transport of peptides and hormones. In the case of KRAS mutations, acinar cells can transform into ductal cells and are considered the origin of PDAC [148]. These changes also occur in ductal cells [149]. In the transformed state, KRAS PDAC cells possess metabolic programs intrinsic to acinar and ductal cells [150].
To adapt to harsh environments, PDAC cells rely on lysosomes for nutrient degradation and regeneration, such as through macropinocytosis [151] and autophagy [152], to obtain sufficient fuel for survival [153, 154]. Macropinosomes can nonselectively internalize and absorb a large amount of extracellular fluid and ultimately metabolize amino acids into central carbon to support the growth of cancer cells [155]. Oncogenic RAS induces macropinocytosis [156] and is regulated by EGFR-Pak signalling [157] and syndecan 1 (SDC1) [158]. Autophagy can degrade cellular macromolecules and organelles, maintain cell homeostasis and survival [159], and maintain PDAC growth [160]. A lack of extracellular supplies enhances autophagy [161], and increased autophagy on ferritin maintains iron availability and thereby promotes tumour progression [162]. Research has shown that autophagy promotes immune evasion in PDAC by degrading MHC-I [163] and that the combination of MEK and autophagy inhibition may inhibit PDAC [164]. Preclinical model results further support the critical role of autophagy in tumor maintenance [160].
Because metabolic reprogramming causes differences in metabolites in cells, epigenetic modifications such as lactylation [165], succinylation [166], glycosylation [167] and SUMOylation [168] may also cause different trends in tumour development. For example, SIRT4 can inhibit tumorigenesis by inducing autophagy [169], while Smarcd3 can establish aggressive lipid metabolism remodelling [170]. The interaction between metabolic reprogramming and epigenetics has gradually become the focus of recent research [171,172,173]. Moreover, some inhibitors have been tested in preclinical models [174].
Recent studies have discovered new forms of regulated cell death resulting from imbalances in cellular metabolism [175]. Copper-dependent signalling pathways were also identified and characterized [176]. Lysosomal function is upregulated in cancer cells and TRPML1 is a cation channel with dual permeability to Ca2+ and Zn2+, but excessive Zn2+ will hinder mitochondrial function and eventually lead to cell death [177]. Disulfide death relies on SLC7A11-mediated cystine transport, which may be resistant to ferroptosis therapy, but is glucose and NADPH dependent [178]. Disruption of intracellular pH balance, such as the selective inhibitor JTC801, triggers alkaline death by blocking OPRL1 and inhibits PDAC growth [179]. Although the role of metabolic cell death is controversial, these may provide insights for novel therapeutic interventions.
Metabolism of the tumour microenvironment
The PDAC microenvironment is highly intricate and heterogeneous. In addition to cancer cells, there is also an extracellular matrix (ECM), stromal cells, immune cells, etc., and their metabolic interactions play a key role in the occurrence and development of PDAC [180].
PDAC cells are surrounded by dense proliferating connective tissue, which is composed of collagen mesh, leading to hypoxia and nutrient deficiency in tumours [181]. In this environment, cancer cells can activate the PI3K/AKT signalling pathway to enhance glycolysis [182] and even use collagen to provide energy [183]. Connexin-43 channels can transport excess lactate produced by glycolysis and maintain a suitable chemical environment [184]. The lactate receptor GPR81 has also been reported to regulate the lactate transport mechanism [185]. Correspondingly, lactic acid also promotes tumour invasiveness through angiogenesis, immune evasion, and cell migration [186].
Pancreatic stellate cells (PSCs), especially activated PSCs (aPSCs) and cancer-associated fibroblasts (CAFs), are the main components of the PDAC stroma [187, 188]. Inflammatory CAFs can promote PDAC progression [189]. Pancreatic CAFs can support the cellular metabolism of PDAC cells in vitro and in vivo by secreting metabolites [190], which is known as the reverse Warburg effect [191]. Metabolic crosstalk between PDAC cells and CAFs can be achieved by alanine uptake by specific transporters, such as the neutral amino acid transporter SLC38A2 [192]. Lau et al. constructed an organoid-fibroblast co-culture system and found that PDAC cells showed increased pyruvate carboxylation [193]. Cancer cells release tryptophan-derived formate, which can be used by PSCs to support purine nucleotide synthesis [194]. CAFs can secrete lipids such as lysophosphatidylcholine (LPA) and promote PDAC cell progression and AKT activation through the autotaxin-LPA axis [195]. Meanwhile, CAFs can secrete pyrimidines and deoxycytidine, inhibiting the effects of gemcitabine [196], and secrete cytokines and chemokines that support PDAC progression [197]. Recent studies have shown that CAFs can provide bioavailable iron to resist autophagy [198] and secrete cysteine to support glutathione synthesis [199], which induces ferroptosis resistance. Metabolic interactions between cancer cells and PSCs also overcome the redox limitations of cell proliferation [200]. Single-cell sequencing revealed a novel subpopulation of CAFs (named mediated CAFs (meCAFs)) with abnormal glucose metabolism that are associated with a poor prognosis but may better respond to immunotherapy [201]. Because fibroblasts have multiple functions that support or suppress PDAC, it is of great significance to understand the mechanisms and develop targeted therapies.
Nerve invasion is a characteristic of PDAC and is related to prognosis [202]. Neurotrophic factors or neurotransmitters secreted by neurons have been shown to have tumour-promoting effects [203], and the secretion of serine is a newly discovered metabolic crosstalk mechanism [204].
Adipocytes can meet high nutritional demands by secreting adipokines and disrupting lipid metabolism and, in addition, have the potential to transdifferentiate into fibroblast-like cells [205]. Adipocytes may also contribute to the malignant progression of PDAC through metabolic interactions [206], such as through glutamine secretion [207] or through interactions with tumour-associated neutrophils [208].
Immune cells are important components of the TME, and continuously activated inflammatory pathways can promote the occurrence of cancer [13]. The known metabolic patterns of immune cells are mainly different: activated immune cell metabolism resembles the Warburg effect, without obvious OXPHOS; glycolysis in effector T cells increases; and resting immune cells mainly use the tricarboxylic acid cycle and OXPHOS [13].
Macrophages are the most abundant cell type in the TME and play a crucial role in immunity and metabolism [209]. The infiltration and activation of tumour-associated macrophages (TAMs) can promote immune evasion and matrix remodelling in PDAC [210]. The metabolic reprogramming of TAMs through collagen turnover can result in a profibrotic profile [211]. The simultaneous exposure to hyperglycaemia and macrophages can increase the migratory potential of PDAC cells through the epithelial-to-mesenchymal transition (EMT) [212]. TAMs can differentiate into M1 or M2 phenotypes to adapt to the microenvironment [213]; for example, lactate promotes the proto-oncogenic M2-like polarization of TAMs, while through metabolic normalization, TAMs can transform into an anticancer M1 phenotype in preclinical models [214, 215]. Conversely, TAMs can also enhance glycolysis in PDAC cells by secreting cytokines, such as IL-8 [216]. Arginase 1 in immunosuppressive macrophages consumes arginine and inhibits T-cell infiltration [217]. Reducing the levels of the immune metabolite itaconic acid through ACOD1 depletion enhances CAR-macrophage function [218].
T cells are the pioneers of adaptive immune responses, and the infiltration of different types of T cell subsets has different effects on tumours [219]. Single-cell sequencing enables the analysis of the metabolic status and metabolite dynamics of T-cell subsets and their interactions with immunity [220]. Type II cytokines secreted by Th2 cells can stimulate cancer cell-intrinsic MYC transcriptional upregulation to drive glycolysis, accelerating tumour growth [221]. Tryptophan and arginine are required for effector function and T-cell survival [222]. IDO is overexpressed in PDAC and inhibits antitumour T cell responses, and it catalyses the conversion of tryptophan to kynurenine [223]. To meet metabolic demands upon glucose deprivation, tumour-infiltrating lymphocytes (TILs) may be forced to enhance OXPHOS, thereby increasing ROS levels and ultimately leading to dysfunction and failure [224]. Regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) are also affected by metabolic reprogramming in the TME, which results in immunosuppression [225]. B cells can induce tumour-promoting and immunosuppressive effects. Tumour-associated neutrophils (TANs) can promote a hypoxic microenvironment and immunosuppression [226]. BHLHE40-driven TANs exhibit hyperactivated glycolysis with pro-oncogenic and immunosuppressive functions [227]. However, current research is limited to the main metabolic pathways involved. The roles of other metabolic pathways, such as cholesterol metabolism, and some immune cells, such as natural killer (NK) and CD4+ T cells, are still unclear, and the role of metabolic reprogramming in immune cells remains to be explored. NK cells can exert cytotoxic effects without antigen pre-sensitization, but some metabolic features in the TME hinder NK cell immunotherapy [228]. NK cell dysfunction may be induced by lactate accumulation [229] and competitively inhibited by the active consumption of vitamin B6 in PDAC [230].
Recently, the link between the microbiome and PDAC has attracted considerable interest [231, 232] and may be useful for PDAC detection [233, 234]. Several microorganisms that act on the adenosine pathway have been found to enhance the efficacy of immune checkpoint blockade (ICB) [235]. The expression of genes involved in the polyamine and nucleotide biosynthetic pathways, which are strongly correlated with host tumorigenesis and can serve as predictive markers for the early detection of PDAC, has been shown to be significantly elevated [236]. Recent research has suggested that the microbiome-derived metabolite trimethylamine N-oxide (TMAO) may be a driver of antitumour immunity [237] and that microbiota-derived 3-IAA can influence chemotherapy efficacy [238]. The aryl hydrocarbon receptor in TAMs can be activated by tryptophan-derived microbial metabolites to suppress antitumour immunity [239]. The role of intratumoural bacteria in PDAC requires further study due to their ability to modulate the host immune system and metabolize drugs [240, 241].
The metabolic regulatory role of small extracellular vesicles (sEVs), such as exosomes, as intercellular communication mediators in tumours should not be ignored [242, 243]. Pancreatic cancer-derived extracellular vesicles may influence lipolysis to induce cancer-related cachexia [244]. The detection of sEVs based on lectin-glycan interactions has potential as an early diagnostic marker [245]. Exosomes derived from the tumour microenvironment can also mediate cancer cell metabolism [246].
Metabolism of the body
In addition to local metabolic reprogramming in tumours, systemic metabolism can also serve as a potential pathogenic mechanism and therapeutic target [247]. A large-scale database study showed that obesity is associated with various cancers [248]. Obesity and diabetes are increasingly recognized as risk factors for PDAC, not only because of the remodelling of the tumour microenvironment but also because of their effects on systemic metabolism [249, 250].
Metabolic syndrome (MetS), a pathological condition characterized by abdominal obesity, insulin resistance, hypertension, and hyperlipidaemia, is associated with the risk of PDAC [251]. In the setting of diet-induced hyperinsulinaemia and obesity, insulin dose-dependently increases the formation of acinar to ductal metaplasia via trypsin and the insulin receptor (InsR) [252]. Stress adaptation through phase-separated organelle stress granules (SGs) mediates the development of PDAC, and obesity may be a driving force behind this process [253]. At the same time, obesity and diabetes are also important factors leading to chemotherapy resistance [254]. Studies have shown that metformin increases the sensitivity of PDAC cells to gemcitabine [255], suggesting the development of new therapies [256]. Clinical trials using related drugs for the treatment of PDAC have been carried out and will be listed later.
There are already many forms and mechanisms of dietary intervention for cancer [257]. However, there is still a long way to go before clinical application. We list several dietary intervention models in Text Box S4.
In addition to diet, physical exercise may reduce the progression of cancer [258]. Moreover, mild cold exposure activates brown fat and hinders glycolysis-based metabolism in cancer cells [259]. Patients with PDAC often experience weight loss and skeletal muscle wasting [260]. Aerobic exercise can promote immune mobilization and exert antitumour effects through the IL-15/IL-15Rα axis [261]. However, due to individual differences in body constitution, caution must be used when applying dietary intervention and physical exercise in cancer treatment.
Cooperation with other antitumour therapies
Radiotherapy
Although there is some controversy regarding the survival benefit of this technique, radiation therapy has been used to treat borderline resectable PDAC, providing the possibility of resection [262]. Additionally, metabolic changes in PDAC can cause radio-resistance [263].
Overexpressed MUC1 enhances nucleotide metabolism, facilitates radiation resistance, and is targeted effectively through glycolytic inhibition [264]. Multiple cholesterol synthesis-related genes are associated with radio-resistance in cell lines [265]. Serine and glycine starvation can inhibit the antioxidant response, nucleotide synthesis, and the TCA cycle and has the potential to become a cancer radio-sensitization strategy [266].
Chemotherapy
Chemotherapy is the most commonly used systemic treatment. Gemcitabine is a type of nucleoside analogue that can interfere with DNA synthesis and block the cell cycle [267].
Gemcitabine-resistant cell lines exhibit increased aerobic glycolysis and reduced ROS levels [268, 269]. Organoid-based metabolomic analysis revealed that PDAC with high glucose metabolism levels is more resistant to chemotherapy and that GLUT1/ALDOB/G6PD axis inhibitors are promising pharmacological agents [270]. The pentose phosphate pathway (PPP) supports DNA replication and RNA production by regulating carbon flux between nucleic acid synthesis and lipogenesis, where key enzymes such as ribulose 5-phosphate isomerase (RPIA) and ribulose-5-phosphate-3-epimerase (RPE) are upregulated to maintain cell proliferation [271]. Studies have shown that S100A11 can enhance transketolase (TKT) synthesis and promote PPP [272]. In addition, the acidic tumor microenvironment can activate the YAP/MMP1 axis, promote the transition of cell metabolism to PPP, and promote tumor progression [273]. Elevated G6PD expression is also an important factor leading to erlotinib resistance [274]. Shukla et al. showed that mucin 1 (MUC1) and hypoxia-inducible factor 1α (HIF1α) increased deoxyCTP (dCTP) pools, thus inducing acquired resistance to gemcitabine through molecular competition [275]. ARNTL2-mediated cellular glycolysis was shown to increase sensitivity to erlotinib treatment through the activation of the PI3K/AKT signalling pathway [276]. Studies have also confirmed that the ERK/E2F1 pathway can cause gemcitabine resistance [277]. In amino acid metabolism, glutamine may contribute to gemcitabine resistance due to its role in controlling ROS and activating mTOR [269]. A nutrient-deficient environment may help activate the RNA-binding protein HuR, thereby upregulating isocitrate dehydrogenase 1 (IDH1) expression, enhancing antioxidant defence, and leading to chemotherapy resistance [278]. Increased glutamine uptake caused by MUC5AC overexpression can cause gemcitabine resistance [279]. The upregulation of FASN expression can induce endoplasmic reticulum stress and promote gemcitabine resistance by enhancing de novo lipid synthesis [92]. The inhibition of cholesterol synthesis may also enhance the effect of anticancer therapy [280], and the use of lipid rafts characterized by Cav-1-cholesterol may be a breakthrough therapeutic approach [281]. By targeting CRABP-II, lipid raft cholesterol accumulation can be reversed to overcome drug resistance [282]. CD36 expression is correlated with antiapoptotic protein expression and may protect PDAC cells from drug-induced cell death [283]. In addition, UBE2T can affect the remodelling of pyrimidine metabolism and confer gemcitabine resistance [284].
Immunotherapy
Immunotherapy has gradually developed into a mature antitumour strategy, but its efficacy in PDAC is limited [285]. Some studies have attempted to predict the response to immunotherapy based on metabolic characteristics [286]. Metabolic disorders of the immune system in the TME are expected to improve the efficacy of metabolism-targeted anticancer strategies [287, 288].
Nucleotide metabolism provides genetic material and energy resources for immune system activation and proliferation [289]. Purine analogues, such as released extracellular ATP or adenosine, activate the purinergic and adenosine receptors of immune cells, thereby promoting or suppressing immune responses [290]. IFN triggers cell cycle arrest in the S phase, resulting in insufficient nucleotide pools and nucleoside efflux, and IFN combined with ATR inhibitors can induce lethal DNA damage and decrease nucleotide biosynthesis, thereby limiting the growth of PDAC [291].
Understanding the influence of metabolic regulation on the efficacy of immunotherapy is still in its infancy. Modulating cell metabolism via the autologous T cell adoptive transfer protocol can promote the generation of T cells with a memory phenotype and improve their antitumour effect [292, 293]. Engineering oncolytic viruses to express leptin can also enhance antitumour responses by increasing FAO and OXPHOS [294]. PD-1 blockade immunotherapy is a widely used therapy, but PDAC patients respond poorly. Targeting glycolysis impacts the accumulation of PD-1+ TILs in PDAC preclinical models [295]. Activating the AMPK and mTOR pathways related to mitochondrial metabolism is also expected to enhance the therapeutic effect of PD-1 blockers [296, 297].
Based on the aforementioned immune-metabolic crosstalk, new therapeutic targets have been investigated. For example, indoleamine 2,3-dioxygenase (IDO), which can degrade tryptophan and inhibit immune responses [298], has been explored in several clinical studies, which we will describe below.
Other therapies
Other drug therapies are gradually being explored through emerging technologies [299]. The use of some nanomedicines can enhance oxidative stress [300]. Reduction-responsive nanoplatforms that can regulate lipid metabolism and polarize macrophages are available for the combined treatment of PDAC [301]. A novel dendrimer nanogel enhances chemoimmunotherapy through endoplasmic reticulum stress [302]. After degrading the ECM, nanoparticle transport can be enhanced, increasing the killing effect on PDAC [303, 304]. Studies have shown that selenoorganic compounds and dihydroartemisinin can induce ferroptosis in PDAC cells [305, 306]. Tailored ruthenium complexes can also affect OXPHOS and exert anticancer effects [307]. Although such investigations are still in the preclinical stage, they are providing new directions for the treatment of PDAC.
Clinical trials targeting metabolism
Metabolic targeted therapy still has a long way to go before being applied as a routine treatment (Table 1). Some metabolic modulators, such as metformin [308] and statins [309], have been used to exert additional cancer-treatment effects. Metformin can affect the malignant behaviour of SMAD4-deficient PDAC cells by inhibiting HNF4G activity [310]; it can also destroy the dense matrix by inhibiting PSC activity, significantly improving the therapeutic efficacy of gemcitabine [311]. Several clinical trials of metformin for the treatment of PDAC, such as NCT02048384, NCT03889795 and NCT02201381, have been carried out. Through in vivo metabolism tracking, it was found that statins can inhibit the synthesis of coenzyme Q and that statins combined with MEK inhibitors can promote oxidative stress and apoptosis [312]. Based on these promising data, phase I and II clinical trials, such as NCT00944463, NCT00584012 and NCT04862260, were conducted. Irbesartan may reverse gemcitabine resistance by inhibiting iron metabolism [313]. In addition, drugs such as digoxin (NCT04141995), dapagliflozin (NCT04542291), omeprazole (NCT04930991), and even combinations of multiple metabolism-regulating drugs (NCT02201381) have been explored.
Clinical trials have begun to explore treatments based on the glycolytic pathway. In the case of NCT00096707, 2-deoxy-D-glucose (2-DG) combined with docetaxel was used for advanced solid tumours and had tolerable adverse effects and certain clinical benefits [314]. The lipoic acid analogue CPI-613 was shown to selectively inhibit PDH activity and inhibit the growth of PDAC [315]. In NCT01835041 and NCT03504423, CPI-613 was combined with chemotherapy for the treatment of metastatic PDAC, yielding a good response rate [316].
Inducing metabolic crises through the antagonism of glutamine inhibitors, such as 6-diazo-5-oxo-L-norleucine (DON) and DRP-104, is a promising strategy in preclinical models [317]. NCT03965845 explored the efficacy of the glutaminase inhibitor telaglenastat (CB-839) in advanced or metastatic solid tumours. In NCT04634539, L-glutamine was combined with standard chemotherapy for advanced PDAC treatment. In recent years, in NCT01523808, NCT02195180, and NCT03665441, the combination of red blood cell-encapsulated asparaginase (such as GRASPA and eryaspase) with chemotherapy has been shown to improve the prognosis of patients, indicating its great potential as a second-line treatment [318]. Calaspargase pegol-mknl, which converts the amino acid L-asparagine into aspartic acid and ammonia, leading to cell death, was recently used in combination with cobimetinib in NCT05034627. The role of tryptophan and IDO1 in the immune microenvironment has recently been emphasized. In NCT00739609, NCT03432676, and NCT03006302, IDO1 inhibitors and PD-1 checkpoint inhibitors caused PDAC to respond to immunotherapy [319], and their efficacy in combination with chemotherapy has been further explored in clinical trials, such as NCT02077881 and NCT03085914. In addition, research on arginine (NCT02101580) and methionine (NCT03435250) has improved the understanding of targeting amino acid metabolism to treat PDAC.
Although lipid synthesis-related genes are overexpressed in a variety of cancers, relevant clinical trials are still exploratory. The application potential of the FASN inhibitor TVB-2640 and an antagonist of PPARα TPST-1120 were explored in NCT02223247 and NCT03829436, respectively. NCT03450018 was conducted to determine whether the CA IX inhibitor SLC-0111 exerts a synergistic anticancer effect by affecting ferroptosis.
There are also several drugs that may exert anticancer effects by affecting redox balance. For example, phase I clinical trials of artesunate and ascorbic acid have been conducted (NCT02353026 and NCT01049880, respectively). Combining β-lapachone (ARQ761) with a GLS1 inhibitor can induce excessive ROS production and selectively lead to PDAC cell death in preclinical models [320]. The potential of ARQ761 combined with gemcitabine/nab-paclitaxel treatment was explored in NCT02514031.
Autophagy plays an important role in the progression of PDAC, and the lysosomal inhibitor hydroxychloroquine may have synergistic effects with the MEK inhibitor trametinib. In this context, numerous clinical trials have been carried out (NCT01777477, NCT03825289, and NCT04132505).
Finally, there are some clinical trials based on dietary interventions, such as NCT01019382 and NCT01419483, but they are all in their early stages. In NCT02336087, a variety of dietary supplements were investigated, among which epigallocatechin-3 gallate (EGCG) may act by inhibiting FASN. However, these interventions are still far from clinical application.
Conclusion and future perspective
In recent years, the metabolic mechanism and translational application of PDAC treatment have become new and challenging research directions [321]. Both in vivo and in vitro experiments have revealed the metabolic dependence of PDAC [322, 323]. The use of stable isotopes has become a key method for exploring metabolic pathways in PDAC [324]. Advanced technology platforms such as organoids are also promoting advancements in the translation of metabolism-related research [325]. Many molecular subtyping methods based on metabolic characteristics are being developed for the treatment and evaluation of different types of PDAC patients [270]. Moreover, the development of single-cell sequencing and spatial omics has made it possible to characterize the metabolic crosstalk of different tissues at the cellular level [326]. Our understanding of the function of cellular metabolism in disease progression has progressed significantly (Fig. 2).

In addition to cellular metabolism such as nucleotide metabolism, carbohydrate metabolism, amino acid metabolism, lipid metabolism and oxidative homeostasis, complex tumor microenvironment metabolism and body metabolism (outer gray circles) together construct the metabolic characteristics of pancreatic cancer. With the development of novel experimental methods, precise tracking detection, integrated multi-omics analysis and personalized metabolic therapy (arrows), therapeutic or sensitization targets (blue regular octagons in the middle layer) have gradually been revealed, which is expected to break through the barriers to pancreatic cancer treatment (innermost), bringing new hope for the diagnosis and treatment of pancreatic cancer.
Given that most of the current understanding of how metabolism supports cell proliferation is based on studies in cancer cells, how metabolism shapes the interactions of different cells needs be explored in more complex ecosystems. This information is expected to help us gain a deeper understanding of the metabolic regulatory network, thereby overcoming the limitations of previous metabolic therapies and providing opportunities for tailor-made treatment plans for patients.
References
Finley LWS. What is cancer metabolism? Cell. 2023;186:1670–88.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–37.
Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022;34:355–77.
Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–49.
Zhang H, Yue X, Chen Z, Liu C, Wu W, Zhang N, et al. Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: new opportunities in cancer immunotherapy and advances in clinical trials. Mol Cancer. 2023;22:159.
Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. 2020;78:1019–33.
Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022;12:31–46.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368:6487.
Zou W, Green DR. Beggars banquet: Metabolism in the tumor immune microenvironment and cancer therapy. Cell Metab. 2023;35:1101–13.
Xiao Y, Yu TJ, Xu Y, Ding R, Wang YP, Jiang YZ, et al. Emerging therapies in cancer metabolism. Cell Metab. 2023;35:1283–303.
Ye Z, Chen W, Li G, Huang J, Lei J. Tissue-derived extracellular vesicles in cancer progression: mechanisms, roles, and potential applications. Cancer Metastasis Rev. 2023;43:575–595.
Fang Z, Meng Q, Xu J, Wang W, Zhang B, Liu J, et al. Signaling pathways in cancer-associated fibroblasts: recent advances and future perspectives. Cancer Commun. 2023;43:3–41.
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28.
Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74:12–49.
Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet. 2020;395:2008–20.
Mullen NJ, Singh PK. Nucleotide metabolism: a pan-cancer metabolic dependency. Nat Rev Cancer. 2023;23:275–94.
Weaver BA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12.
Wu HL, Gong Y, Ji P, Xie YF, Jiang YZ, Liu GY. Targeting nucleotide metabolism: a promising approach to enhance cancer immunotherapy. J Hematol Oncol. 2022;15:45.
Kimmelman AC, White E. Autophagy and Tumor Metabolism. Cell Metab. 2017;25:1037–43.
Madsen HB, Peeters MJ, Straten PT, Desler C. Nucleotide metabolism in the regulation of tumor microenvironment and immune cell function. Curr Opin Biotechnol. 2023;84:103008.
Warburg O, Wind F, Negelein E. The Metabolism Of Tumors In The Body. J Gen Physiol. 1927;8:519–30.
Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64.
Cao L, Wu J, Qu X, Sheng J, Cui M, Liu S, et al. Glycometabolic rearrangements-aerobic glycolysis in pancreatic cancer: causes, characteristics and clinical applications. J Exp Clin Cancer Res. 2020;39:267.
Yang J, Ren B, Yang G, Wang H, Chen G, You L, et al. The enhancement of glycolysis regulates pancreatic cancer metastasis. Cell Mol Life Sci. 2020;77:305–21.
Yan L, Tu B, Yao J, Gong J, Carugo A, Bristow CA, et al. Targeting Glucose Metabolism Sensitizes Pancreatic Cancer to MEK inhibition. Cancer Res. 2021;81:4054–65.
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70.
Huang C, Li Y, Li Z, Xu Y, Li N, Ge Y, et al. LIMS1 promotes pancreatic cancer cell survival under oxygen-glucose deprivation conditions by enhancing HIF1A protein translation. Clin Cancer Res. 2019;25:4091–103.
Hirschey MD, DeBerardinis RJ, Diehl AME, Drew JE, Frezza C, Green MF, et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol. 2015;35:S129–S150.
Zhang L, Jiang C, Zhong Y, Sun K, Jing H, Song J, et al. STING is a cell-intrinsic metabolic checkpoint restricting aerobic glycolysis by targeting HK2. Nat Cell Biol. 2023;25:1208–22.
Huangyang P, Li F, Lee P, Nissim I, Weljie AM, Mancuso A, et al. Fructose-1,6-Bisphosphatase 2 Inhibits Sarcoma progression by restraining mitochondrial biogenesis. Cell Metab. 2020;31:1032.
Minchenko OH, Tsuchihara K, Minchenko DO, Bikfalvi A, Esumi H. Mechanisms of regulation of PFKFB expression in pancreatic and gastric cancer cells. World J Gastroenterol. 2014;20:13705–17.
Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. 2016;73:377–92.
He D, Feng H, Sundberg B, Yang J, Powers J, Christian AH, et al. Methionine oxidation activates pyruvate kinase M2 to promote pancreatic cancer metastasis. Mol Cell. 2022;82:3045–3060.e3011.
Hillis AL, Lau AN, Devoe CX, Dayton TL, Danai LV, Di Vizio D, et al. PKM2 is not required for pancreatic ductal adenocarcinoma. Cancer Metab. 2018;6:17.
Cui Y, Tian J, Wang Z, Guo H, Zhang H, Wang Z, et al. Fructose-induced mTORC1 activation promotes pancreatic cancer progression through inhibition of autophagy. Cancer Res. 2023;83:4063–79.
Ji S, Zhang B, Liu J, Qin Y, Liang C, Shi S, et al. ALDOA functions as an oncogene in the highly metastatic pancreatic cancer. Cancer Lett. 2016;374:127–35.
Butera G, Pacchiana R, Mullappilly N, Margiotta M, Bruno S, Conti P, et al. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim Biophys Acta Mol Cell Res. 2018;1865:1914–23.
Shao F, Yang X, Wang W, Wang J, Guo W, Feng X, et al. Associations of PGK1 promoter hypomethylation and PGK1-mediated PDHK1 phosphorylation with cancer stage and prognosis: a TCGA pan-cancer analysis. Cancer Commun. 2019;39:54.
Wen CL, Huang K, Jiang LL, Lu XX, Dai YT, Shi MM, et al. An allosteric PGAM1 inhibitor effectively suppresses pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA. 2019;116:23264–73.
Principe M, Borgoni S, Cascione M, Chattaragada MS, Ferri-Borgogno S, Capello M, et al. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J Hematol Oncol. 2017;10:16.
Comandatore A, Franczak M, Smolenski RT, Morelli L, Peters GJ, Giovannetti E. Lactate Dehydrogenase and its clinical significance in pancreatic and thoracic cancers. Semin Cancer Biol. 2022;86:93–100.
Cui J, Shi M, Xie D, Wei D, Jia Z, Zheng S, et al. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin Cancer Res. 2014;20:2595–606.
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010;107:2037–42.
Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24:2482–90.
Ozturk H, Cingoz H, Tufan T, Yang J, Adair SJ, Tummala KS, et al. ISL2 is a putative tumor suppressor whose epigenetic silencing reprograms the metabolism of pancreatic cancer. Dev Cell. 2022;57:1331–1346.e1339.
Zhou C, Sun H, Zheng C, Gao J, Fu Q, Hu N, et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis. 2018;9:161.
Fu S, Xu S, Zhang S. The role of amino acid metabolism alterations in pancreatic cancer: From mechanism to application. Biochim Biophys Acta Rev Cancer. 2023;1878:188893.
Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: Metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10:10111564.
Helmlinger G, Sckell A, Dellian M, Forbes NS, Jain RK. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin Cancer Res. 2002;8:1284–91.
Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–34.
Bott AJ, Shen J, Tonelli C, Zhan L, Sivaram N, Jiang YP, et al. Glutamine anabolism plays a critical role in pancreatic cancer by coupling carbon and nitrogen metabolism. Cell Rep. 2019;29:1287–1298.e1286.
Herner A, Sauliunaite D, Michalski CW, Erkan M, De Oliveira T, et al. Glutamate increases pancreatic cancer cell invasion and migration via AMPA receptor activation and Kras-MAPK signaling. Int J Cancer. 2011;129:2349–59.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5.
Hu T, Shukla SK, Vernucci E, He C, Wang D, King RJ, et al. Metabolic rewiring by loss of Sirt5 Promotes Kras-induced pancreatic cancer progression. Gastroenterology. 2021;161:1584–1600.
Nelson BS, Lin L, Kremer DM, Sousa CM, Cotta-Ramusino C, Myers A, et al. Tissue of origin dictates GOT1 dependence and confers synthetic lethality to radiotherapy. Cancer Metab. 2020;8:1.
Tsai PY, Lee MS, Jadhav U, Naqvi I, Madha S, Adler A, et al. Adaptation of pancreatic cancer cells to nutrient deprivation is reversible and requires glutamine synthetase stabilization by mTORC1. Proc Natl Acad Sci USA. 2021;118:e2003014118.
Teixeira E, Silva C, Martel F. The role of the glutamine transporter ASCT2 in antineoplastic therapy. Cancer Chemother Pharm. 2021;87:447–64.
Wang VM, Ferreira RMM, Almagro J, Evan T, Legrave N, Zaw Thin M, et al. CD9 identifies pancreatic cancer stem cells and modulates glutamine metabolism to fuel tumour growth. Nat Cell Biol. 2019;21:1425–35.
De Santis MC, Gozzelino L, Margaria JP, Costamagna A, Ratto E, Gulluni F, et al. Lysosomal lipid switch sensitises to nutrient deprivation and mTOR targeting in pancreatic cancer. Gut. 2023;72:360–71.
Encarnacion-Rosado J, Sohn ASW, Biancur DE, Lin EY, Osorio-Vasquez V, Rodrick T, et al. Targeting pancreatic cancer metabolic dependencies through glutamine antagonism. Nat Cancer. 2024;5:85–99.
Recouvreux MV, Grenier SF, Zhang Y, Esparza E, Lambies G, Galapate CM, et al. Glutamine mimicry suppresses tumor progression through asparagine metabolism in pancreatic ductal adenocarcinoma. Nat Cancer. 2024;5:100–13.
Casero RA Jr, Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer. 2018;18:681–95.
Holbert CE, Cullen MT, Casero RA Jr, Stewart TM. Polyamines in cancer: integrating organismal metabolism and antitumour immunity. Nat Rev Cancer. 2022;22:467–80.
Phanstiel Ot. An overview of polyamine metabolism in pancreatic ductal adenocarcinoma. Int J Cancer. 2018;142:1968–76.
Wang H, Li QF, Chow HY, Choi SC, Leung YC. Arginine deprivation inhibits pancreatic cancer cell migration, invasion and EMT via the down regulation of Snail, Slug, Twist, and MMP1/9. J Physiol Biochem. 2020;76:73–83.
Awad D, Lyssiotis CA. An unexpected pathway to polyamines in pancreatic cancer. Mol Cell. 2023;83:1765–6.
Lee MS, Dennis C, Naqvi I, Dailey L, Lorzadeh A, Ye G, et al. Ornithine aminotransferase supports polyamine synthesis in pancreatic cancer. Nature. 2023;616:339–47.
Fahy E, Cotter D, Sud M, Subramaniam S. Lipid classification, structures and tools. Biochim Biophys Acta. 2011;1811:637–47.
Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018;38:27.
Yin X, Xu R, Song J, Ruze R, Chen Y, Wang C, et al. Lipid metabolism in pancreatic cancer: emerging roles and potential targets. Cancer Commun. 2022;42:1234–56.
Pirhonen J, Szkalisity A, Hagstrom J, Kim Y, Migh E, Kovacs M, et al. Lipid Metabolic Reprogramming Extends Beyond Histologic Tumor Demarcations In Operable Human Pancreatic Cancer. Cancer Res. 2022;82:3932–49.
Wolrab D, Jirasko R, Cifkova E, Horing M, Mei D, Chocholouskova M, et al. Lipidomic profiling of human serum enables detection of pancreatic cancer. Nat Commun. 2022;13:124.
Koundouros N, Poulogiannis G. Reprogramming of fatty acid metabolism in cancer. Br J Cancer. 2020;122:4–22.
Acharya R, Shetty SS, Kumari NS. Fatty acid transport proteins (FATPs) in cancer. Chem Phys Lipids. 2023;250:105269.
Wang J, Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. 2019;9:4893–908.
McKillop IH, Girardi CA, Thompson KJ. Role of fatty acid binding proteins (FABPs) in cancer development and progression. Cell Signal. 2019;62:109336.
Tanase C, Gheorghisan-Galateanu AA, Popescu ID, Mihai S, Codrici E, Albulescu R, et al. CD36 and CD97 in pancreatic cancer versus other malignancies. Int J Mol Sci. 2020;21:21165656.
Li Z, Kang Y. Lipid metabolism fuels cancer’s spread. Cell Metab. 2017;25:228–30.
Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–55.
Rozeveld CN, Johnson KM, Zhang L, Razidlo GL. KRAS controls pancreatic cancer cell lipid metabolism and invasive potential through the Lipase HSL. Cancer Res. 2020;80:4932–45.
Guertin DA, Wellen KE. Acetyl-CoA metabolism in cancer. Nat Rev Cancer. 2023;23:156–72.
Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71.
Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–61.
Verschueren KHG, Blanchet C, Felix J, Dansercoer A, De Vos D, Bloch Y, et al. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature. 2019;568:571–5.
Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710–30.
Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol. 2018;14:452–63.
Agar S, Akkurt B, Ulukaya E. The inhibition mechanism of pancreatic ductal Adenocarcinoma via LXR Receptors: A multifaceted approach integrating molecular docking, molecular dynamics and post-MD inter-molecular contact analysis. Asian Pac J Cancer Prev. 2023;24:4103–9.
Hunkeler M, Hagmann A, Stuttfeld E, Chami M, Guri Y, Stahlberg H, et al. Structural basis for regulation of human acetyl-CoA carboxylase. Nature. 2018;558:470–4.
Bruning U, Morales-Rodriguez F, Kalucka J, Goveia J, Taverna F, Queiroz KCS, et al. Impairment of Angiogenesis by fatty acid synthase inhibition involves mTOR Malonylation. Cell Metab. 2018;28:866–880.e815.
Kubota CS, Espenshade PJ. Targeting Stearoyl-CoA desaturase in solid tumors. Cancer Res. 2022;82:1682–8.
Chen L, Duan Y, Wei H, Ning H, Bi C, Zhao Y, et al. Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors. Expert Opin Investig Drugs. 2019;28:917–30.
Tadros S, Shukla SK, King RJ, Gunda V, Vernucci E, Abrego J, et al. De Novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017;77:5503–17.
Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, et al. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci USA. 2013;110:8882–7.
Sen U, Coleman C, Sen T. Stearoyl coenzyme A desaturase-1: multitasker in cancer, metabolism, and ferroptosis. Trends Cancer. 2023;9:480–9.
Xiao M, Xu J, Wang W, Zhang B, Liu J, Li J, et al. Functional significance of cholesterol metabolism in cancer: from threat to treatment. Exp Mol Med. 2023;55:1982–95.
Zheng S, Lin J, Pang Z, Zhang H, Wang Y, Ma L, et al. Aberrant cholesterol metabolism and wnt/beta-catenin signaling coalesce via Frizzled5 in supporting cancer growth. Adv Sci. 2022;9:e2200750.
Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2020;21:225–45.
Sah RP, Sharma A, Nagpal S, Patlolla SH, Sharma A, Kandlakunta H, et al. Phases of metabolic and soft tissue changes in months preceding a diagnosis of pancreatic ductal Adenocarcinoma. Gastroenterology. 2019;156:1742–52.
Acier A, Godard M, Gassiot F, Finetti P, Rubis M, Nowak J, et al. LDL receptor-peptide conjugate as in vivo tool for specific targeting of pancreatic ductal adenocarcinoma. Commun Biol. 2021;4:987.
Fan J, Lin R, Xia S, Chen D, Elf SE, Liu S, et al. Tetrameric Acetyl-CoA Acetyltransferase 1 is important for tumor growth. Mol Cell. 2016;64:859–74.
Lu XY, Shi XJ, Hu A, Wang JQ, Ding Y, Jiang W, et al. Feeding induces cholesterol biosynthesis via the mTORC1-USP20-HMGCR axis. Nature. 2020;588:479–84.
Chua NK, Coates HW, Brown AJ. Squalene monooxygenase: a journey to the heart of cholesterol synthesis. Prog Lipid Res. 2020;79:101033.
Oni TE, Biffi G, Baker LA, Hao Y, Tonelli C, Somerville TDD, et al. SOAT1 promotes mevalonate pathway dependency in pancreatic cancer. J Exp Med. 2020;217:20192389.
Lu J, Chen S, Bai X, Liao M, Qiu Y, Zheng LL, et al. Targeting cholesterol metabolism in Cancer: From molecular mechanisms to therapeutic implications. Biochem Pharm. 2023;218:115907.
Delk SC, Chattopadhyay A, Escola-Gil JC, Fogelman AM, Reddy ST. Apolipoprotein mimetics in cancer. Semin Cancer Biol. 2021;73:158–68.
Gabitova-Cornell L, Surumbayeva A, Peri S, Franco-Barraza J, Restifo D, Weitz N, et al. Cholesterol pathway inhibition induces TGF-beta signaling to promote basal differentiation in pancreatic cancer. Cancer Cell. 2020;38:567–583.e511.
Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11–31.
Lee JS, Oh SJ, Choi HJ, Kang JH, Lee SH, Ha JS, et al. ATP production relies on fatty acid oxidation rather than glycolysis in pancreatic ductal Adenocarcinoma. Cancers. 2020;12:12092477.
Patra KC, Kato Y, Mizukami Y, Widholz S, Boukhali M, Revenco I, et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. 2018;20:811–22.
Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science. 2019;363:644–9.
Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian CN, et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med. 2016;22:666–71.
Yu M, Liu H, Duan Y, Zhang D, Li S, Wang F. Four types of fatty acids exert differential impact on pancreatic cancer growth. Cancer Lett. 2015;360:187–94.
Nishizawa H, Matsumoto M, Chen G, Ishii Y, Tada K, Onodera M, et al. Lipid peroxidation and the subsequent cell death transmitting from ferroptotic cells to neighboring cells. Cell Death Dis. 2021;12:332.
Snaebjornsson MT, Janaki-Raman S, Schulze A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 2020;31:62–76.
Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18:33–50.
Liu J, van der Hoeven R, Kattan WE, Chang JT, Montufar-Solis D, Chen W, et al. Glycolysis regulates KRAS plasma membrane localization and function through defined glycosphingolipids. Nat Commun. 2023;14:465.
Chen Z, Ho IL, Soeung M, Yen EY, Liu J, Yan L, et al. Ether phospholipids are required for mitochondrial reactive oxygen species homeostasis. Nat Commun. 2023;14:2194.
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9.
Weiss-Sadan T, Ge M, Hayashi M, Gohar M, Yao CH, de Groot A, et al. NRF2 activation induces NADH-reductive stress, providing a metabolic vulnerability in lung cancer. Cell Metab. 2023;35:722.
Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014;2:535–62.
Ge M, Papagiannakopoulos T, Bar-Peled L. Reductive stress in cancer: coming out of the shadows. Trends Cancer. 2024;10:103–12.
Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget. 2015;6:31151–63.
Kremer DM, Nelson BS, Lin L, Yarosz EL, Halbrook CJ, Kerk SA, et al. GOT1 inhibition promotes pancreatic cancer cell death by ferroptosis. Nat Commun. 2021;12:4860.
Kerk SA, Lin L, Myers AL, Sutton DJ, Andren A, Sajjakulnukit P, et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife. 2022;11:73245.
Do BT, Vander Heiden MG. GOT2 consider the tumor microenvironment. Trends Cancer. 2022;8:884–6.
Mijit M, Kpenu E, Chowdhury NN, Gampala S, Wireman R, Liu S, et al. In vitro and In vivo evidence demonstrating chronic absence of Ref-1 Cysteine 65 impacts Ref-1 folding configuration, redox signaling, proliferation and metastasis in pancreatic cancer. Redox Biol. 2024;69:102977.
Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol. 2018;217:2291–8.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620.
Wang X, Li Y, Li Z, Lin S, Wang H, Sun J, et al. Mitochondrial calcium uniporter drives metastasis and confers a targetable cystine dependency in pancreatic cancer. Cancer Res. 2022;82:2254–68.
Julian-Serrano S, Yuan F, Wheeler W, Benyamin B, Machiela MJ, Arslan AA, et al. Hepcidin-regulating iron metabolism genes and pancreatic ductal adenocarcinoma: a pathway analysis of genome-wide association studies. Am J Clin Nutr. 2021;114:1408–17.
Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82:2215–27.
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–89.
Zhang YY, Han Y, Li WN, Xu RH, Ju HQ. Tumor iron homeostasis and immune regulation. Trends Pharm Sci. 2024;45:145–56.
Tao W, Wang N, Ruan J, Cheng X, Fan L, Zhang P, et al. Enhanced ROS-boosted phototherapy against pancreatic cancer via Nrf2-mediated stress-defense pathway suppression and ferroptosis induction. ACS Appl Mater Interfaces. 2022;14:6404–16.
Sandoval-Acuna C, Torrealba N, Tomkova V, Jadhav SB, Blazkova K, Merta L, et al. Targeting mitochondrial iron metabolism suppresses tumor growth and metastasis by inducing mitochondrial dysfunction and mitophagy. Cancer Res. 2021;81:2289–303.
Yamasaki T, Terai S, Sakaida I. Deferoxamine for advanced hepatocellular carcinoma. N. Engl J Med. 2011;365:576–8.
Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L, et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature. 2024;626:411–8.
Freitas FP, Alborzinia H, Dos Santos AF, Nepachalovich P, Pedrera L, Zilka O, et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. 2024;626:401–10.
Yang X, Wang Z, Samovich SN, Kapralov AA, Amoscato AA, Tyurin VA, et al. PHLDA2-mediated phosphatidic acid peroxidation triggers a distinct ferroptotic response during tumor suppression. Cell Metab. 2024;36:762–77.
Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab. 2019;1:404–15.
Ju HQ, Lin JF, Tian T, Xie D, Xu RH. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Target Ther. 2020;5:231.
Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative stress in cancer. Cancer Cell. 2020;38:167–97.
Lee J, Kim H, Lee JE, Shin SJ, Oh S, Kwon G, et al. Selective cytotoxicity of the NAMPT inhibitor FK866 toward gastric cancer cells with markers of the epithelial-mesenchymal transition, due to loss of NAPRT. Gastroenterology. 2018;155:799–814.e713.
Lucena-Cacace A, Otero-Albiol D, Jimenez-Garcia MP, Munoz-Galvan S, Carnero A. NAMPT is a potent oncogene in colon cancer progression that modulates cancer stem cell properties and resistance to therapy through Sirt1 and PARP. Clin Cancer Res. 2018;24:1202–15.
Chini CCS, Zeidler JD, Kashyap S, Warner G, Chini EN. Evolving concepts in NAD(+) metabolism. Cell Metab. 2021;33:1076–87.
Lv H, Lv G, Chen C, Zong Q, Jiang G, Ye D, et al. NAD(+) metabolism maintains inducible PD-L1 expression to drive tumor immune evasion. Cell Metab. 2021;33:110–127.e115.
Kerk SA, Papagiannakopoulos T, Shah YM, Lyssiotis CA. Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nat Rev Cancer. 2021;21:510–25.
Seino T, Kawasaki S, Shimokawa M, Tamagawa H, Toshimitsu K, Fujii M, et al. Human pancreatic tumor organoids reveal loss of stem cell niche factor dependence during disease progression. Cell Stem Cell. 2018;22:454–467.e456.
Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–89.
Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353:1161–5.
Zhang Y, Commisso C. Macropinocytosis in cancer: a complex signaling network. Trends Cancer. 2019;5:332–4.
Santana-Codina N, Mancias JD, Kimmelman AC. The role of autophagy in cancer. Annu Rev Cancer Biol. 2017;1:19–39.
Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5.
Elliott IA, Dann AM, Xu S, Kim SS, Abt ER, Kim W, et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc Natl Acad Sci USA. 2019;116:6842–7.
Garcia-Bermudez J, Badgley MA, Prasad S, Baudrier L, Liu Y, La K, et al. Adaptive stimulation of macropinocytosis overcomes aspartate limitation in cancer cells under hypoxia. Nat Metab. 2022;4:724–38.
Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497:633–7.
Lee SW, Zhang Y, Jung M, Cruz N, Alas B, Commisso C. EGFR-Pak signaling selectively regulates glutamine deprivation-induced macropinocytosis. Dev Cell. 2019;50:381–392 e385.
Yao W, Rose JL, Wang W, Seth S, Jiang H, Taguchi A, et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature. 2019;568:410–4.
Amaravadi RK, Kimmelman AC, Debnath J. Targeting autophagy in cancer: recent advances and future directions. Cancer Discov. 2019;9:1167–81.
Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018;8:276–87.
Saliakoura M, Sebastiano MR, Nikdima I, Pozzato C, Konstantinidou G. Restriction of extracellular lipids renders pancreatic cancer dependent on autophagy. J Exp Clin Cancer Res. 2022;41:16.
Santana-Codina N, Del Rey MQ, Kapner KS, Zhang H, Gikandi A, Malcolm C, et al. NCOA4-Mediated Ferritinophagy is a pancreatic cancer dependency via maintenance of iron bioavailability for iron-sulfur cluster proteins. Cancer Discov. 2022;12:2180–97.
Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature. 2020;581:100–5.
Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628–40.
Wang T, Ye Z, Li Z, Jing DS, Fan GX, Liu MQ, et al. Lactate-induced protein lactylation: A bridge between epigenetics and metabolic reprogramming in cancer. Cell Prolif. 2023;56:e13478.
Liu Z, Wang R, Wang Y, Duan Y, Zhan H. Targeting succinylation-mediated metabolic reprogramming as a potential approach for cancer therapy. Biomed Pharmacother. 2023;168:115713.
Lumibao JC, Tremblay JR, Hsu J, Engle DD. Altered glycosylation in pancreatic cancer and beyond. J Exp Med. 2022;219:e20211505.
Zheng S, Tian Q, Yuan Y, Sun S, Li T, Xia R, et al. Extracellular vesicle-packaged circBIRC6 from cancer-associated fibroblasts induce platinum resistance via SUMOylation modulation in pancreatic cancer. J Exp Clin Cancer Res. 2023;42:324.
Li J, Zhan H, Ren Y, Feng M, Wang Q, Jiao Q, et al. Sirtuin 4 activates autophagy and inhibits tumorigenesis by upregulating the p53 signaling pathway. Cell Death Differ. 2023;30:313–26.
Ferguson LP, Gatchalian J, McDermott ML, Nakamura M, Chambers K, Rajbhandari N, et al. Smarcd3 is an epigenetic modulator of the metabolic landscape in pancreatic ductal adenocarcinoma. Nat Commun. 2023;14:292.
Ren J, Ren B, Liu X, Cui M, Fang Y, Wang X, et al. Crosstalk between metabolic remodeling and epigenetic reprogramming: A new perspective on pancreatic cancer. Cancer Lett. 2024;587:216649.
Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877–919.
Ge T, Gu X, Jia R, Ge S, Chai P, Zhuang A, et al. Crosstalk between metabolic reprogramming and epigenetics in cancer: updates on mechanisms and therapeutic opportunities. Cancer Commun (Lond). 2022;42:1049–82.
Kumar S, Schoonderwoerd MJA, Kroonen JS, de Graaf IJ, Sluijter M, Ruano D, et al. Targeting pancreatic cancer by TAK-981: a SUMOylation inhibitor that activates the immune system and blocks cancer cell cycle progression in a preclinical model. Gut. 2022;71:2266–83.
Mao C, Wang M, Zhuang L, Gan B. Metabolic cell death in cancer: Ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. 2024.
Ge EJ, Bush AI, Casini A, Cobine PA, Cross JR, DeNicola GM, et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22:102–13.
Du W, Gu M, Hu M, Pinchi P, Chen W, Ryan M, et al. Lysosomal Zn(2+) release triggers rapid, mitochondria-mediated, non-apoptotic cell death in metastatic melanoma. Cell Rep. 2021;37:109848.
Liu X, Zhuang L, Gan B Disulfidptosis: disulfide stress-induced cell death. Trends Cell Biol. 2023;34:327–37.
Song X, Zhu S, Xie Y, Liu J, Sun L, Zeng D, et al. JTC801 induces pH-dependent death specifically in cancer cells and slows growth of tumors in mice. Gastroenterology. 2018;154:1480–93.
Rebelo R, Xavier CPR, Giovannetti E, Vasconcelos MH. Fibroblasts in pancreatic cancer: molecular and clinical perspectives. Trends Mol Med. 2023;29:439–53.
Encarnacion-Rosado J, Kimmelman AC. Harnessing metabolic dependencies in pancreatic cancers. Nat Rev Gastroenterol Hepatol. 2021;18:482–92.
Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun. 2017;8:16031.
Tung JC, Barnes JM, Desai SR, Sistrunk C, Conklin MW, Schedin P, et al. Tumor mechanics and metabolic dysfunction. Free Radic Biol Med. 2015;79:269–80.
Dovmark TH, Saccomano M, Hulikova A, Alves F, Swietach P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene. 2017;36:4538–50.
Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S, et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014;74:5301–10.
San-Millan I, Brooks GA. Reexamining cancer metabolism: lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis. 2017;38:119–33.
Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174–86.
Helms E, Onate MK, Sherman MH. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov. 2020;10:648–56.
Picard FSR, Lutz V, Brichkina A, Neuhaus F, Ruckenbrod T, Hupfer A, et al. IL-17A-producing CD8(+) T cells promote PDAC via induction of inflammatory cancer-associated fibroblasts. Gut. 2023;72:1510–22.
Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–83.
Chen S, Chen X, Shan T, Ma J, Lin W, Li W, et al. MiR-21-mediated metabolic alteration of cancer-associated fibroblasts and its effect on pancreatic cancer cell behavior. Int J Biol Sci. 2018;14:100–10.
Parker SJ, Amendola CR, Hollinshead KER, Yu Q, Yamamoto K, Encarnacion-Rosado J, et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 2020;10:1018–37.
Lau AN, Li Z, Danai LV, Westermark AM, Darnell AM, Ferreira R, et al. Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. Elife. 2020;9:56782.
Newman AC, Falcone M, Huerta Uribe A, Zhang T, Athineos D, Pietzke M, et al. Immune-regulated IDO1-dependent tryptophan metabolism is source of one-carbon units for pancreatic cancer and stellate cells. Mol Cell. 2021;81:2290–2302.e2297.
Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019;9:617–27.
Dalin S, Sullivan MR, Lau AN, Grauman-Boss B, Mueller HS, Kreidl E, et al. Deoxycytidine release from pancreatic stellate cells promotes Gemcitabine resistance. Cancer Res. 2019;79:5723–33.
Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell. 2015;162:259–70.
Mukhopadhyay S, Encarnacion-Rosado J, Lin EY, Sohn ASW, Zhang H, Mancias JD, et al. Autophagy supports mitochondrial metabolism through the regulation of iron homeostasis in pancreatic cancer. Sci Adv. 2023;9:eadf9284.
Zhu Y, Fang S, Fan B, Xu K, Xu L, Wang L, et al. Cancer-associated fibroblasts reprogram cysteine metabolism to increase tumor resistance to ferroptosis in pancreatic cancer. Theranostics. 2024;14:1683–1700.
Datta R, Sivanand S, Lau AN, Florek LV, Barbeau AM, Wyckoff J, et al. Interactions with stromal cells promote a more oxidized cancer cell redox state in pancreatic tumors. Sci Adv. 2022;8:eabg6383.
Wang Y, Liang Y, Xu H, Zhang X, Mao T, Cui J, et al. Single-cell analysis of pancreatic ductal adenocarcinoma identifies a novel fibroblast subtype associated with poor prognosis but better immunotherapy response. Cell Discov. 2021;7:36.
Liebl F, Demir IE, Mayer K, Schuster T, D’Haese JG, Becker K, et al. The impact of neural invasion severity in gastrointestinal malignancies: a clinicopathological study. Ann Surg. 2014;260:900–7. discussion 907-908
Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405.
Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M, et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell. 2020;183:1202–1218 e1225.
Lin YC, Hou YC, Wang HC, Shan YS. New insights into the role of adipocytes in pancreatic cancer progression: paving the way towards novel therapeutic targets. Theranostics. 2023;13:3925–42.
Cai Z, Liang Y, Xing C, Wang H, Hu P, Li J, et al. Cancer‑associated adipocytes exhibit distinct phenotypes and facilitate tumor progression in pancreatic cancer. Oncol Rep. 2019;42:2537–49.
Meyer KA, Neeley CK, Baker NA, Washabaugh AR, Flesher CG, Nelson BS, et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem Biophys Rep. 2016;7:144–9.
Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016;6:852–69.
Yang J, Li Y, Sun Z, Zhan H. Macrophages in pancreatic cancer: An immunometabolic perspective. Cancer Lett. 2021;498:188–200.
Bulle A, Lim KH. Beyond just a tight fortress: contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct Target Ther. 2020;5:249.
LaRue MM, Parker S, Puccini J, Cammer M, Kimmelman AC, Bar-Sagi D. Metabolic reprogramming of tumor-associated macrophages by collagen turnover promotes fibrosis in pancreatic cancer. Proc Natl Acad Sci USA. 2022;119:e2119168119.
Otto L, Rahn S, Daunke T, Walter F, Winter E, Moller JL, et al. Initiation of pancreatic cancer: the interplay of hyperglycemia and macrophages promotes the acquisition of malignancy-associated properties in pancreatic ductal epithelial cells. Int J Mol Sci. 2021;22:22105086.
Ajam-Hosseini M, Heydari R, Rasouli M, Akhoondi F, Asadi Hanjani N, Bekeschus S, et al. Lactic acid in macrophage polarization: A factor in carcinogenesis and a promising target for cancer therapy. Biochem Pharm. 2024;222:116098.
Ye H, Zhou Q, Zheng S, Li G, Lin Q, Wei L, et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018;9:453.
Edderkaoui M, Chheda C, Soufi B, Zayou F, Hu RW, Ramanujan VK, et al. An Inhibitor of GSK3B and HDACs kills pancreatic cancer cells and slows pancreatic tumor growth and metastasis in mice. Gastroenterology. 2018;155:1985–1998 e1985.
Zhong Z, Yang K, Li Y, Zhou S, Yao H, Zhao Y, et al. Tumor-associated macrophages drive glycolysis through the IL-8/STAT3/GLUT3 signaling pathway in pancreatic cancer progression. Cancer Lett. 2024;588:216784.
Menjivar RE, Nwosu ZC, Du W, Donahue KL, Hong HS, Espinoza C, et al. Arginase 1 is a key driver of immune suppression in pancreatic cancer. Elife. 2023;12:80721.
Wang X, Su S, Zhu Y, Cheng X, Cheng C, Chen L, et al. Metabolic Reprogramming via ACOD1 depletion enhances function of human induced pluripotent stem cell-derived CAR-macrophages in solid tumors. Nat Commun. 2023;14:5778.
Huber M, Brehm CU, Gress TM, Buchholz M, Alashkar Alhamwe B, von Strandmann EP, et al. The immune microenvironment in pancreatic cancer. Int J Mol Sci 2020, 21.
Huang L, Li H, Zhang C, Chen Q, Liu Z, Zhang J, et al. Unlocking the potential of T-cell metabolism reprogramming: Advancing single-cell approaches for precision immunotherapy in tumour immunity. Clin Transl Med. 2024;14:e1620.
Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H, et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov. 2020;10:608–25.
Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829–842.e813.
Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bosse D, et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun. 2019;10:4346.
Yu YR, Imrichova H, Wang H, Chao T, Xiao Z, Gao M, et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat Immunol. 2020;21:1540–51.
Madden MZ, Rathmell JC. The Complex Integration of T-cell Metabolism and Immunotherapy. Cancer Discov. 2021;11:1636–43.
Munir H, Jones JO, Janowitz T, Hoffmann M, Euler M, Martins CP, et al. Stromal-driven and Amyloid beta-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat Commun. 2021;12:683.
Wang L, Liu Y, Dai Y, Tang X, Yin T, Wang C, et al. Single-cell RNA-seq analysis reveals BHLHE40-driven pro-tumour neutrophils with hyperactivated glycolysis in pancreatic tumour microenvironment. Gut. 2023;72:958–71.
Miao L, Lu C, Zhang B, Li H, Zhao X, Chen H, et al. Advances in metabolic reprogramming of NK cells in the tumor microenvironment on the impact of NK therapy. J Transl Med. 2024;22:229.
Ge W, Meng L, Cao S, Hou C, Zhu X, Huang D, et al. The SIX1/LDHA axis promotes lactate accumulation and leads to NK cell dysfunction in pancreatic cancer. J Immunol Res. 2023;2023:6891636.
He C, Wang D, Shukla SK, Hu T, Thakur R, Fu X, et al. Vitamin B6 competition in the tumor microenvironment hampers antitumor functions of NK cells. Cancer Discov. 2024;14:176–93.
Canale FP, Basso C, Antonini G, Perotti M, Li N, Sokolovska A, et al. Metabolic modulation of tumours with engineered bacteria for immunotherapy. Nature. 2021;598:662–6.
Pandya G, Kirtonia A, Singh A, Goel A, Mohan CD, Rangappa KS, et al. A comprehensive review of the multifaceted role of the microbiota in human pancreatic carcinoma. Semin Cancer Biol. 2022;86:682–92.
Oldfield L, Costello E. Where the metabolome meets the microbiome for pancreatic cancer detection. Cell Rep. Med. 2023;4:101011.
Nagata N, Nishijima S, Kojima Y, Hisada Y, Imbe K, Miyoshi-Akiyama T, et al. Metagenomic identification of microbial signatures predicting pancreatic cancer from a multinational study. Gastroenterology. 2022;163:222–38.
Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science. 2020;369:1481–9.
Mendez R, Kesh K, Arora N, Di Martino L, McAllister F, Merchant N, et al. Microbial dysbiosis and polyamine metabolism as predictive markers for early detection of pancreatic cancer. Carcinogenesis. 2020;41:561–70.
Mirji G, Worth A, Bhat SA, El Sayed M, Kannan T, Goldman AR, et al. The microbiome-derived metabolite TMAO drives immune activation and boosts responses to immune checkpoint blockade in pancreatic cancer. Sci Immunol. 2022;7:eabn0704.
Wang Y, Yang G, You L, Yang J, Feng M, Qiu J, et al. Role of the microbiome in occurrence, development and treatment of pancreatic cancer. Mol Cancer. 2019;18:173.
Hezaveh K, Shinde RS, Klotgen A, Halaby MJ, Lamorte S, Ciudad MT, et al. Tryptophan-derived microbial metabolites activate the aryl hydrocarbon receptor in tumor-associated macrophages to suppress anti-tumor immunity. Immunity. 2022;55:324–340.e328.
Panebianco C, Ciardiello D, Villani A, Maiorano BA, Latiano TP, Maiello E, et al. Insights into the role of gut and intratumor microbiota in pancreatic ductal adenocarcinoma as new key players in preventive, diagnostic and therapeutic perspective. Semin Cancer Biol. 2022;86:997–1007.
Capula M, Peran M, Xu G, Donati V, Yee D, Gregori A, et al. Role of drug catabolism, modulation of oncogenic signaling and tumor microenvironment in microbe-mediated pancreatic cancer chemoresistance. Drug Resist Updat. 2022;64:100864.
Li S, Dong R, Kang Z, Li H, Wu X, Li T. Exosomes: Another intercellular lipometabolic communication mediators in digestive system neoplasms? Cytokine Growth Factor Rev. 2023;73:93–100.
Anastasi F, Botto A, Immordino B, Giovannetti E, McDonnell LA. Proteomics analysis of circulating small extracellular vesicles: Focus on the contribution of EVs to tumor metabolism. Cytokine Growth Factor Rev. 2023;73:3–19.
Shibata C, Otsuka M, Seimiya T, Kishikawa T, Ishigaki K, Fujishiro M. Lipolysis by pancreatic cancer-derived extracellular vesicles in cancer-associated cachexia via specific integrins. Clin Transl Med. 2022;12:e1089.
Choi Y, Akyildiz K, Seong J, Lee Y, Jeong E, Park JS, et al. Dielectrophoretic capture of cancer-derived small-extracellular-vesicle-bound Janus nanoparticles via lectin-glycan interaction. Adv Health Mater. 2024;13:e2302313.
Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250.
Ruze R, Song J, Yin X, Chen Y, Xu R, Wang C, et al. Mechanisms of obesity- and diabetes mellitus-related pancreatic carcinogenesis: a comprehensive and systematic review. Signal Transduct Target Ther. 2023;8:139.
Rask-Andersen M, Ivansson E, Hoglund J, Ek WE, Karlsson T, Johansson A. Adiposity and sex-specific cancer risk. Cancer Cell. 2023;41:1186–1197.e1184.
Ruze R, Chen Y, Xu R, Song J, Yin X, Wang C, et al. Obesity, diabetes mellitus, and pancreatic carcinogenesis: Correlations, prevention, and diagnostic implications. Biochim Biophys Acta Rev Cancer. 2023;1878:188844.
Jain T, Dudeja V. The war against pancreatic cancer in 2020 - advances on all fronts. Nat Rev Gastroenterol Hepatol. 2021;18:99–100.
Park JH, Han K, Hong JY, Park YS, Hur KY, Kang G, et al. Changes in metabolic syndrome status are associated with altered risk of pancreatic cancer: a nationwide cohort study. Gastroenterology. 2022;162:509–520.e507.
Zhang AMY, Xia YH, Lin JSH, Chu KH, Wang WCK, Ruiter TJJ, et al. Hyperinsulinemia acts via acinar insulin receptors to initiate pancreatic cancer by increasing digestive enzyme production and inflammation. Cell Metab. 2023;35:2119–2135.e2115.
Fonteneau G, Redding A, Hoag-Lee H, Sim ES, Heinrich S, Gaida MM, et al. Stress Granules Determine the Development of Obesity-Associated Pancreatic Cancer. Cancer Discov. 2022;12:1984–2005.
Cascetta P, Cavaliere A, Piro G, Torroni L, Santoro R, Tortora G, et al. Pancreatic cancer and obesity: molecular mechanisms of cell transformation and chemoresistance. Int J Mol Sci. 2018;19:19113331.
Chai X, Chu H, Yang X, Meng Y, Shi P, Gou S. Metformin increases sensitivity of pancreatic cancer cells to Gemcitabine by reducing CD133+ cell populations and suppressing ERK/P70S6K signaling. Sci Rep. 2015;5:14404.
Pothuraju R, Rachagani S, Junker WM, Chaudhary S, Saraswathi V, Kaur S, et al. Pancreatic cancer associated with obesity and diabetes: an alternative approach for its targeting. J Exp Clin Cancer Res. 2018;37:319.
Xiao YL, Gong Y, Qi YJ, Shao ZM, Jiang YZ. Effects of dietary intervention on human diseases: molecular mechanisms and therapeutic potential. Signal Transduct Target Ther. 2024;9:59.
Wang Q, Zhou W. Roles and molecular mechanisms of physical exercise in cancer prevention and treatment. J Sport Health Sci. 2021;10:201–10.
Seki T, Yang Y, Sun X, Lim S, Xie S, Guo Z, et al. Brown-fat-mediated tumour suppression by cold-altered global metabolism. Nature. 2022;608:421–8.
Babic A, Rosenthal MH, Sundaresan TK, Khalaf N, Lee V, Brais LK, et al. Adipose tissue and skeletal muscle wasting precede clinical diagnosis of pancreatic cancer. Nat Commun. 2023;14:4317.
Kurz E, Hirsch CA, Dalton T, Shadaloey SA, Khodadadi-Jamayran A, Miller G, et al. Exercise-induced engagement of the IL-15/IL-15Ralpha axis promotes anti-tumor immunity in pancreatic cancer. Cancer Cell. 2022;40:720–737.e725.
Badiyan SN, Molitoris JK, Chuong MD, Regine WF, Kaiser A. The role of radiation therapy for pancreatic cancer in the adjuvant and neoadjuvant settings. Surg Oncol Clin N. Am. 2017;26:431–53.
Seshacharyulu P, Baine MJ, Souchek JJ, Menning M, Kaur S, Yan Y, et al. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim Biophys Acta Rev Cancer. 2017;1868:69–92.
Gunda V, Souchek J, Abrego J, Shukla SK, Goode GD, Vernucci E, et al. MUC1-mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin Cancer Res. 2017;23:5881–91.
Souchek JJ, Baine MJ, Lin C, Rachagani S, Gupta S, Kaur S, et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br J Cancer. 2014;111:1139–49.
Falcone M, Uribe AH, Papalazarou V, Newman AC, Athineos D, Stevenson K, et al. Sensitisation of cancer cells to radiotherapy by serine and glycine starvation. Br J Cancer. 2022;127:1773–86.
Amrutkar M, Gladhaug IP. Pancreatic cancer chemoresistance to Gemcitabine. Cancers. 2017;9:157.
Zhao H, Duan Q, Zhang Z, Li H, Wu H, Shen Q, et al. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J Cell Mol Med. 2017;21:2055–67.
Feng M, Xiong G, Cao Z, Yang G, Zheng S, Qiu J, et al. LAT2 regulates glutamine-dependent mTOR activation to promote glycolysis and chemoresistance in pancreatic cancer. J Exp Clin Cancer Res. 2018;37:274.
Li Y, Tang S, Shi X, Lv J, Wu X, Zhang Y, et al. Metabolic classification suggests the GLUT1/ALDOB/G6PD axis as a therapeutic target in chemotherapy-resistant pancreatic cancer. Cell Rep. Med. 2023;4:101162.
Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat Commun. 2018;9:4945.
Zeng X, Guo H, Liu Z, Qin Z, Cong Y, Ren N, et al. S100A11 activates the pentose phosphate pathway to induce malignant biological behaviour of pancreatic ductal adenocarcinoma. Cell Death Dis. 2022;13:568.
Chen S, Ning B, Song J, Yang Z, Zhou L, Chen Z, et al. Enhanced pentose phosphate pathway activity promotes pancreatic ductal adenocarcinoma progression via activating YAP/MMP1 axis under chronic acidosis. Int J Biol Sci. 2022;18:2304–16.
Sharma N, Bhushan A, He J, Kaushal G, Bhardwaj V. Metabolic plasticity imparts erlotinib-resistance in pancreatic cancer by upregulating glucose-6-phosphate dehydrogenase. Cancer Metab. 2020;8:19.
Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71–87.e77.
Ge W, Wang Y, Quan M, Mao T, Bischof EY, Xu H, et al. Activation of the PI3K/AKT signaling pathway by ARNTL2 enhances cellular glycolysis and sensitizes pancreatic adenocarcinoma to erlotinib. Mol Cancer. 2024;23:48.
Xia G, Wang H, Song Z, Meng Q, Huang X, Huang X. Gambogic acid sensitizes gemcitabine efficacy in pancreatic cancer by reducing the expression of ribonucleotide reductase subunit-M2 (RRM2). J Exp Clin Cancer Res. 2017;36:107.
Zarei M, Lal S, Parker SJ, Nevler A, Vaziri-Gohar A, Dukleska K, et al. Posttranscriptional upregulation of IDH1 by HuR establishes a powerful survival phenotype in pancreatic cancer cells. Cancer Res. 2017;77:4460–71.
Ganguly K, Bhatia R, Rauth S, Kisling A, Atri P, Thompson C, et al. Mucin 5AC serves as the Nexus for beta-Catenin/c-Myc interplay to promote glutamine dependency during pancreatic cancer chemoresistance. Gastroenterology. 2022;162:253–268.e213.
Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2015;112:2473–8.
Gupta VK, Sharma NS, Kesh K, Dauer P, Nomura A, Giri B, et al. Metastasis and chemoresistance in CD133 expressing pancreatic cancer cells are dependent on their lipid raft integrity. Cancer Lett. 2018;439:101–12.
Yu S, Wang L, Che D, Zhang M, Li M, Naito M, et al. Targeting CRABP-II overcomes pancreatic cancer drug resistance by reversing lipid raft cholesterol accumulation and AKT survival signaling. J Exp Clin Cancer Res. 2022;41:88.
Kubo M, Gotoh K, Eguchi H, Kobayashi S, Iwagami Y, Tomimaru Y, et al. Impact of CD36 on chemoresistance in pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2020;27:610–9.
Jiang X, Ma Y, Wang T, Zhou H, Wang K, Shi W, et al. Targeting UBE2T Potentiates Gemcitabine Efficacy in Pancreatic Cancer by Regulating Pyrimidine Metabolism and Replication Stress. Gastroenterology. 2023;164:1232–47.
Bandi DSR, Sarvesh S, Farran B, Nagaraju GP, El-Rayes BF. Targeting the metabolism and immune system in pancreatic ductal adenocarcinoma: Insights and future directions. Cytokine Growth Factor Rev. 2023;71–72:26–39.
Guo Y, Wang R, Shi J, Yang C, Ma P, Min J, et al. Machine learning-based integration develops a metabolism-derived consensus model for improving immunotherapy in pancreatic cancer. J Immunother Cancer. 2023;11:007466.
Bantug GR, Hess C. The immunometabolic ecosystem in cancer. Nat Immunol. 2023;24:2008–20.
Lian X, Yang K, Li R, Li M, Zuo J, Zheng B, et al. Immunometabolic rewiring in tumorigenesis and anti-tumor immunotherapy. Mol Cancer. 2022;21:27.
Ishii KJ, Akira S. Potential link between the immune system and metabolism of nucleic acids. Curr Opin Immunol. 2008;20:524–9.
Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177–92.
Abt ER, Le TM, Dann AM, Capri JR, Poddar S, Lok V, et al. Reprogramming of nucleotide metabolism by interferon confers dependence on the replication stress response pathway in pancreatic cancer cells. Cell Rep. 2022;38:110236.
Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123:4479–88.
Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76.
Rivadeneira DB, DePeaux K, Wang Y, Kulkarni A, Tabib T, Menk AV, et al. Oncolytic viruses engineered to enforce leptin expression reprogram tumor-infiltrating T cell metabolism and promote tumor clearance. Immunity. 2019;51:548–560.e544.
Cortese N, Capretti G, Barbagallo M, Rigamonti A, Takis PG, Castino GF, et al. Metabolome of pancreatic juice delineates distinct clinical profiles of pancreatic cancer and reveals a link between glucose metabolism and PD-1(+) Cells. Cancer Immunol Res. 2020;8:493–505.
Wolpin BM, Hezel AF, Abrams T, Blaszkowsky LS, Meyerhardt JA, Chan JA, et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J Clin Oncol. 2009;27:193–8.
Kordes S, Klumpen HJ, Weterman MJ, Schellens JH, Richel DJ, Wilmink JW. Phase II study of capecitabine and the oral mTOR inhibitor everolimus in patients with advanced pancreatic cancer. Cancer Chemother Pharm. 2015;75:1135–41.
Manuel ER, Chen J, D’Apuzzo M, Lampa MG, Kaltcheva TI, Thompson CB, et al. Salmonella-based therapy targeting Indoleamine 2,3-Dioxygenase coupled with enzymatic depletion of tumor hyaluronan induces complete regression of aggressive pancreatic tumors. Cancer Immunol Res. 2015;3:1096–107.
Anthiya S, Ozturk SC, Yanik H, Tavukcuoglu E, Sahin A, Datta D, et al. Targeted siRNA lipid nanoparticles for the treatment of KRAS-mutant tumors. J Control Release. 2023;357:67–83.
Liu F, Xiang Q, Luo Y, Luo Y, Luo W, Xie Q, et al. A hybrid nanopharmaceutical for specific-amplifying oxidative stress to initiate a cascade of catalytic therapy for pancreatic cancer. J Nanobiotechnol. 2023;21:165.
Cao S, Saw PE, Shen Q, Li R, Liu Y, Xu X. Reduction-responsive RNAi nanoplatform to reprogram tumor lipid metabolism and repolarize macrophage for combination pancreatic cancer therapy. Biomaterials. 2022;280:121264.
Zhang G, Zhan M, Zhang C, Wang Z, Sun H, Tao Y, et al. Redox-Responsive Dendrimer Nanogels enable ultrasound-enhanced chemoimmunotherapy of pancreatic cancer via endoplasmic reticulum stress amplification and macrophage polarization. Adv Sci (Weinh). 2023;10:e2301759.
Wang L, Dou J, Jiang W, Wang Q, Liu Y, Liu H, et al. Enhanced intracellular transcytosis of nanoparticles by degrading extracellular matrix for deep tissue radiotherapy of pancreatic adenocarcinoma. Nano Lett. 2022;22:6877–87.
Yang XY, Zhang JG, Zhou QM, Yu JN, Lu YF, Wang XJ, et al. Extracellular matrix modulating enzyme functionalized biomimetic Au nanoplatform-mediated enhanced tumor penetration and synergistic antitumor therapy for pancreatic cancer. J Nanobiotechnol. 2022;20:524.
Noe R, Inglese N, Romani P, Serafini T, Paoli C, Calciolari B, et al. Organic Selenium induces ferroptosis in pancreatic cancer cells. Redox Biol. 2023;68:102962.
Wang Y, Chen F, Zhou H, Huang L, Ye J, Liu X, et al. Redox Dyshomeostasis with dual stimuli-activatable dihydroartemisinin nanoparticles to potentiate ferroptotic therapy of pancreatic cancer. Small Methods. 2023;7:e2200888.
Alcala S, Villarino L, Ruiz-Canas L, Couceiro JR, Martinez-Calvo M, Palencia-Campos A, et al. Targeting cancer stem cell OXPHOS with tailored ruthenium complexes as a new anti-cancer strategy. J Exp Clin Cancer Res. 2024;43:33.
Bhaw-Luximon A, Jhurry D. Metformin in pancreatic cancer treatment: from clinical trials through basic research to biomarker quantification. J Cancer Res Clin Oncol. 2016;142:2159–71.
Huang BZ, Chang JI, Li E, Xiang AH, Wu BU. Influence of statins and cholesterol on mortality among patients with pancreatic cancer. J Natl Cancer Inst. 2017;109:djw275.
Wang C, Zhang T, Liao Q, Dai M, Guo J, Yang X, et al. Metformin inhibits pancreatic cancer metastasis caused by SMAD4 deficiency and consequent HNF4G upregulation. Protein Cell. 2021;12:128–44.
Han H, Hou Y, Chen X, Zhang P, Kang M, Jin Q, et al. Metformin-induced stromal depletion to enhance the penetration of Gemcitabine-loaded magnetic nanoparticles for pancreatic cancer targeted therapy. J Am Chem Soc. 2020;142:4944–54.
Cordes T, Metallo CM. Statins limit coenzyme Q synthesis and metabolically synergize with MEK inhibition in pancreatic tumors. Cancer Res. 2020;80:151–2.
Zhou T, Xie Y, Hou X, Bai W, Li X, Liu Z, et al. Irbesartan overcomes gemcitabine resistance in pancreatic cancer by suppressing stemness and iron metabolism via inhibition of the Hippo/YAP1/c-Jun axis. J Exp Clin Cancer Res. 2023;42:111.
Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharm. 2013;71:523–30.
Zachar Z, Marecek J, Maturo C, Gupta S, Stuart SD, Howell K, et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl). 2011;89:1137–48.
Alistar A, Morris BB, Desnoyer R, Klepin HD, Hosseinzadeh K, Clark C, et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017;18:770–8.
Pillai R, Papagiannakopoulous T. DON of hope: Starving pancreatic cancer by glutamine antagonism. Cancer Res. 2024;84:349–50.
Hammel P, Fabienne P, Mineur L, Metges JP, Andre T, De La Fouchardiere C, et al. Erythrocyte-encapsulated asparaginase (eryaspase) combined with chemotherapy in second-line treatment of advanced pancreatic cancer: An open-label, randomized Phase IIb trial. Eur J Cancer. 2020;124:91–101.
Powderly JD, Klempner SJ, Naing A, Bendell J, Garrido-Laguna I, Catenacci DVT, et al. Epacadostat Plus Pembrolizumab and chemotherapy for advanced solid tumors: results from the Phase I/II ECHO-207/KEYNOTE-723 Study. Oncologist. 2022;27:905–e848.
Chakrabarti G, Moore ZR, Luo X, Ilcheva M, Ali A, Padanad M, et al. Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ss-lapachone. Cancer Metab. 2015;3:12.
Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: Advances and challenges. Cell. 2023;186:1729–54.
Biancur DE, Kapner KS, Yamamoto K, Banh RS, Neggers JE, Sohn ASW, et al. Functional genomics identifies metabolic vulnerabilities in pancreatic cancer. Cell Metab. 2021;33:199–210.e198.
Zhu XG, Chudnovskiy A, Baudrier L, Prizer B, Liu Y, Ostendorf BN, et al. Functional genomics in vivo reveal metabolic dependencies of pancreatic cancer cells. Cell Metab. 2021;33:211–221.e216.
Bartman CR, Faubert B, Rabinowitz JD, DeBerardinis RJ. Metabolic pathway analysis using stable isotopes in patients with cancer. Nat Rev Cancer. 2023;23:863–78.
Duan X, Zhang T, Feng L, de Silva N, Greenspun B, Wang X, et al. A pancreatic cancer organoid platform identifies an inhibitor specific to mutant KRAS. Cell Stem Cell. 2024;31:71–88.e78.
Dart A. Spatial metabolic patterns. Nat Rev Cancer. 2024;24:3.
Funding
Supported by the National Natural Science Foundation of China (Grant number: 82172727), Nature Science Foundation of Beijing (Grant number: 7202164), CAMS Innovation Fund for Medical Sciences (CIFMS) (Grant number: 2021-I2M-1-002) National High-Level Hospital Clinical Research Funding (Grant number: 2022-PUMCH-D-001, and 2022-PUMCH-B-004).
Author information
Authors and Affiliations
Contributions
All authors helped to perform the research; Hao Wu and Ziwen Liu were involved in the conception and design; Hao Wu and Mengdi Fu were involved in the drafting of the paper or revising it critically for intellectual content; Qiyao Zhang assisted in retrieving and summarizing ongoing clinical trials; Mengwei Wu assisted in logical consistency and language polishing; Zhen Cao assisted in creation and modification of figures and charts; Ziwen Liu was involved in the final approval of the version to be published. All authors agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Francesca Pentimalli
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, H., Fu, M., Wu, M. et al. Emerging mechanisms and promising approaches in pancreatic cancer metabolism. Cell Death Dis 15, 553 (2024). https://doi.org/10.1038/s41419-024-06930-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-024-06930-0
- Springer Nature Limited