
Abstract
Molecular mechanisms underlying luteinization (terminal differentiation of granulosa and theca cells after ovulation) and luteolysis (demise of corpus luteum) are poorly understood in human ovary. Here we report that activin-A, after binding to its cognate receptors induces a functional luteolytic state and reverses luteinization phenotype by downregulating the expression of the steroidogenic enzymes, LH receptor and VEGF and reducing estradiol (E2) progesterone (P4) production and upregulating FSH receptor and cyclin D1 expression in human primary luteinized granulosa cells. Further, this action of activin-A involves downregulation of JNK signaling pathway and is opposite to that of human chorionic gonadotropin (hCG), which acts as a luteotropic hormone and improves luteal function through the activation of JNK pathway in the same cell type. Reversal of luteinization phenotype in luteal granulosa cells by activin-A potentially makes this hormone an attractive candidate for use under certain clinical situations, where induction of luteolysis and rapid reduction of endogenous sex steroid levels are beneficial such as ovarian hyperstimulation syndrome (OHSS), in which the ovaries hyper-respond to gonadotropin stimulation by producing too many growing follicles along with development of ascites, pleural effusion, and hemo-concentrations as a result of increased vascular permeability and leakage of intravascular volume into third spaces. Our work unveils a previously undefined role for activin-A and JNK signaling pathway in human corpus luteum biology, that might have a direct clinical impact in assisted reproductive technologies.
Similar content being viewed by others

Introduction
Corpus luteum is a transient endocrine organ that is composed of different compartments and cell types1. It is the main source of progesterone production during luteal phase of the menstrual cycle that is required for the preparation of endometrium for implantation. If pregnancy occurs CL is rescued from undergoing demise (luteolysis) and continues to produce progesterone (P4) hormone by the stimulatory action of human chorionic gonadotropin (hCG) produced by the trophoblasts of the embryo until luteo-placental shift occurs and placenta becomes the major source of P4 production for the remainder of pregnancy2. Luteinization process and luteolysis are complex event that are poorly understood in human. While the former is characterized by a marked tissue remodeling, marked changes in intracellular signaling, gene regulations, extracellular matrix adhesion proteins, angiogenesis, cell cycle regulators3,4,5 the latter can be defined as loss of function, decrease in P synthesis and output, tissue break-down, degradation of extracellular matrix proteins, infiltration of macrophages, and subsequent involution of the luteal structures and vessels and increased cell death1,6.
There are three important issues regarding the luteal function in human assisted reproduction as follows: First, it is well documented that the functions of corpus luteum are defective in patients who undergo ovarian stimulation with gonadotropin hormones during assisted reproduction7. This necessitates progesterone and/or hCG administration known as luteal phase support in order to improve clinical pregnancy rates and prevent miscarriage8,9. Second important issue is the risk of ovarian hyperstimulation syndrome (OHSS) during assisted reproduction in which the ovaries hyper-respond to gonadotropin stimulation by producing too many growing follicles along with development of ascites, pleural effusion, and hemoconcentrations as a result of increased vascular permeability and leakage of intravascular volume into third spaces10,11,12. As a third issue there are some clinical circumstances in which early luteolysis and rapid reduction in serum estrogen level are indicated such as IVF patients with estrogen sensitive tumors such as breast cancer13. Therefore, identification of hormones signaling pathways involved in the luteinization and luteolysis are of paramount importance from the perspective of clinical practice in IVF.
Activin-A is a member of TGF-β superfamily and has a number of actions defined in human reproductive functioning such as stimulation of the production of FSHβ subunit from the gonadotropes in the anterior pituitary, mitogenic effect on granulosa cells (GCs) of the growing follicles, upregulation of their FSH receptor expression and oocyte maturation14,15. It was also documented that GCs continue to express activin-A and its cognate receptors after luteinization process15. However, available data is limited and somewhat controversial regarding the biological role of activin-A in these cells. This works unveils a novel mechanism that link activin-A and JNK signaling pathway in corpus luteum biology using human luteal granulosa cells obtained from IVF patients undergoing assisted reproduction.
Results
Validation experiments
It was previously documented that human luteal granulosa cells (GCs) continue to have steroidogenic activity in vitro16,17,18,19,20,21,22. In agreement, we first confirm that these cells have a certain basal steroidogenic activity that produce detectable amounts of E2 and P4 even under serum-free culture condition. When fetal bovine serum (FBS) was added to the culture medium, E2 and P4 output of the cells were further enhanced in proportion with increasing concentration of FBS (Supplementary Figs. 1A and 1B). Next, we analyzed and validated the expression of activin receptors type 1 and type 2 (ACVR1, ACR2) in these cells with qRT-PCR method (Supplementary Fig. 1C). Treatment of the cells with activin-A at 5, 25, and 100 ng/mL concentrations for 30 min induced phosphorylation of Smad2 as shown by a dose-dependent increase in the expression of phospho-Smad2Ser465/Ser467 in western blotting. This effect was abolished when the cells were treated with activin-A and a receptor blocker (Supplementary Fig. 1D). Taken together, these validation experiments confirm as a proof of concept the presence of the steroidogenic function, the expression of activin receptors and their responsiveness to exogenously administered activin-A in human luteal GCs in vitro.
Activin-A downregulates the expression of steroidogenic enzymes and reduces E2 and P4 production in human luteal GCs
Having confirmed the expression and biological activity of activin receptors in human luteal GCs, we next analyzed the effect of activin-A on the expression of the steroidogenic enzymes, gonadotropin receptors, and hormone production in these cells. Incubation of the cells with activin-A at four different concentrations (1, 5, 25, and 100 ng/ml) for 24 h resulted in a significant downregulation in the expression of the steroidogenic enzymes StAR, P450 SCC, 3β-HSD, and 17β-HSD in a dose-dependent fashion as shown both on qRT-PCR (Fig. 1a) and western blotting (Fig. 1b, c). However, such a notable change was not observed in the aromatase expression after activin-A treatment either at transcriptional or translational level (Fig. 1a, b). In line with downregulated expression of the steroidogenic enzymes, in vitro E2 and P4 production of the cells gradually decreased following treatment with activin-A at incremental concentration (Fig. 1d). We also observed that the suppressive effect of activin-A on the steroidogenic function of the cells persisted and their E2 and P4 production continued to decrease progressively when culture period was extended up to 96 h (Supplementary Fig. 2).

Activin-A treatment is associated with significant decrease in the expression of steroidogenic enzymes StAR, P450 SCC, 3β-HSD, and 17β-HSD on qRT-PCR (a) and western blotting (b, c) along with decreased production of E2 and P4 (d). Note that aromatase expression did not change meaningfully after activin-A treatment either at transcriptional or translational level. The expression of phospho-c-JunSer73 and phospho-JNKSerThr183/Tyr185 expression were gradually diminished along with the steroidogenic enzymes in western blotting (b, c). FSH receptor expression was significantly upregulated and LH receptor was down-regulated in the cells in a dose-dependent fashion after activin-A treatment on qRT-PCR (a).
In order to rule out the possibility that declining E2 and P4 production of the cells after activin-A treatment is due to reduced viability rather than suppression of steroidogenic activity, we compared the viability of the cells using intravital carbocyanine uptake assay, and could not find any difference in the vitality of the cells before and after activin-A treatment (Supplementary Fig. 3).
Since these results were obtained in the luteal GCs retrieved from the patients stimulated with GnRH antagonist protocol, we repeated the experiments in the GCs of natural cycle and GnRH agonist stimulation protocol and obtained similar findings (Supplementary Fig. 4).
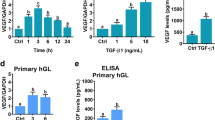
We also observed that FSH receptor expression was significantly upregulated and LH receptor was down-regulated in the cells in a dose-dependent fashion after activin-A treatment on qRT-PCR (Fig. 1a). Increased vascularization and decreased expression of cyclin D1 are other hallmarks of luteinization in the granulosa cells5,6,23. We therefore compared the expression of these markers in another set of experiments and found that activin-A significantly downregulated the expression of LH receptor, VEGF and upregulated FSH receptor and cyclin D1, while hCG as a luteotropic hormone exerted the opposite effects (Supplementary Fig. 5).
Activin-A downregulates the steroidogenic enzymes along with JNK pathway in human luteal granulosa cells
Our findings so far provided supporting evidence that activin-A treatment reverses luteinization phenotype in the luteal GCs, as evidenced by decreased expression of steroidogenic enzymes particularly StAR and 3β-HSD, LH receptor, VEGF-A, cyclin D1, and reduced E2/P4 production. We have recently shown in another study that hCG improves luteal function by upregulating the expression of steroidogenic enzymes and enhancing P4 output through activation of JNK pathway in human luteal GCs24. Furthermore, we also observed in that study that hCG stimulation of the cells after silencing of JNK via siRNA failed to improve the luteal defect and increase P4 output, suggestive of a pivotal role for JNK in the regulation of steroidogenesis in these cells. Given that hCG and activin-A have opposed effects on the luteinization characteristics of the GCs, we therefore, hypothesized if hCG induced the improvement of luteal function involves upregulation of JNK pathway in luteal GCs, activin-A induced reversal of luteinization might involve downregulation of JNK pathway in these cells. To test our hypothesis, we conducted a series of experiment. First, we simultaneously analyzed the effect of hCG and activin-A on the steroidogenic function and JNK pathway in these cells. As shown in the Fig. 2, hCG (10 IU/ml) significantly upregulated the expression of StAR, 3β-HSD, and phospho-c-Jun along with increased P4 production of the cells. By contrast, activin-A (25 ng/ml) produced opposite effects on these cells by downregulating the expression of these enzymes, phospho-c-Jun and reducing their P4 production. When the cells were treated with activin-A at different concentrations (1, 5, 25, and 100 ng/ml) rather than a single dose we obtained the same findings. The expression of phospho-c-JunSer73 and phospho-JNKSerThr183/Tyr185 were diminished gradually. However total JNK and c-Jun expression did not change, suggestive of a change in the JNK activity after activin-A (Fig. 1b, c). To confirm this observation, we next carried out a kinase assay and found that endogenous kinase activity of JNK was diminished in the cells treated with activin-A as evidenced by reduced phosphorylation of recombinant c-Jun at Ser73 residue (Fig. 3a, b).

hCG treatment (10IU/ml) significantly upregulated StAR, 3β-HSD expression along with phospho-c-Jun (a, b) in western blotting and increased P4 output in the cells (c). By contrast, activin-A (25 ng/ml) exerted opposite effects and reduced P4 production. FSH (25 mIU/ml) increased the expression of aromatase (a, b) and increased E2 production (c) but did not change the levels of total or phosphorylated c-Jun.

Activin-A treatment caused a dose-dependent decrease in the kinase activity of JNK as evidenced by decreased phosphorylation of c-JunSer63. Total c-Jun expression remained stable. Vinculin is used as a loading control.
In another set of experiments, the cells were treated with activin-A with and without a receptor blocker to analyze the impact of blocking activin-A signaling on the steroidogenic function and JNK expression. We observed that the suppressive effects of activin-A on the steroidogenic enzymes and JNK pathway were relieved and the expression of StAR, 3β-HSD, phospho-c-JunSer73, and phospho-JNKSerThr183/Tyr185 increased together with increased E2 and P4 production of the cells when the biological actions of activin-A was antagonized with a selective receptor blocker (Fig. 4a, b).

When the biological actions of activin-A was inhibited with a selective receptor blocker the suppressive effects of activin-A on the steroidogenic enzymes and JNK pathway were reversed and the expression of StAR, 3β-HSD, phospho-c-JunSer73, and phospho-JNKSerThr183/Tyr185 were increased along with E2 and P production of the cells (a, b). Aromatase expression did not change meaningfully after activin-A treatment with and without the receptor blocker (a).
Consistent with western blot results, the confocal images showed that the percentages of the cells immuno-stained positively for StAR and phospho-c-JunSer73 were significantly decreased after treatment with activin-A in comparison to control and hCG-treated cells. While control cells have a relatively faint cytoplasmic staining for StAR, hCG-treated cells are characterized by a diffuse and intense cytoplasmic staining pattern (Fig. 5a, c, e). By contrast, there was almost no signal for StAR in the activin-treated cells (Fig. 5a, c). The staining pattern for phospho-c-Jun was predominantly nuclear, which was further increased and decreased after treatment with hCG and activin-A, respectively (Fig. 5b, d, e).

The intensity of staining for StAR (green signal, a high magnification, c low magnification) and phospho-c-Jun (green signal, b high magnification, d low magnification) are substantially increased and decreased after treatment with hCG and activin-A respectively. In accordance with this the percentage of the cells stained positive for these markers were significantly decreased in the activin-treated cells in comparison to control and hCG-treated ones (e). Nuclei were highlighted in blue with DAPI staining. Oil Red O staining shows neutral lipid droplets in the cells as red dots. Scale bar is 10 μm.
Culture of luteal GCs with activin-A in follicular fluid yields similar results
The cell culture experiments presented so far were carried out in the culture medium supplemented with FBS. We repeated the experiments with follicular fluid (FF) to see if activin-A will have the same suppressive effect on steroidogenesis when culture media is supplemented with androgen-rich FF instead of FBS. For this purpose, we first measured the concentrations of testosterone, E2, P4, hCG and lipids in the follicular fluids as shown in the Table 1. The mean concentrations of E2, P4, and testosterone were much higher in the FF samples compared to the FBS and culture media. Similar to the results of the experiment conducted with FBS, the expression of StAR was substantially decreased and increased in the cells after treatment with activin-A and hCG respectively, when they were cultured in culture medium with supplemented with FF (Fig. 6a). In line with that, P4 production of the cells decreased after activin-A treatment (Fig. 6b). Since the concentrations of E2 were extremely high in the FF samples (ranging from 141,935 to 305,000 pg/ml), it masked daily endogenous E2 production of the cells at baseline and after treatment with activin-A even when FF was used at 1–2% concentrations (Fig. 6c).

Note the substantial decrease in the staining intensity of StAR and reduced P4 output after activin-A treatment similar to what it is observed in the cells cultured with FBS. hCG was associated significant increase in the StAR expression and P4 production, confirming its well-known luteotropic effects in both types of culture condition.
Discussion
We showed in this study that activin-A downregulates the expression of steroidogenic enzymes StAR, P450 SCC, 3β-HSD, 17β-HSD, LH receptor, VEGF-A, and upregulates cyclin D1 and FSH receptor expression in human luteal granulosa cells. E2 and P4 production of the cells dramatically decreased after activin-A treatment. StAR is a rate-limiting enzyme in ovarian steroidogenesis that regulates cholesterol transfer from outer to inner mitochondrial membrane. P450 SCC catalyzes the conversion of cholesterol to pregnenolone and 3β-HSD converts pregnenolone to progesterone. 17β-HSD catalyzes the conversion of weak estrogen estrone (E1) to more potent estradiol (E2)25. Therefore, the suppression of these enzymes by activin-A resulted in decreased E2 and P4 production of the cells. Midcycle LH surge activates a intriguingly complex network of signaling cascades that will not only cause the rupture of the follicle and the release of the oocyte, but also triggers the release of many mediators of LH-induced signaling pathways. The latter is responsible for initiation of luteinization process and the formation of corpus luteum26. Luteinization process in fact, is characterized by upregulated expression of LH receptor, VEGF, increased vascularization and enhanced P4 output along with activation of inflammatory and proangiogenic responses26. Downregulation of FSH receptor and degradation of cyclin D1 (required for mitotic exit) are other hallmarks of luteinization event23,26. In accordance with this, we demonstrated in this study that activin-A downregulated the expression of steroidogenic enzymes, VEGF, LH receptor, and upregulated FSH receptor and cyclin D1 in human luteal GCs while hCG as a well-known luteotropic hormone exerted the opposite effect. Taken together, these actions of activin-A are consistent with reversal of luteinization phenotype or functional luteolysis in human luteal GCs.
Limited data is available regarding the physiological actions of activin-A and its receptor expressed on human luteal granulosa cells. While existing data is more consistent regarding the effects of activin-A on the expression of StAR and 3β-HSD and P4 production18,19,20,21,22, there is a controversy in literature regarding the effect of activin-A on aromatase expression and estrogen production in human luteal granulosa cells21,22. Our results are consistent with the findings of Dr. Jaffe’s team in 1991 that activin-A suppresses both P4 and E2 secretion in cultured human luteal granulosa cells22. In contrast to the findings of Chang et al.21, we did not observe any meaningful change in the aromatase expression in luteal granulosa cells when activin-A was administered at the same concentrations that they used. As a novel finding, we have shown in this study that activin-A downregulates JNK pathway along with steroidogenic enzymes in human luteal GCs. We recently showed that hCG-induced improvement in luteal function is associated with activation of JNK pathway. When JNK pathway was inhibited pharmacologically or knocked-down with siRNA luteal function was compromised and P4 production declined along with the expression of StAR and 3β-HSD in the cells24. It is worthwhile to note that the observed changes in the steroidogenic function of luteal GCs after JNK inhibition either pharmacologically or via siRNA are similar to those we observed after activin-A treatment. Taken together these data suggest that JNK signaling pathway play a pivotal role in the regulation of luteal function in human GCs by hCG and activin-A. In other words, luteotropic hCG improves luteal function by activating JNK pathway whereas activin-A reverses it by downregulating JNK pathway in human luteal GCs in vitro. StAR as a gatekeeper enzyme in steroidogenesis was upregulated and downregulated along with 3β-HSD, when JNK is activated by hCG and inhibited by activin-A respectively, in luteal GCs, indicating that StAR is closely associated with JNK pathway. In line with this, it was previously shown in a mouse Leydig cell tumor model that c-Jun binds to activator protein-1 (AP-1) motif in the promoter region of StAR and increases both StAR transcription and P4 synthesis27.
JNK as a member of mitogen-activated protein kinases (MAP kinases) is activated by a wide range of stimuli such as mitogenic agents, genomic damage, inflammation, and UV irradiation28. AP-1 transcription factors (Jun and Fos family) are the major downstream targets of JNK signaling pathway28. Limited data is available on the biological role of this pathway in human ovary. We previously documented the critical roles of JNK pathway in the regulation of FSH induced follicle growth and cell cycle progression of granulosa cells in mouse model29,30, in the pathogenesis of granulosa cell tumor in human31, and very recently in the hCG-induced improvement of luteal function in human luteal GCs24. Albeit not human a study on rat ovary and granulosa cells demonstrated that the expression patterns of Jun and Fos family members in response to FSH and LH were distinct. JunB, c-Jun, c-Fos, and Fra2 were rapidly but transiently induced by FSH in immature granulosa cells. JunD and Fra2 were induced by LH and maintained as granulosa cells terminally differentiated into luteal cells32.
Activin-A like the other members of TGF-β family act through the serine/threonine kinase receptors and Smad signaling15. However, previous studies documented that activin-A can also act via the mitogen-activated protein (MAP) kinase signaling pathway, which is generally induced in inflammatory states33. To the best of our knowledge, our study is the first one to identify a link between activin-A and JNK signaling in human granulosa cells. Activin-A and its receptors are widespread through female reproductive system and has diverse biological functions in the ovary, uterus, and during pregnancy15. Although biological functions of activins on granulosa cells were extensively studied, limited data is available regarding the biological roles of activin isoforms on human granulosa cells. Apparently, the action of this hormone varies depending upon whether or not the granulosa cells are luteinized or not. In nonluteinized human granulosa cells activin-A suppresses basal and FSH induced estradiol production34 and induces follicle growth by a direct mitogenic effect on granulosa cells by binding to its cognate receptors, and by enhancing FSH responsiveness of the follicle by upregulating the expression of FSH receptor and aromatase enzyme in human granulosa tumor cell line KGN21,35,36,37. Activin-rich intraovarian milieu characteristic of early follicular phase is later replaced by inhibin dominant environment during the processes of FSH induced further growth of a selected cohort of antral follicles and dominant follicle selection36,38. However, luteinized granulosa cells but not theca cells continue to express activin receptors in corpus luteum39. While serum activin-A level increases progressively during transition from mid to late luteal phase in human it is bounded and neutralized by follistatin, raising questions about its relative bioavailability for action on peripheral target cells40. Previous studies showed that activin-A is also found in the follicular fluid at the concentrations varying from <10 ng/ml to >200 ng/ml in patients undergoing IVF although no conclusive data exist regarding the biological role of this form of activin-A in luteal function or oocyte maturation41,42,43,44. Unfortunately we were not able to measure activin-A level in the follicular fluid samples that comes with mural luteal granulosa cells during oocyte retrieval procedure. However, activin-A was still able to downregulate the expression of StAR and reduce P4 output of the cells when they were cultured in androgen-rich follicular fluid. This observation is particularly important because granulosa cells lack 17-α hydroxylase and 17/20 lyase enzyme systems, which constitute the biological basis of the two-cell and two-gonadotrophins theory in human ovary45,46,47,48. Therefore, they rely on theca cell-derived androgens for conversion into estrogens by aromatase enzyme even after ovulation process luteinization event. Even though the FBS, that used in this study to supplement the culture medium, was a single batch and contained detectable amounts of testosterone, it was unknown until this experiment if activin-A would have the same suppressive effect on steroidogenesis when culture media is supplemented with androgen-rich FF instead of fetal bovine serum. Taken together activin-A consistently downregulates the expression of steroidogenic enzymes and reduces P4 production of granulosa cells after luteinization regardless of the origin of luteal granulosa cells (natural vs. stimulated IVF cycles), stimulation protocol and culture environment. This is the strength of our study.
Luteinization process and luteolysis are complex event that are poorly understood in human. While the former is characterized by a marked tissue remodeling, marked changes in intracellular signaling, gene regulations, extracellular matrix adhesion proteins, angiogenesis, and cell cycle regulators3,4,5 the latter can be defined as loss of function, decrease in P4 synthesis and output, tissue break-down, degradation of extracellular matrix proteins, infiltration of macrophages, and subsequent involution of the luteal structures and vessels and increased cell death1,6. There are certain clinical situations, where rapid regression of corpus luteum and reduction in sex steroid output and production of vasoactive and angiogenesis promoting factors like VEGF are of paramount importance such as ovarian hyperstimulation syndrome (OHSS). This syndrome is characterized by excess number of growing follicles >20 as a result of ovarian hyper-response to gonadotropin stimulation, enhanced ovarian neo-angiogenesis, supraphysiological serum levels of estrogen, increased vascular permeability and a fluid shift from intravascular space to the third space and edema due to elevated levels of vasoactive substances secreted by the ovaries such as VEGF, interleukins, tumor necrosis factor-alpha, and endothelin-149. The current strategy to avoid this syndrome is to trigger ovulation with a GnRH agonist rather than hCG and freeze all embryos for transfer later because the latter has a longer half-life and promotes luteinization by upregulating the expression of the steroidogenic enzymes, VEGF, LH, and enhancing P4 output. hCG, which is either administrated exogenously for ovulation trigger; or produced endogenously by trophoblastic cells of the ensuing embryo if conception occurs deteriorates the clinical picture50. In this regard, activin-A as an hormone having opposing effects of hCG might at least help to reduce the severity of the syndrome if clinical studies confirm its beneficial effects in-vivo. In fact, the animal models of OHSS could be a good starting point before launching clinical studies.
Our study has a number of limitations. First, corpus luteum is a solid and dynamic organ composed of many different cell types and has as early, mid, and late phases. Mural luteal GC is only one of those cell types and represent early stage of the phase. Therefore, the biological actions of activin-A that we observed in these cells in vitro might not be true for the rest of the luteal phase. Second, luteal regression and luteolysis defined as the demise of corpus luteum is involves a complex interaction of immune cells with steroidogenic and stromal cells of the corpus luteum characterized by tissue break-down, degradation of extracellular matrix proteins, infiltration of macrophages, and subsequent involution of the luteal structures and vessels and increased cell death1,6. Thus, it is not a simple apoptotic death of the cells with subsequent reduction in steroidogenesis. Therefore, the observed in vitro actions of activin-A is not necessarily the same for real in vivo intraovarian environment.
Conclusion
Activin-A appears to reverse luteinization phenotype in human luteal granulosa cells and JNK silencing through activin-A seems to be involved in the intricate mechanisms controlling the steroidogenic activity of corpus luteum in human. A better understanding of luteal functions at the molecular level might have a direct clinical impact. Identification of the molecular mechanisms in the formation of corpus luteum, luteinization, and luteolysis might facilitate the efforts to develop strategies against luteal phase pathologies commonly observed in assisted reproductive technologies.
Materials and methods
This study was approved by the Institutional Review Board of Koc University (IRB# 2017.141.IRB2.069).
Patients
Luteal granulosa cells were obtained from follicular aspirates during oocyte retrieval procedure in a total of 50 patients undergoing assisted reproduction treatment. Demographic and IVF characteristics of the patients are provided in Table 2. In stimulated IVF cycles, 225–300 IU recombinant FSH (Gonal F, Merck-Serono, Darmstadt, Germany) was initiated at early follicular phase to achieve multifollicular growth. In 32 patients, a GnRH antagonist (cetrorelix acetate, Cetrotide, Merck-Serono, Darmstadt, Germany) was started to prevent premature LH surge and ovulation when the dominant follicle reached 10–12 mm in size. In 12 patients, pituitary downregulation was induced using a GnRH agonist (leuprolide acetate, Lucrin, Abbott, IL, USA) from 7 days prior to the anticipated day of menstrual bleeding to the day of final oocyte maturation. In six natural cycle IVF patients, no ovarian stimulation was given and spontaneous follicle growth was achieved with endogenously produced FSH. In all cases, ovulation was triggered with 250 μg recombinant HCG (Ovitrelle; Merck-Serono, Darmstadt, Germany) when a leading follicle of ≥19 mm and two or more trailing follicles of ≥17 mm was recorded. Oocyte pick-up was performed 36 h after ovulation trigger.
Isolation and culture of human luteal GCs
Luteal GCs were isolated during oocyte retrieval procedure from follicular aspirates and cultured according to the procedures we previously described16,24. The cells were pooled for the experiment for most of the time only when the patients were comparable for age, infertility etiology, and stimulation protocol. If not, they were used individually rather than pooling should the number of cells recovered deemed sufficient for the experiments. Pooling not only provided us sufficient number of cells required for the experiments, but also allowed homogenous distribution of the cells among the experiments. However, the experiments were repeated in the cells coming from a single patient when the number of cells were sufficient.
The number of cells harvested varied among the protocols. Since there is only one naturally grown ovulatory follicle in the natural cycles, the number of cells obtained from these cycles was low (100,000–200,000 cells) compared to the stimulated cycles in which millions of cells could be harvested from each patient. In brief, recovered cells were cultured in 6-well format culture plates at a density of 500,000 cells per well using DMEM-F12 culture medium supplemented with 10% fetal bovine serum. For natural cycle, approximately 25,000 cells were plated for each well. For culture with follicular fluid (FF), the follicular aspirates obtained during oocyte retrieval procedure were spun down at 1000×g for 5 min. The supernatant was filtered through 0.22 µm filter and added to the culture medium at the indicated concentration. The cells were cultured at 37 °C and 5% CO2. One day after plating, the cells were treated with the hormones or chemicals at the indicated concentrations for 24 h. In brief, activin-A was used at 1–5–25–100 ng/ml, FSH 50 mIU/ml, and hCG 5 IU/ml concentrations.
Chemicals and reagents
All cell culture materials were obtained from Gibco Inc. Recombinant Human Activin-A (GFH6) was purchased from Cell Guidance Systems. RepSox (#72392), a cell permeable, selective inhibitor of the TGF-β type 1 receptor (TGFβRI) ALK5 was obtained from Stemcell Technologies. Recombinant forms of FSH (Gonal-F) and hCG (Ovitrelle) was purchased from Merck Global (Darmstadt, Germany). SAPK/JNK Kinase Assay Kit (#8794, nonradioactive), Hoechst 33342 (#4082), anti c-Jun (#9165), anti-phospho-c-JunSer73 (#3270S), Anti-Phospho-SAPK/JNKThr183/Tyr185 (#9251), Smad2 (#3122) and Phospho-SMAD2Ser465/Ser467 (#18338) antibodies were obtained from Cell Signaling. Oil Red O was purchased from Sigma Inc. (USA). All western blotting buffers and reagents were purchased from Bio-Rad. Anti-Vinculin Antibody (V9264) was purchased from Sigma-Aldrich. Mouse antihuman monoclonal antibodies were purchased from Santa Cruz Biotechnology for the detection of human 3β-HSD Type II (sc-100466), 17β-HSD type-I (sc-376719), StAR (sc-166821), and P450 SCC (CYP11A1, sc-292456). Aromatase (CYP19A1, ab34193) monoclonal mouse antibody was from Abcam Inc. YO-PRO®-1 Iodide (491/509) was obtained from Life Technologies. Texas Red™-X Phalloidin was obtained from Thermo Fisher Scientific.
SAPK/JNK kinase assay
Endogenous JNK activity was measured by the nonradioactive assay kit instructions that described previously31. Briefly, the cells were harvested under nondenaturing conditions, lysed on ice and centrifuged. Then, 200 μl of cell/tissue lysates was incubated with 20 μl phospho-JNK rabbit monoclonal antibody linked to agarose beads to precipitate JNK enzyme. After addition of necessary buffers, c-Jun substrate and adenosine triphosphate, reaction mixture was incubated for 30 min at 30 °C, optimal reaction condition that allows c-Jun substrate to be phosphorylated by precipitated JNK. The reaction was stopped by adding 4× SDS sample buffer and the samples were loaded onto 10% polyacrylamide gel. Immunoblotting protocol was performed using monoclonal antibodies to measure JNK-induced phosphorylation of c-Jun substrate at Ser73.
Viability assay
Live/dead cell assay was performed with YO-PRO (1 mM), a green-fluorescent carbocyanine nucleic acid stain absorbed by only apoptotic cells, whereas live cells are impermeant to it. Hoechst 33342 (1 mg/ml) was used for counterstaining. Live/dead cell imaging of the cells were undertaken under appropriate channels using an IF microscope (Olympus IX71, Japan).
Gene expression analysis
RNA isolation was performed with Quick-RNA MicroPrep Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. RNA was quantified with spectrophotometric read at 260 nm by Nanodrop 2000 (Thermo Fisher Scientific, MA, USA) and 500 ng cDNA was prepared by using M-MLV Reverse Transcriptase (Invitrogen). Quantitative real-time expressions of mRNAs of genes of interest were detected and compared by using Light Cycler 480 SYBR Green I Master (Roche, Germany). All primers that were used in this study are listed in the Table 3 below. The mean Cts and their SDs for each target gene were calculated from three technical replicates within the same quantitative RT-PCR (qRT-PCR) assay. We have used ΔΔCt method for relative quantitation of target genes45,51. The cDNA of the ovary used for the positive control of activin receptor expression in qRT-PCR experiment (Supplementary Fig. 1B) was already in our library and had been obtained from a 23 year patient undergoing laparoscopic excision of benign ovarian cyst within which some cortical piece was embedded.
Hormone assays
E2, P4, testosterone, and hCG levels in culture media were determined by using electrochemiluminescence immunoassay “ECLIA” via the Roche Cobas-6000 analyzer (Roche, Mannheim, Germany). Lower detection limits of E2, P4, testosterone and hCG were 5.00 pg/ml (18.4 pmol/ml), 0.05 ng/ml (0.159 nmol/ml), 0.025 ng/ml (0.087 nmol/l) and 0.1 mIU/ml, respectively. The intra-assay CVs % of E2, P4, testosterone and hCG were, 2.4%, 2.3%, 2.9%, and 1.8%, respectively. The inter-assay CVs% of E2, P4, testosterone and hCG were 3.8%, 3.2%, 3.1%, and 4.6% respectively. Total cholesterol and LDL-C levels were measured by enzymatic, colorimetric methods, through the Roche Cobas-6000 analyzer (Roche, Mannheim, Germany). The intra-assay CVs% of total cholesterol and LDL-C and triglyceride were 1.1% and 0.7%, respectively. The interassay CVs% of total cholesterol and LDL-C were 1.6% and 2.3%, respectively.
Western blotting
Proteins were extracted from samples using radioimmunoprecipitation assay buffer (Sigma-Aldrich). Protein concentration was quantified by standard BCA Assay method. Equal amounts of proteins for every sample (20 µg) were separated on SDS-PAGE and transferred onto Immuno-Blot® PVDF membrane using BioRad semi-dry blotting system. After blocking of nonspecific proteins, the membranes were incubated with primary antibodies overnight at 4 °C at dilutions instructed by the manufacturers. Following washing steps, membranes were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature (RT). Anti-Vinculin was used the loading control. Following final washes and incubation with enhanced chemiluminescence (ECL), the signals on blots were visualized by the ChemiDoc MP Imaging System (Bio-Rad Laboratories, Inc).
Immunofluorescence staining
Oil Red O (Sigma, USA) as a fat-soluble dye is used for staining of natural triglycerides and lipid droplets of the cells. Oil Red O working solution (0.5%) was prepared by boiling of 0.5 g Oil Red O in 100 ml 100% isopropanol. Cells were washed with Dulbecco’s phosphate-buffered saline (DPBS) and fixed with 4% paraformaldehyde (PFA) for 20 min at RT. Following washing with DPBS, cells rinsed at 60% isopropanol and stained with Oil Red O (Sigma, USA) for 20 min at RT. Subsequently, cells were rinsed by 60% isopropanol and running tap wate,r respectively and prepared for immunofluorescence staining steps. Permeabilization was performed in 0.2% Triton X-100 containing DPBS for 20 min at RT. Blocking of nonspecific epitopes was performed by incubation in Super Block (ScyTek, USA) medium for 20 min at RT. Thereafter, the cells were incubated with primary antibodies for overnight at 4 °C. Cells were washed three times with DPBS-Tween (0.01%), then incubated with secondary antibodies for 1 h at 37 °C. Cells were washed three times, then covered with Fluoroshield mounting medium with DAPI (Abcam, UK) and images were taken using a confocal microscope (Leica, DMI8).
Statistical analysis
The samples size required for statistical significance and proper interpretation of the results was calculated according to the experimental methodologies used in the study as we previously described24. mRNA levels of the target genes used in the qRT-PCR assay (steroidogenic enzymes, LH and FSH receptor) and hormone levels are continuous variables therefore, expressed as the mean ± SD. ANOVA/ Bonferroni or Kruskal–Wallis/Dunn post-hoc tests were applied to compare the groups if data is parametric or nonparametric, respectively. The percentages of viable and apoptotic cells were compared between the groups using Fisher’ exact test. Significance level was set at 5% (P < 0.05). SPSS statistical program (version 22) was used to analyze the data.
References
Duncan, W. C. The human corpus luteum: remodelling during luteolysis and maternal recognition of pregnancy. Rev. Reprod. 5, 12–17 (2000).
Csapo, A. The luteo-placental shift, the guardian of pre-natal life. Postgrad. Med. J. 45, 57–64 (1969).
Russell, D. L. & Robker, R. L. Molecular mechanisms of ovulation: co-ordination through the cumulus complex. Hum. Reprod. Update 13, 289–312 (2007).
Honda, T. et al. Integrin alpha5 is expressed on human luteinizing granulosa cells during corpus luteum formation, and its expression is enhanced by human chorionic gonadotrophin in vitro. Mol. Hum. Reprod. 3, 979–984 (1997).
Robker, R. L. & Richards, J. S. Hormone-induced proliferation and differentiation of granulosa cells: a coordinated balance of the cell cycle regulators cyclin D2 and p27Kip1. Mol. Endocrinol. 12, 924–940 (1998).
Devoto, L. et al. The human corpus luteum: life cycle and function in natural cycles. Fertil. Steril. 92, 1067–1079 (2009).
Fatemi, H. M. The luteal phase after 3 decades of IVF: what do we know? Reprod. Biomed. Online 19, 4331 (2009).
van der Linden, M., Buckingham, K., Farquhar, C., Kremer, J. A. & Metwally, M. Luteal phase support for assisted reproduction cycles. Cochrane Database Syst. Rev. https://doi.org/10.1002/14651858.CD009154.pub2 (2011).
Pritts, E. A. & Atwood, A. K. Luteal phase support in infertility treatment: a meta-analysis of the randomized trials. Hum. Reprod. 17, 2287–2299 (2002).
Mascarenhas, M. & Balen, A. H. The high responder: a review of pathophysiology and outcomes during IVF treatment. Hum. Fertil. 20, 155–167 (2017).
Humaidan, P., Quartarolo, J. & Papanikolaou, E. G. Preventing ovarian hyperstimulation syndrome: guidance for the clinician. Fertil. Steril. 94, 389–400 (2010).
Humaidan, P. et al. GnRHa trigger and individualized luteal phase hCG support according to ovarian response to stimulation: two prospective randomized controlled multi-centre studies in IVF patients. Hum. Reprod. 28, 2511–2521 (2013).
Oktay, K., Turkcuoglu, I. & Rodriguez-Wallberg, K. A. GnRH agonist trigger for women with breast cancer undergoing fertility preservation by aromatase inhibitor/FSH stimulation. Reprod. Biomed. Online 20, 783–788 (2010).
Alexander, J. M., Jameson, J. L., Bikkal, H. A., Schwall, R. H. & Klibanski, A. The effects of activin on follicle-stimulating hormone secretion and biosynthesis in human glycoprotein hormone-producing pituitary adenomas. J. Clin. Endocrinol. Metab. 72, 1261–1267 (1991).
Wijayarathna, R. & de Kretser, D. M. Activins in reproductive biology and beyond. Hum. Reprod. Update 22, 342–357 (2016).
Bildik, G. et al. Luteal granulosa cells from natural cycles are more capable of maintaining their viability, steroidogenic activity and LH receptor expression than those of stimulated IVF cycles. Hum. Reprod. 34, 345–355 (2019).
Bildik, G. et al. hCG improves luteal function and promotes progesterone output through the activation of JNK pathway in the luteal granulosa cells of the stimulated IVF cyclesdagger. Biol. Reprod. 102, 1270–1280 (2020).
Myers, M., Gay, E., McNeilly, A. S., Fraser, H. M. & Duncan, W. C. In vitro evidence suggests activin-A may promote tissue remodeling associated with human luteolysis. Endocrinology 148, 3730–3739 (2007).
Di Simone, N. et al. Effect of activin-A on progesterone synthesis in human luteal cells. Fertil. Steril. 62, 1157–1161 (1994).
Myers, M., van den Driesche, S., McNeilly, A. S. & Duncan, W. C. Activin A reduces luteinisation of human luteinised granulosa cells and has opposing effects to human chorionic gonadotropin in vitro. J. Endocrinol. 199, 201–212 (2008).
Chang, H. M., Cheng, J. C., Klausen, C., Taylor, E. L. & Leung, P. C. Effects of recombinant activins on steroidogenesis in human granulosa-lutein cells. J. Clin. Endocrinol. Metab. 99, E1922–E1932 (2014).
Cataldo, N. A., Rabinovici, J., Fujimoto, V. Y. & Jaffe, R. B. Follistatin antagonizes the effects of activin-A on steroidogenesis in human luteinizing granulosa cells. J. Clin. Endocrinol. Metab. 79, 272–277 (1994).
Hampl, A., Pachernik, J. & Dvorak, P. Levels and interactions of p27, cyclin D3, and CDK4 during the formation and maintenance of the corpus luteum in mice. Biol. Reprod. 62, 1393–1401 (2000).
Bildik, G. et al. hCG improves luteal function and promotes progesterone output through the activation of JNK pathway in the luteal granulosa cells of the stimulated IVF cycles. Biol. Reprod. https://doi.org/10.1093/biolre/ioaa034 (2020).
Miller, W. L. & Auchus, R. J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 (2011).
Duffy, D. M., Ko, C., Jo, M., Brannstrom, M. & Curry, T. E. Ovulation: parallels with inflammatory processes. Endocr. Rev. 40, 369–416 (2019).
Manna, P. R. & Stocco, D. M. The role of JUN in the regulation of PRKCC-mediated STAR expression and steroidogenesis in mouse Leydig cells. J. Mol. Endocrinol. 41, 329–341 (2008).
Weston, C. R. & Davis, R. J. The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 (2007).
Oktem, O., Buyuk, E. & Oktay, K. Preantral follicle growth is regulated by c-Jun-N-terminal kinase (JNK) pathway. Reprod. Sci. 18, 269–276 (2011).
Oktay, K., Buyuk, E., Oktem, O., Oktay, M. & Giancotti, F. G. The c-Jun N-terminal kinase JNK functions upstream of Aurora B to promote entry into mitosis. Cell Cycle 7, 533–541 (2008).
Bildik, G. et al. Endogenous c-Jun N-terminal kinase (JNK) activity marks the boundary between normal and malignant granulosa cells. Cell Death Dis. 9, 421 (2018).
Sharma, S. C. & Richards, J. S. Regulation of AP1 (Jun/Fos) factor expression and activation in ovarian granulosa cells. Relation of JunD and Fra2 to terminal differentiation. J. Biol. Chem. 275, 33718–33728 (2000).
Hedger, M. P. & de Kretser, D. M. The activins and their binding protein, follistatin-Diagnostic and therapeutic targets in inflammatory disease and fibrosis. Cytokine Growth Factor Rev. 24, 285–295 (2013).
Alak, B. M. et al. Activin A stimulates meiotic maturation of human oocytes and modulates granulosa cell steroidogenesis in vitro. Fertil. Steril. 70, 1126–1130 (1998).
Nakamura, M. et al. Effect of an activin A on follicle-stimulating hormone (FSH) receptor messenger ribonucleic acid levels and FSH receptor expressions in cultured rat granulosa cells. Endocrinology 133, 538–544 (1993).
Oktem, O. & Urman, B. Understanding follicle growth in vivo. Hum. Reprod. 25, 2944–2954 (2010).
Nomura, M. et al. Activin stimulates CYP19A gene expression in human ovarian granulosa cell-like KGN cells via the Smad2 signaling pathway. Biochem. Biophys. Res. Commun. 436, 443–448 (2013).
Knight, P. G., Satchell, L. & Glister, C. Intraovarian roles of activins and inhibins. Mol. Cell Endocrinol. 359, 53–65 (2012).
Roberts, V. J., Barth, S., el-Roeiy, A. & Yen, S. S. Expression of inhibin/activin subunits and follistatin messenger ribonucleic acids and proteins in ovarian follicles and the corpus luteum during the human menstrual cycle. J. Clin. Endocrinol. Metab. 77, 1402–1410 (1993).
Muttukrishna, S., Fowler, P. A., George, L., Groome, N. P. & Knight, P. G. Changes in peripheral serum levels of total activin A during the human menstrual cycle and pregnancy. J. Clin. Endocrinol. Metab. 81, 3328–3334 (1996).
Lau, C. P., Ledger, W. L., Groome, N. P., Barlow, D. H. & Muttukrishna, S. Dimeric inhibins and activin A in human follicular fluid and oocyte-cumulus culture medium. Hum. Reprod. 14, 2525–2530 (1999).
Cupisti, S. et al. Correlations between anti-mullerian hormone, inhibin B, and activin A in follicular fluid in IVF/ICSI patients for assessing the maturation and developmental potential of oocytes. Eur. J. Med. Res. 12, 604–608 (2007).
Fujiwara, T. et al. Analysis of follicular fluid hormone concentrations and granulosa cell mRNA levels for the inhibin-activin-follistatin system: relation to oocyte and embryo characteristics. Fertil. Steril. 74, 348–355 (2000).
Wen, X. et al. Follicular fluid levels of inhibin A, inhibin B, and activin A levels reflect changes in follicle size but are not independent markers of the oocyte’s ability to fertilize. Fertil. Steril. 85, 1723–1729 (2006).
Oktem, O. et al. FSH stimulation promotes progesterone synthesis and output from human granulosa cells without luteinization. Hum. Reprod. 32, 643–652 (2017).
Patel, S. S., Beshay, V. E., Escobar, J. C., Suzuki, T. & Carr, B. R. Molecular mechanism for repression of 17alpha-hydroxylase expression and androstenedione production in granulosa cells. J. Clin. Endocrinol. Metab. 94, 5163–5168 (2009).
Hillier, S. G., Whitelaw, P. F. & Smyth, C. D. Follicular oestrogen synthesis: the ‘two-cell, two-gonadotrophin’ model revisited. Mol. Cell Endocrinol. 100, 51–54 (1994).
Ryan, K. J. & Petro, Z. Steroid biosynthesis by human ovarian granulosa and thecal cells. J. Clin. Endocrinol. Metab. 26, 46–52 (1966).
Gomez, R. et al. Physiology and pathology of ovarian hyperstimulation syndrome. Semin. Reprod. Med. 28, 448–457 (2010).
Kumar, P., Sait, S. F., Sharma, A. & Kumar, M. Ovarian hyperstimulation syndrome. J. Hum. Reprod. Sci. 4, 70–75 (2011).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Acknowledgements
The authors also gratefully acknowledge the use of services and facilities of the Koç University Research Center for Translational Medicine (KUTTAM). Findings, opinions, or points of view expressed on this article do not necessarily represent the official position or policies of the Ministry of Development. Funded by the School of Medicine, the Graduate School of Health Sciences of Koc University and Koç University Research Center for Translational Medicine (KUTTAM), equally funded by the Republic of Turkey Ministry of Development Research Infrastructure Support Program.
Funding
Funded by the School of Medicine, the Graduate School of Health Sciences of Koc University and Koç University Research Center for Translational Medicine (KUTTAM), equally funded by the Republic of Turkey Ministry of Development Research Infrastructure Support Program.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Alessandro Rufini
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bildik, G., Akin, N., Esmaeilian, Y. et al. Terminal differentiation of human granulosa cells as luteinization is reversed by activin-A through silencing of Jnk pathway. Cell Death Discov. 6, 93 (2020). https://doi.org/10.1038/s41420-020-00324-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-020-00324-9
- Springer Nature Limited
This article is cited by
-
Inhibin subunit beta B (INHBB): an emerging role in tumor progression
Journal of Physiology and Biochemistry (2024)
-
Autophagy regulates sex steroid hormone synthesis through lysosomal degradation of lipid droplets in human ovary and testis
Cell Death & Disease (2023)