
Abstract
Using genetic information to develop and implement conservation programs is vital for maintaining biodiversity and ecosystem resilience. Evaluation of the genetic variability within and among remnant populations can inform management of both natural and translocated populations to maximise species’ adaptive potential, mitigate negative impacts of inbreeding, and subsequently minimise risk of extinction. Here we use reduced representation sequencing to undertake a genetic assessment of the golden bandicoot (Isoodon auratus), a threatened marsupial endemic to Australia. The currently recognised taxon consists of three subspecies distributed among multiple natural and translocated populations. After confirming the genetic distinctiveness of I. auratus from two closely related taxa, I. fusciventer and I. macrourus, we identified four genetic clusters within I. auratus. These clusters exhibited substantial genetic differentiation (pairwise FST values ranging from 0.18 to 0.65, pairwise DXY ranging from 0.1 to 0.168), reflecting long-term isolation of some populations on offshore islands and the influence of genetic drift. Mainland natural populations in the Kimberley region had the highest genetic diversity and the largest contribution to overall allelic and gene diversity compared to both natural and translocated island populations. A population translocated to Guluwuru Island in the Northern Territory had the lowest genetic diversity. Our data suggest that island populations can appear genetically unique due to genetic drift and this needs to be taken into account when considering genetic diversity in conservation efforts to maintain overall genetic diversity of the species. We effectively demonstrate how genomic information can guide practical conservation planning, especially when declining species are represented by multiple isolated populations.
Similar content being viewed by others

Introduction
Genetic diversity is a fundamental element of biodiversity and helps drive ecosystem resilience, stability, and services (Raffard et al. 2019; Reynolds et al. 2012). Low genetic diversity can increase the risk of extinction, and management actions to maintain or increase genetic variation are increasingly advocated for in the conservation of threatened and keystone species (Hoban et al. 2021; Hoffmann et al. 2015; Weeks et al. 2011). Despite a previous emphasis on demographic concerns in threatened species management, the integration of genetic considerations into species conservation has grown substantially (Haig et al. 2016; Hoban et al. 2021; Kershaw et al. 2022; McDonald et al. 2015; Ottewell and Byrne 2022). Leveraging genetic information is pivotal for addressing the loss of genetic variation and subsequent adaptive potential, mitigating inbreeding effects, and predicting species’ resilience to demographic, environmental and/or genetic stochasticity (Frankham et al. 2017; Hoban et al. 2021; Ralls et al. 2018). Knowledge of genetic variability of remnant populations provides information as a baseline for ongoing management (e.g. von Takach et al. 2023), identifying populations requiring supplementation (Pacioni et al. 2020; Undin et al. 2021; Weeks et al. 2017; White et al. 2020), selecting optimal sources for translocations (Robinson et al. 2021; Weeks et al. 2011), and evaluating management outcomes (Ottewell et al. 2014; Rick et al. 2019; Thavornkanlapachai et al. 2019). International guidelines now advocate protecting at least 90% of genetic diversity within species (Hoban et al. 2020, 2021), but it is not always clear how best this can be achieved in relation to threatened species management.
Remnant populations, particularly those found on islands, often exhibit high levels of genetic and phenotypic divergence (Robertson et al. 2014). Evidence of strong population structure, which can arise rapidly in small and fragmented populations, can lead conservation managers to treat these as independent units, which can unintentionally reinforce the effects of isolation and subsequent extinction risk (Weeks et al. 2016). While population-specific management may be useful for populations that exhibit adaptive differences, such as those termed Evolutionarily Significant Units (ESUs) (Casacci et al. 2014; Coates et al. 2018; Moritz 1994), it is important to consider the degree to which observed population divergence is adaptive versus the result of stochastic processes (e.g. founder effects, genetic drift, etc.). Further, infraspecific units (e.g. subspecies) are often delimited based on morphology and do not always reflect underlying population genetic structure, which may overinflate the evolutionary significance of the infraspecific unit relative to other populations and impede species-level conservation actions (Robertson et al. 2014; Wolf and Ellegren 2017). This recognition is leading to a paradigm shift, with improved understanding that the loss of genetic diversity, the accumulation of genetic load and inbreeding are more proximal threats to species persistence than the risks of mixing genetically distinct populations (Frankham et al. 2017; Hoffmann et al. 2021; Ralls et al. 2018; Weeks et al. 2016). Consequently, there is a growing appreciation among conservation managers to prioritise management of these threats rather than being prescriptive in maintaining separate management of fragmented populations.
Australia faces a severe mammal extinction crisis (Woinarski et al. 2015, 2019), largely as a result of the introduction of feral cats (Felix catus) and red foxes (Vulpes vulpes) (Woinarski et al. 2015). The conservation of surviving mammals often relies heavily on insurance populations on islands or in predator-free fenced reserve “havens” (Legge et al. 2018). The latter approach has proven to be highly successful in establishing populations of Australian mammal species extirpated from their historic mainland ranges (Legge et al. 2018; Woinarski et al. 2023). Despite the popularity of utilising islands as sources for translocations this can also lead to many conservation challenges. The physical isolation of islands and fenced areas can subject these populations to founder effects, genetic bottlenecks, small population sizes, and limited dispersal, leading to the potential erosion of genetic diversity and demographic stochasticity. Thus, it is necessary to prioritise genetic management of remnant populations and to include genetic information in strategic decision-making for species’ recovery.
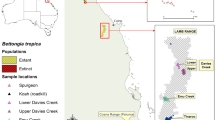
Australia recently released a national ten-year (2022–2032) plan for threatened species conservation and recovery, the “Threatened Species Action Plan: Towards Zero Extinctions”. The plan indicates that evidence-based conservation actions will be needed to achieve recovery targets. Genetic management will be integral to strengthening species’ resilience and adaptive capacity, as well as to ensure genetic diversity is maintained in insurance populations, particularly those in havens. In light of this ambition, here we conducted a genetic assessment of the threatened golden bandicoot Isoodon auratus (Ramsay 1887), a small (300–850 g) omnivorous marsupial. The remnant distribution of this species is heavily fragmented due to recent range collapse to near extinction on the mainland, attributed primarily to feral cat and fox predation and exacerbated by changed fire regimes and habitat loss (Woinarski et al. 2014, 2015). The naturally remnant populations of I. auratus are found on mainland Australia in the Kimberley and on offshore islands, including Barrow Island and Middle Island in the Pilbara region, Lachlan Island, Augustus Island, Storr Island and Uwins Island in the Kimberley, and Marchinbar Island in the Northern Territory (Woinarski et al. 2014) (Fig. 1a). Multiple single-source translocations of this species have occurred from Barrow Island to a fenced reserve (Matuwa Kurrara Kurrara National Park) (Lohr et al. 2021) and to Hermite Island and Doole Island in the Pilbara (Dunlop et al. 2021) (Fig. 1b). A captive population at Alice Springs Desert Park, Northern Territory, and a recently reintroduced population to Wild Deserts, New South Wales have been secondarily sourced from the Matuwa population. Populations have also been established on Guluwuru Island and Raragala Island using animals from Marchinbar Island. At least one more site is proposed for translocation: Newhaven in the Northern Territory (Fig. 1a).

(a) Distribution of I. auratus (golden bandicoot), I. macrourus (northern brown bandicoot) and I. fusciventer (quenda) across Australia with the number of samples used for each population represented by the size of the circle; and (b) translocation history of I. auratus (see maps (a) for an explanation of acronyms). Image of I. auratus sourced from Creazilla under an Attribution 4.0 International (CC BY 4.0) license. Image can be found at https://creazilla.com/nodes/64034-golden-bandicoot-clipart.
The taxonomy of the species at both the specific and subspecific level has been in flux for several decades (summarised in Cooper et al. 2018; Jackson and Groves 2015; Thavornkanlapachai et al. 2021; Warburton and Travouillon 2016). For this study, we adhere to the delineation of three subspecies: I. a. auratus from the Kimberley region, I. a. arnhemensis from the Northern Territory and I. a. barrowensis from Barrow Island, Pilbara region (Jackson and Groves 2015). However, we note that I. a. arnhemensis (Lyne and Mort 1981) is variously recognised as a separate subspecies or merged with I. a. auratus in different treatments (Westerman et al. 2012), with only two subspecies, I. a. barrowensis (Thomas 1901) and I. a. auratus (merged with I. a. arnhemensis) listed independently as Vulnerable under the Environment Protection and Biodiversity Conservation (EPBC) Act 1999. Furthermore, recent molecular analysis suggests a pattern of intermediate polyphyly of I. auratus and I. fusciventer (Gray 1841) in mitochondrial DNA (Cooper et al. 2018; Thavornkanlapachai et al. 2021).
For I. auratus, decisions around population prioritisation and whether mixing within the three subspecies should be attempted are critical issues faced by conservation managers and organisations wishing to support the species’ recovery via reintroductions. Consequently, we aimed to (1) estimate the degree of genetic differentiation between each I. auratus sampling locality (hereafter referred to as population) and interpret it in an evolutionary context by comparing to other Isoodon taxa; (2) quantify the existing genetic variation within each population; (3) determine whether genetic diversity has been conserved in translocated populations, and (4) prioritise populations for conservation based on genetic contributions to within-species diversity. We expected to find strong structure between subspecies- and species-level taxa. Within species, we expected island populations to show low genetic diversity, particularly when established via translocations, and to be strongly differentiated from higher diversity mainland populations. Given the close relationship of I. fusciventer and I. auratus, we include samples from this species as well as I. macrourus (Gould 1842) (outgroup) to provide evolutionary context to patterns detected within I. auratus and to assist in establishing the relative scale of genetic differentiation between I. auratus subspecies, especially as strong structure is expected due to island isolation.
Methods
Sample collection, DNA sequencing, read assembly and filtering
Tissue samples were collected from 245 individuals and included 222 I. auratus, 9 I. fusiventer, and 14 I. macrourus samples (Fig. 1a, Table S1, and Supplementary Text).
DNA extraction procedures are detailed in the Supplementary Text. Library and ddRAD sequencing were performed at the Australian Genome Research Facility (AGRF) in Melbourne, Victoria. Illumina libraries were built on 280–375-bp DNA fragments and sequenced on the Illumina NextSeq 600 system (Supplementary Text).
Sequenced reads were cleaned, demultiplexed and trimmed to 125-bp with a phred quality score ≥30 using process_radtags module in Stacks v2.59 (Catchen et al. 2013). Samples with less than 400,000 reads (n = 28) were discarded. Parameters for the Stacks de novo pipeline were chosen on a subset of 100 random samples following the r80 optimisation approach of Paris et al. (2017), detailed in Supplementary Text. Two datasets were obtained through the de novo pipeline; the first including natural populations of I. auratus, I. fusciventer and I. macrourus (hereafter, the ‘Isoodon’ dataset, n = 134) and the second including all natural and translocated populations of I. auratus (hereafter, ‘auratus’ dataset, n = 222). Each dataset was run through the Populations module in Stacks with the following parameters: loci needed to be present in a single population, 50% of samples were required to process a locus, a maximum observed heterozygosity of 70%, and pruning to only a single SNP per locus to account for short distance linkage disequilibrium. The resulting VCF was filtered in R v4.0.2 using a modified script from Wright et al. (2019) and von Takach et al. (2020) with average allelic depth >2.5×, only retaining loci with a coverage difference between the reference and SNP allele <80%, iteratively filtering samples and SNPs with increasing thresholds reaching a final call rate of 90% in individuals and 95% in SNPs, minor allele count > = 3, and removing any loci with <100% reproducibility between technical replicates (see Fig. S1 for summary). Closely related individuals were estimated using the beta.dosage function in the hierfstat R package with pairwise kinship values > 0.25 considered closely related and removed (n = 7). Additional details on read assembly and data filtering methods are described in the Supplementary Text. Note that datasets were filtered in different ways to meet assumptions of analysis methods, as detailed in Fig. S1.
Investigating population structure
Due to the apparent complex evolutionary histories of Isoodon species and the uncertainty in the taxonomic status (Cooper et al. 2018; Thavornkanlapachai et al. 2021), we investigated population structure using both the ‘Isoodon’ and ‘auratus’ datasets. We inferred population structure without prior knowledge of demographic history via principal coordinate analyses (PCoA) and Discriminant Analysis of Principle Components (DAPC). We used the gl.pcoa function in the dartR package (Gruber et al. 2018) to run the PCoA and the find.clusters function in the adegenet R package to run the DAPC (Jombart 2008). For DAPC, the number of components (PCs) was initially set to allow 90% of cumulative variance to be retained (50 PCs for ‘Isoodon’ dataset and 100 PCs for ‘auratus’ dataset) and then optimised using cross validation to choose the optimal number of PCs to retain (10 PCs and 30 PCs for the ‘Isoodon’ and ‘auratus’ datasets respectively). Successive DAPC analyses were run from K values 2–10 and the K value selected based on the lowest Bayesian information criterion (BIC) and after the largest decrease in BIC (Jombart et al. 2010). For the ‘auratus’ dataset, we also present analyses when K equalled the number of sampling locations (K = 15).
We estimated the proportion of an individual’s genome belonging to ancestral K gene pools using the sNMF function in the R package LEA (Frichot et al. 2014; Frichot and François 2015). We ranged the number of clusters from 1–10 with 1000 iterations and 100 repetitions, assessing the most likely K based on cross-validation and the entropy criterion (an informatic theoretic measure which reflects the number of ancestral populations that best explains the genotypic data). Since unequal population sizes can influence the estimation of ancestry proportions, we also re-ran the model subsampling each population to have ≤10 individuals (ranging from 1–10).
We further explored the evolutionary relationships among populations of the ‘Isoodon’ dataset as a maximum likelihood bifurcating tree using TreeMix version 1.13 (Pickrell and Pritchard 2012). This approach draws inferences from covariance in allele frequencies among populations by reconstructing population histories and whether populations represent independent divergence events. We first ran TreeMix 10 times for varying number of migration events (m) ranging from 0–10 (-global -k 500 -noss) using I. macrourus as an outgroup to root the tree. The optimal number of migration events was selected using the OptM R package (Fitak 2021) by estimating change points from threshold models (Sonderegger et al. 2009) and the variance explained by each migration event where the threshold of 99.8% of the variance explained is recommended (Pickrell and Pritchard 2012). We then ran TreeMix 50 times for each chosen migration event (m = 0 as null model, m = 1 and m = 6) and obtained a consensus tree and bootstrap values using the BITE R package (Milanesi et al. 2017). The residual covariance matrix was also estimated for each chosen migration event and the consensus tree using TreeMix.
We quantified the degree of genetic differentiation (FST) and genetic divergence (Dxy) between populations using the software pixy (Korunes and Samuk 2021). The ‘Isoodon’ and ‘auratus’ datasets were reparsed through the Populations module of Stacks (-p = 1, -r = 0.5) to produce a VCF consisting of both variant and invariant sites (--vcf-all) which was further filtered in VCFtools version 0.1.16 (Danecek et al. 2011) allowing 10% missing data (--max-missing = 0.9) and mean depth of loci between five and 100 (--min-meanDP = 5, --max-meanDP = 100). Pairwise summary statistics were computed in 10 kb windows. Populations with small sample size (n < 6) were excluded.
Using the ‘auratus’ dataset, including translocated populations, we tested for an isolation by distance model of differentiation by plotting geographic distance against FST/(1-FST) (Rousset 1997) and performed a Mantel test using the function gl.ibd in the dartR package.
Assessing genetic variation of Isoodon auratus
To assess genetic diversity within populations, we first calculated the mean values of standard diversity parameters for each population with a sample size >6 using the ‘auratus’ dataset. Allelic richness (AR) was estimated using the allelic.richness functions in the hierfstat R package and the number of alleles which only occur in a single population (PA) using the private_alleles function in the poppr R package (Kamvar et al. 2014). Both metrics were standardised for unequal sample size. We followed the recommendations in Schmidt et al. (2021) when estimating heterozygosity to reduce biases associated with filtering, sample sizes and differences in allele frequencies among populations. This was achieved by re-calling variants in each population independently using individuals with less than 10% missing data (i.e. 90% genotyped) and 100% of individuals across all populations required to process a locus (-R = 100), thereby allowing no missing data across loci. Observed heterozygosity (HO), expected heterozygosity (HE) and nucleotide diversity (π) values were then extracted from Populations in Stacks summary statistics output. These independent datasets were also used to identify temporal changes in our translocated populations by calculating diversity metrics as described above across time periods. Due to only a single sample available in 2016 from Raragala Island, this population was excluded from this analysis. Autosomal heterozygosity estimates were calculated for the Guluwuru Island population as this method is more robust to small sample size (Schmidt et al. 2021), however, AR was not calculated as sample sizes were <6.
Prioritising populations of Isoodon auratus for genetic conservation
Using the ‘auratus’ dataset we investigated how genetic diversity was partitioned among the currently recognised subspecies as well as within and between populations using the QDiver function (Smouse et al. 2017) in GenAlEx v6.51b2 (Peakall and Smouse 2006, 2012). This method computes standardised genetic diversity metrics partitioned into hierarchical strata, and allows for evaluation of homo-/heterogeneity of within-stratum diversity components using a ‘Q’ metric derived from Rao’s Quadratic Entropy. The QDiver function creates a ‘diversity cascade’ that includes the total diversity of the species (γ), the diversity among subspecies (δ), the within-subspecies diversity (σ), the among-population diversity within each subspecies (β), and the within-population diversity within each subspecies (α). As this function does not allow for missing data, a dataset with a final call rate of 100% in SNPs was generated using the custom R script described above.
Secondly, we used the program Metapop2 (López-Cortegano et al. 2019) to assess the relative contribution of specific populations to both gene and allelic diversity across all populations sampled. This approach provides a statistically robust way to identify which populations contribute most to the overall genetic diversity of the species based on both local variation and genetic differentiation. The contribution of each population was estimated by removing that population and re-calculating the changes in within-population diversity (AS, HS), among-population diversity (DA, DG) and total allelic (AT) and gene (HT) diversity (López-Cortegano et al. 2019). In this instance, total allelic diversity is not the total number of alleles but is calculated as the average number of different alleles available in each pairwise grouping of populations (López-Cortegano et al. 2019). A positive value for a population means that on average, more genetic diversity is lost in the metapopulation when that population is removed while a negative value for a population indicates a gain in genetic diversity in the metapopulation when that population is removed. Using the Metapop2 program, we also simulated the optimum contribution of each population to a synthetic pool of 1000 individuals (e.g. a hypothetical translocation) to maximise heterozygosity (H) or the number of alleles (k). To account for unequal sample size between populations, we randomly subsampled each population (n = 6) and repeated the analysis 50 times, taking the average result across all runs.
Thirdly, we used the systematic planning approach of MARXAN (Watts et al. 2009) to identify combinations of extant populations that would best represent genetic diversity in the species (von Takach et al. 2021). In the absence of specific costed conservation options for each population, we allocated an equal unit cost of one to conserve each population and identified the optimal combination of populations to maximise allelic richness in the species, identifying optimal solutions for scenarios of 1–11 ‘protected’ populations using the R package ‘prioritizr’ (Hanson et al. 2022) and the SYMPHONY integer linear programming solver (Ralphs and Güzelsoy 2005). Using this method, each allele is considered a feature to be conserved, and each population is considered a planning unit. For each of 100 iterations, we randomly sampled six individuals per population, calculated the total number of alleles across all combined populations and identified a conservation solution for a maximum coverage (of alleles) objective for scenarios of 1–11. We tallied the number of configurations across the 100 replicates, as well as the resulting total allele count for each solution.
Results
Bioinformatic pipeline and filtering
Using the systematic evaluation of Stack parameters, both the number of polymorphic loci and number of SNPs increased with increasing M and n values; however, this increase reached an asymptote at M = n = 4 (Fig. S2) and therefore we chose this value for our de novo assembly. We ensured that coverage thresholds were on average greater than 25x to increase the robustness of our dataset to variation in sequence quality. Iterating m values resulted in mean coverage thresholds that ranged from 40.3× to 51.5× and therefore we deemed m = 3 to be suitable for our de novo assembly (Fig. S3).
Running the 134 Isoodon samples through the de novo pipeline (m = 3, M = 4, n = 4), our ‘Isoodon’ dataset retained 124,079 SNPs, with a mean read depth of 32.491 and 17.444 and a median read depth of 14.182 and 8.091 for the reference and alternative allele, respectively. After filtering using the custom R script, 117 unique individuals were retained with 8552 SNPs. When all missing data were removed for the TreeMix analysis, 117 individuals and 3043 SNPs were retained. Running 203 I. auratus samples that passed quality control (19 samples failed) through the de novo pipeline, 121,863 SNPs were retained, with a mean read depth of 26.088 and 11.436 and a median read depth of 11.282 and 5.222 for the reference and alternate allele, respectively. After filtering using the custom R script, 176 unique individuals and 8244 SNPs were retained, with an overall level of missing data for the filtered SNP by sample matrix of 1.3%. When all missing data were removed for the QDiver analysis, 172 unique individuals and 2495 SNPs were retained.
Hierarchical population structure
Strong structure was observed between currently recognised subspecies and species nomenclature for both the ‘Isoodon’ and ‘auratus’ dataset in ‘sNMF’ analysis (Figs. 2a, b; S5), DAPC analysis (Fig. S6) and PCoA (Fig. S7). sNMF and DAPC analyses both resolved K = 6 and K = 4 for the ‘Isoodon’ and ‘auratus’ datasets respectively (Figs. S8, S9), although additional K values also revealed meaningful hierarchical population structuring (Fig. 2). There was some complexity in the way genetic relationships were visualised across analyses as would be expected with hierarchically structured populations. Isoodon macrourus was clearly distinct from I. auratus and I. fusciventer in sNMF and PCoA, and I. fusciventer showed a closer genetic relationship with I. auratus, as expected from their established taxonomic relationships. The relative placement of these species differed in DAPC analysis with the primary axis discriminating I. auratus and I. fusciventer (Fig. S6a) and minimising variation with I. auratus and I. macrourus. Island populations and Kimberley mainland populations (Artesian Range, Mitchell Plateau and Yampi Sound) from the ‘auratus’ dataset were consistently separated in all analyses, with the first two PC axes explaining 49% for the PCoA (Fig. S7) and 86% variation for the DAPC (Fig. S6b). Genetic differentiation of Marchinbar Island from remaining I. auratus was detected in sNMF and PCoA, yet minimised in DAPC. Augustus Island was consistently separated from the other populations, forming its own genetic cluster. Kimberley mainland populations clustered together and translocated populations consistently grouped with their source populations in the ‘sNMF’, PCoA and DAPC analyses (Figs. 2; S6c; S7).

Estimates of admixture coefficients for an individual in each population using the ‘sNMF’ function in the R package LEA at different K values when using (a) the ‘Isoodon’ dataset and (b) the ‘auratus’ dataset, where each population was subset to contain a maximum of 10 individuals. Populations include Barrow Island (BWI), Doole Island (DOOL), Hermite Island (HERM), Matuwa (MATU), Alice Springs Desert Park (ASDP), Augustus Island (AUG), Lachlan Island (LACH), Storr Island (STOR), Uwins Island (UWIN), Yampi Sound (YAMP), Artesian Range (ART), Mitchell Plateau (MITC), Marchinbar Island (MARC), Guluwuru Island (GULU), Raragala Island (RARA), I. macrourus (IM) and I. fusciventer (IF). Translocated populations are indicated by an asterisk (*).
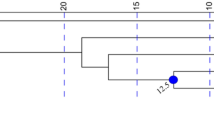
Topology inferred from the TreeMix maximum likelihood tree with no migration events (Fig. 3a) was concordant with population clustering (Fig. 2). Six migration events were chosen to be the optimal value, explaining 99.77% of variation with a possible suboptimal number of a single migration event (Fig. S10). However, one migration event only explained 99.14% of variation (Fig. S10b). Tree topology did not change significantly with the addition of subsequent migration events (Figs. 3b, S11) and migration events had low weights (likely reflecting historic gene flow). Branch lengths supported strong drift in allele frequencies in the island populations, in contrast to the shorter branch lengths in the Kimberley mainland populations (Figs. 3, S11). Residuals from all models (m = 0, m = 1, m = 6; Figs. 3, S11b) revealed populations which may not fit a strict tree model where strongly positive residuals among some pairs indicated that these may be more closely related than they appear in the consensus tree and are potential candidates for admixture events.

(a) no migration edges (m = 0) and (b) six migration edges (m = 6), inferred as the best topology. Branch lengths on the horizontal axis represent the amount of genetic drift that has occurred along each branch. Bootstrap supports for each node are indicated. Below each tree is the associated residual fit of the observed versus the predicted squared allele frequency difference, expressed as the number of SE of the deviation. SE values are represented by colours according to the palette on the right. Residuals above zero indicate populations that are more closely related to each other in the data than in the best-fit tree and have potentially undergone admixture. Negative residuals represent populations that are less closely related in the data than represented in the best-fit tree. Populations include Barrow Island (BWI), Augustus Island (AUG), Lachlan Island (LACH), Storr Island (STOR), Uwins Island (UWIN), Yampi Sound (YAMP), Artesian Range (ART), Mitchell Plateau (MITC), Marchinbar Island (MARC), I. macrourus (IM) and I. fusciventer (IF).
Genetic differentiation (pairwise FST) and nucleotide divergence (Dxy) were variable between different named taxa as well as allopatric populations within taxa, as expected (Fig. S12). Translocated populations were not differentiated from their source population or other populations established from the same source (FST < 0.03, Dxy < 0.017), consistent with their clustering in the ‘sNMF’ analysis (Fig. 2b). For both the ‘Isoodon’ and ‘auratus’ datasets, pairwise genetic differentiation was greatest between Marchinbar Island and Augustus Island (FST = 0.65 and 0.57, Dxy = 0.112 and 0.125 for each dataset respectively). These islands also had the longest branch lengths in the TreeMix tree (Fig. 3). Isoodon macrourus had highest nucleotide divergence (Dxy = 0.159–0.168; Fig. S12a) followed by I. fusciventer (Dxy = 0.113–0.122). Isoodon a. arnhemensis had higher pairwise FST values (FST = 0.39–0.65) than I. fusciventer (FST = 0.2–0.54) and I. macrourus (FST = 0.33–0.57). Pairwise FST values of Augustus Island were higher than the other Kimberley mainland populations, consistent with clustering analyses (Figs. 2, S6, S7), however this was not reflected in the nucleotide divergence estimates which were very similar between each I. a. auratus population (Fig. S12). Increasing genetic differentiation among I. auratus populations was correlated with increasing geographic distance as supported by the mantel test for the isolation by distance analysis (r = 0.48, p = 0.003, 999 permutations; Fig. S13).
Genetic diversity within populations
Mainland I. auratus populations had the highest genetic diversity (encompassing allelic richness, observed heterozygosity, expected heterozygosity and nucleotide diversity) as well as the highest number of private alleles, whilst the lowest diversity was observed in I. a. arnhemensis populations, which also had the lowest number of private alleles (Table 1, Fig. S14). Of all the natural island populations, Barrow Island had the highest genetic diversity, yet Augustus Island had the most private alleles (Table 1). Diversity metrics were similar between the two sampling time points in Doole Island, Hermite Island and Matuwa (Table S2), but were higher than Barrow Island (although note the difference in total number of sites called; Table 1). An increase was observed in heterozygosity and nucleotide diversity in the Guluwuru Island translocated population three years after this population was established (2009– 2011), despite low statistical power (Table S2).
Populations of high priority for genetic conservation
Partitioning genetic diversity into hierarchical strata via a standardised ‘Q’ diversity metric using the QDiver analysis revealed the unequal distribution of genetic diversity across the three recognised subspecies of I. auratus (Table 2; among ‘regions’ δ = 0.014—less than 15% of the grand total γ = 0.102). Isoodon a. auratus had statistically higher within-region diversity (σ = 0.123) and among-population diversity (β = 0.023) than the other subspecies restricted to offshore islands (I. a. barrowensis σ = 0.001 and β = 0.001; I. a. arnhemensis σ = 0.04 and β = 0; Bartlett’s test for homogeneity p = 0.001). Within populations, Artesian Range (α = 0.128) had slightly more diversity than Mitchell Plateau (α = 0.124), which was not detected in diversity metrics (Table 1). Augustus Island had a significantly lower level of diversity than the other I. auratus mainland populations (α = 0.082 in comparison to α = 0.117, α = 0.128, α = 0.124 for Yampi Peninsula, Artesian Range and Mitchell Plateau, respectively; Bartlett’s test for homogenous p = 0.047).
The rank of contributions (from the MetaPop2 analysis) to total diversity of the species was similar for gene and allelic diversity, except for Augustus Island which had a negative contribution to allelic (AT) diversity but a positive contribution to gene (HT) diversity (Fig. 4a). As AT reflects the number and distribution of alleles across populations (López-Cortegano et al. 2019), this decrease likely reflects the private alleles present in Augustus Island (Table 1) that would be lost if this population is removed. There were profound differences between these two diversity components across populations, with mainland I. a. auratus populations having the greatest contribution to within-population allelic (AS) and gene (HS) diversity (Fig. 4a), consistent with within-region diversity of the QDiver analysis (Table 2). These populations, as well as Augustus Island, also had the greatest contribution to between-population allelic diversity (DA) which coincided with these populations having the greatest number of private alleles (Table 1). Populations of I. a. arnhemensis had a substantial negative contribution to within-population diversity (AS, HS), consistent with low estimates of genetic diversity metrics (Table 1) and no among-population diversity in the QDiver analysis (Table 2). Conversely, these populations, as well as Augustus Island, contributed most significantly to between-population gene (DG) diversity, which reflected their higher pairwise FST values (Fig. S12). When the optimal contribution (%) of individuals to a synthetic pool was computed, the contribution to the number of alleles (k) was similar between populations but I. a. auratus mainland populations had the largest contribution to heterozygosity (H) (Fig. 4b).

Contributions to (a) within- (AS, HS; light blue), between (DA, DG; dark blue) and total (AT, HT; black dot) allelic and gene diversity, respectively, of I. auratus populations: Barrow Island (BWI), Doole Island (DOOL), Hermite Island (HERM), Matuwa (MATU), Augustus Island (AUG), Yampi Sound (YAMP), Artesian Range (ART), Mitchell Plateau (MITC), Marchinbar Island (MARC), Guluwuru Island (GULU), Raragala Island (RARA); and (b) percentage of individuals from each population to a synthetic pool of individuals to maximise gene diversity (H) and/or number of alleles (k).
Our MARXAN approach suggested that either the Artesian Range or Mitchell Plateau populations from the mainland were the most effective single populations to conserve allelic richness across the species, representing 79% of all alleles detected (67% and 33% of iterations selected these populations respectively; Fig. 5; Table S3). At least three populations were necessary to conserve 90% of alleles across the species (Fig. 5), with populations from I. a. auratus and I. a. barrowensis always being selected (Table S3). Populations from the Artesian Range, Mitchell Plateau and Barrow Island together retained 94% of alleles. Conserving five to ten of the I. auratus populations sampled in this study consistently retained 98% of alleles (Fig. 5), with populations from the Kimberley mainland always being selected (Table S3). Despite Augustus Island never being selected in an optimal scenario (Table S3), all 11 populations would need to be conserved to retain 100% of remaining genetic diversity of I. auratus, reflecting some unique diversity present within Augustus Island (Table 1).

Populations include Barrow Island (BWI), Doole Island (DOOL), Hermite Island (HERM), Matuwa (MATU), Alice Springs Desert Park (ASDP), Augustus Island (AUG), Lachlan Island (LACH), Storr Island (STOR), Uwins Island (UWIN), Yampi Sound (YAMP), Artesian Range (ART), Mitchell Plateau (MITC), Marchinbar Island (MARC), Guluwuru Island (GULU), and Raragala Island (RARA).
Discussion
Here we confirmed genetic distinction of I. auratus from I. fusciventer and found that the three subspecies of I. auratus, representing the remnant Kimberley mainland, Barrow Island and Marchinbar Island populations, showed strong genetic differentiation as anticipated due to their long-term geographic isolation. A fourth genetic cluster, Augustus Island in the Kimberley, adjacent to the only remnant mainland population, was also resolved within I. auratus that had not been previously recognised. Consistent with our expectations, all island populations had lower genetic diversity relative to the mainland populations, contributing to their genetic distinctiveness, as genetic differentiation metrics are influenced by the level of within-population variance. Hierarchical diversity analyses indicated that the remnant Kimberley mainland population of I. auratus represents the greatest reservoir of genetic diversity within the species, with secondary contribution from Barrow Island. As such, these populations are a high priority to safeguard against future decline and to ensure the preservation of >90% of remaining species-level diversity. Our findings show the importance of understanding the genetic relationships among populations, particularly remnant island populations, and the selection of appropriate combinations of source animals for reintroduction and ecosystem restoration programs to maximise genetic diversity and adaptive potential.
Conserving genetic diversity in highly structured populations
As genetic diversity is not evenly distributed across populations, it is essential to consider patterns of population genetic structure when developing strategic frameworks for the management of threatened species to ensure that most genetic diversity within a species is conserved. Our prioritization analyses indicate that populations most important for conserving genetic diversity of I. auratus consisted primarily of I. a. auratus ancestry, highlighting the importance of the Kimberley mainland populations as a major reservoir of genetic diversity for this species. Furthermore, as diversity loss tends to be higher on islands (Leigh et al. 2019), well connected, large populations on the mainland may better support long-term retention of genetic diversity for this species. Currently, the spatial extent of these populations across the Kimberley region, and their population sizes, are unknown, although survey work is underway (Sayers et al. 2022).
Introduced predators, inappropriate fire regimes, and habitat degradation and fragmentation are known threats to populations of I. auratus (Woinarski et al. 2014) and numerous other Australian species in northern Australia (Carwardine et al. 2012; Geary et al. 2019; Kearney et al. 2019; von Takach et al. 2020). Yampi Sound is recognised as a ‘priority place’ in Australia’s national Threatened Species Action Plan, acknowledging a focus on threatened species which share the same habitat to support recovery of multiple species. As the majority of currently known I. auratus populations already exist within conservation land tenures and Indigenous Protected Areas, ongoing management in collaboration with Traditional Custodians to mitigate the impact of these threatening processes on the Australian mainland are a high priority (Carwardine et al. 2012; von Takach et al. 2020; von Takach et al. 2022a), with a particular emphasis on protecting critical habitat and maintaining its connectivity.
In the past, northern Australia has been considered to provide a level of conservation security for biodiversity, particularly for mammals, yet population declines are being observed (Davies et al. 2018; Woinarski et al. 2011). This region offers the only remaining refugia for I. auratus on the mainland, given the species’ disappearance from 95% of its extensive historic distribution (Woinarski et al. 2014) (Fig. 1a). The large geographic contraction and subsequent population declines have likely led to a massive loss in historical genetic diversity, potentially exceeding 33% (see Exposito-Alonso et al. 2022). This loss, in conjunction with compounding impacts of ongoing threats, may hinder the species’ ability to recolonize large areas of its fundamental niche (Woinarski et al. 2011). This pattern aligns with previous findings for Australian rodents, where extinct populations with large geographic distributions exhibited notably higher heterozygosity compared to extant species with small/restricted remnant populations, such as the Shark Bay mouse (Pseudomys gouldii) (Roycroft et al. 2021). Given the role that I. auratus Kimberley mainland populations play as a crucial repository of genetic diversity, it is imperative to safeguard these populations to ensure the long-term survival of the species.
In this study, island populations showed lower levels of genetic variability relative to mainland Kimberley populations, suggesting that islands have suffered a loss of genetic variation through genetic drift and population bottlenecks caused by their geographic isolation. This is particularly evident for I. a. arnhemensis populations in the Northern Territory that have substantially lower diversity relative to the other populations sampled and make a limited contribution to the overall diversity of the species (Table 1; Fig. 4). Similar results have been found for the Marchinbar Island population of the northern quoll (Dasyurus hallucatus), which displayed extremely low levels of genetic diversity compared to the mainland (von Takach et al. 2022b). Small populations on islands are susceptible to losing genetic diversity due to strong drift and are more likely to accumulate deleterious mutations (Lohr and Haag 2015; Weeks et al. 2016). However, populations with low genetic diversity are also known to have persisted for long periods of time (e.g. Westbury et al. 2019). Additional monitoring on these Northern Territory islands could assist in understanding whether the observed low diversity is indicative of demographic decline or may result in reduced population fitness.
While the relative importance of I. a. arnhemensis populations for the preservation of neutral genomic diversity as represented by these genomic SNPs at the species level is somewhat limited, the loss of these populations could result in the loss of a possible subspecies and the loss of ecological function on remnant islands. Furthermore, I. auratus has already undergone a large range contraction and possibly already suffered a loss in diversity exceeding the preservation targets of international guidelines. Conservation efforts should aim to retain all remaining genetic diversity of the species, which would entail the conservation of populations across all three subspecies, with a precedence in conserving the mainland populations.
Incorporating genetic information into species’ management
To preserve the long-term viability, resilience, and adaptive potential of I. auratus, genetic mixing between islands or between mainland and islands is increasingly being considered (Hoffmann et al. 2021; Liddell et al. 2021). The option of genetic mixing, even between subspecies, could be a worthy endeavour to accentuate adaptive potential to keep pace with changing environments (Brauer et al. 2023; Chan et al. 2019; Zecherle et al. 2021). This has been shown to be successful in a range of species and advocated as a potentially valuable conservation tool (Brauer et al. 2023; Chan et al. 2019; Harrisson et al. 2016; Rick et al. 2019; Taylor and Larson 2019; Undin et al. 2021; Weeks et al. 2017). Given that the Kimberly mainland (Artesian Range and Mitchell Plateau) as well as Barrow Island were key to retaining >90% allelic diversity in the species (Fig. 5), we recommend that mixing these populations should be considered for future reintroductions. While there are potential risks when crossing highly diverged populations, namely outbreeding depression and genetic swamping (Edmands 2007; Frankham et al. 2011; Muhlfeld et al. 2009), these risks may be exaggerated (Frankham 2015; Liddell et al. 2021; Ralls et al. 2018; Weeks et al. 2016). At a minimum, future translocations of I. auratus into mainland reserves or feral predator-free islands should consider sourcing from the Kimberley mainland to ensure that the important genetic diversity from this region is preserved within the conservation ‘safe havens’ network, within which only a single population (Barrow Island) is currently represented.
Given the success of reintroductions of this species to date, translocations will continue to be an important conservation tool to reduce the risk of extinction for threatened species and can ensure persistence of important genetic variation (Weeks et al. 2011). Petroleum engineering companies operating on Barrow Island have funded multiple translocations of mammals to found insurance populations elsewhere, via environmental offsets linked to environmental approvals (Dunlop et al. 2021). Our analyses indicate genome-wide diversity of founder groups has been maintained (>98%) in the translocated populations on Hermite and Doole Islands and at Matuwa, exceeding recommendations in translocation guidelines (Frankham et al. 2014; Weeks et al. 2011) and expectations from populations viability analyses (Ottewell et al. 2014). This is likely the consequence of relatively large founder sizes (92–160 individuals; Fig. 1b), consistent with multiple studies highlighting the importance of establishing new populations with large numbers (n > 100; Ottewell et al. 2014; Weeks et al. 2011).
Nevertheless, ongoing monitoring is necessary to ensure that genetic diversity is conserved over time in translocated populations, as changes in population size, structure, and selection pressure can lead to genetic changes or erosion over the long-term. Periodic monitoring (recommended at least every five years based on PVA modelling, see Ottewell et al. 2014) will be required to ensure that genetic diversity within these populations persists, with more frequent monitoring recommended after catastrophic events (i.e. drought or disease) and when sourcing animals for translocations. While we attempted to investigate temporal changes in the Guluwuru Island, we were limited by small sample size between years. Further sampling of the populations translocated to Raragala and Guluwuru Islands would be valuable to assess the population trajectories. Regular monitoring of the Northern Territory populations may also be useful to ensure that the relatively low genetic diversity observed in these populations does not impact their health (e.g. declining population size due to inbreeding effects). Furthermore, periodic genetic monitoring across the Kimberley mainland would serve as a valuable metric, not only to ensure genetic diversity is maintained in these populations but also as a useful indicator of whether other management actions are successfully maintaining occupancy and connectivity across the Kimberley.
Australia’s heavy reliance on offshore islands for threatened species management, whilst crucial for the persistence of multiple mammal species (Legge et al. 2018), leads to many challenges when planning conservation efforts across a species more generally. Even when populations on islands are relatively large and stable, declines are still observed (Davies et al. 2018) and subsequently their persistence is questionable without the intervention of active conservation actions. Foremost of interest when considering genetic management, is that island populations are consistently deemed to be unique lineages despite insufficient evidence that any differentiation is adaptive (Wolf and Ellegren 2017). While island populations of I. auratus appear to be genetically distinct from one another and the mainland, genetic differentiation measures are often heavily influenced by changes in allele frequencies and within population variance (Weeks et al. 2016). Subsequently we observed populations with the lowest diversity metrics to be the most genetically ‘distinct’, namely Marchinbar Island. The population structure observed in our analyses support the premise that islands have experienced independent histories to such an extent that allele frequencies are clearly differentiated. However, the low genetic divergence (DXY) observed between I. auratus populations and long branch lengths in the TreeMix analysis indicated a strong influence of population-specific genetic drift. Other Kimberley Islands in closer proximity to the mainland (Lachlan Island, Storr Island and Uwins Island), although excluded from most analyses due to small sample size, were not distinguished in the clustering analyses. Therefore, divergence time of islands from the mainland and ecologically relevant phenotypic variation should also be considered. The identified hierarchical structure generally coincides with the three subspecies of I. auratus, but this structure primarily reflects geographic isolation and thus should not constrain management options by defining discrete units. In the case of reintroduction programs, prioritizing the conservation of populations for their genetic diversity (with the aim of maintaining >90% of remaining diversity), and thus their adaptive potential, should take precedence over preserving the perceived genetic uniqueness of island populations. This approach ensures the conservation of the species’ overall genetic variability, resilience, and adaptive capacity.
We still lack an understanding of how genomic differences translate directly into population dynamics in natural populations, especially when translocating them to novel environments (Seaborn et al. 2021). Genetic mixing and associated eco-evolutionary feedbacks can be unpredictable, thereby data-driven approaches are needed to guide best practice (Aisya et al. 2022; Frankham et al. 2011; Hoffmann et al. 2015; Rossetto et al. 2021; Seaborn et al. 2021). An adaptive management framework would therefore be appropriate in undertaking specific conservation actions, including mixing of divergent populations, and scientifically evaluating their outcomes to inform future conservation approaches. Our work provides an excellent model to encourage conservation managers to embrace the complexity of integrating multiple practices, including genomics, in decision-making frameworks for an adaptive management approach.
Concluding remarks
While remnant island populations harbour a proportion of the genetic diversity present within I. auratus, this differentiation should be considered in the context of genetic processes. Genetic drift and within-population variance are likely driving the apparent differentiation among these geographically isolated islands. In this aspect, separate genetic management of populations may hinder the species’ ability to adapt to future environmental change and thus conservation goals for species recovery should be targeted at the species level. Our findings emphasize the critical contribution of mainland Kimberley populations in conserving the genetic diversity of I. auratus. Future translocations should aim to safeguard sufficient genetic diversity (>90%) at the species level, with insurance populations containing representatives of each lineage to spatially spread the risk of cumulative threats and catastrophes. Overall, our study highlights the importance of understanding population genetic structure when considering the loss of genetic diversity across genetically diverged and fragmented populations, particularly islands, and how such information is crucial to incorporate into conservation strategies and management of threatened species.
Data availability
All raw sequencing data has been uploaded to the Oz Mammal Genomics Initiative data portal (https://data.bioplatforms.com/organization/about/bpa-omg). All bioinformatics and R scripts along with relevant metadata have been uploaded to the Mendeley Data Repository (https://data.mendeley.com/datasets/32kz25fzy9/1).
References
Aisya Z, White DJ, Thavornkanlapachai R, Friend JA, Rick K, Mitchell NJ (2022) Using PVA and captive breeding to balance trade-offs in the rescue of the island dibbler onto a new island ark. Sci Rep https://doi.org/10.1038/s41598-022-14150-9
Brauer CJ, Sandoval-Castillo J, Gates K, Hammer MP, Unmack PJ, Bernatchez L et al. (2023) Natural hybridization reduces vulnerability to climate change. Nat Clim Change https://doi.org/10.1038/s41558-022-01585-1
Carwardine J, O’Connor T, Legge S, Mackey B, Possingham HP, Martin TG (2012) Prioritizing threat management for biodiversity conservation. Conserv. Lett. https://doi.org/10.1111/j.1755-263X.2012.00228.x
Casacci LP, Barbero F, Balletto E (2014) The “Evolutionarily Significant Unit” concept and its applicability in biological conservation. Italian J Zoology https://doi.org/10.1080/11250003.2013.870240
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: An analysis tool set for population genomics. Mol. Ecol. https://doi.org/10.1111/mec.12354
Chan WY, Hoffmann AA, van Oppen MJH (2019) Hybridization as a conservation management tool. Conserv Lett https://doi.org/10.1111/conl.12652
Coates DJ, Byrne M, Moritz C (2018) Genetic diversity and conservation units: Dealing with the species-population continuum in the age of genomics. Front Ecol Evolut https://doi.org/10.3389/fevo.2018.00165
Cooper SJB, Ottewell K, MacDonald A J, Adams M, Byrne M, Carthew SM et al. (2018) Phylogeography of southern brown and golden bandicoots: Implications for the taxonomy and distribution of endangered subspecies and species. Aust J Zool https://doi.org/10.1071/ZO19052
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA et al (2011). The variant call format and VCFtools. Bioinformatics https://doi.org/10.1093/bioinformatics/btr330
Davies HF, McCarthy MA, Firth RSC, Woinarski JCZ, Gillespie GR, Andersen AN et al. (2018) Declining populations in one of the last refuges for threatened mammal species in northern Australia. Austral Ecology https://doi.org/10.1111/aec.12596
Dunlop J, Smith A, Burbidge AH, Thomas N, Hamilton NA, Morris K (2021) Industry environmental offset funding facilitates a large multi-species fauna translocation program. Pacific Conserv Biol https://doi.org/10.1071/PC20036
Edmands S (2007) Between a rock and a hard place: Evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol Ecol https://doi.org/10.1111/j.1365-294X.2006.03148.x
Exposito-Alonso M, Booker TR, Czech L, Gillespie L, Hateley S, Kyriazis CC et al. (2022) Genetic diversity loss in the Anthropocene. Science https://doi.org/10.1126/science.abn5642
Fitak RR (2021) OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biol MethodsProtocols https://doi.org/10.1093/biomethods/bpab017
Frankham R, Ballou JD, Eldridge MDB, Lacy RC, Ralls K, Dudash MR et al. (2011) Predicting the probability of outbreeding depression. Conserv Biol 25:465–475
Frankham R (2015) Genetic rescue of small inbred populations: Meta-analysis reveals large and consistent benefits of gene flow. Mol Ecol https://doi.org/10.1111/mec.13139
Frankham R, Ballou JD, Ralls K, Eldridge M, Dudash MR, Fenster CB et al. (2017) Genetic Management of Fragmented Animal and Plant Populations. Oxford University Press
Frankham R, Bradshaw CJA, Brook BW (2014). Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol Conserv https://doi.org/10.1016/j.biocon.2013.12.036
Frichot E, François O (2015) LEA: An R package for landscape and ecological association studies. Methods Ecol Evolut https://doi.org/10.1111/2041-210X.12382
Frichot E, Mathieu F, Trouillon T, Bouchard G, François O (2014) Fast and efficient estimation of individual ancestry coefficients. Genetics https://doi.org/10.1534/genetics.113.160572
Geary WL, Nimmo DG, Doherty TS, Ritchie EG, Tulloch AIT (2019) Threat webs: Reframing the co-occurrence and interactions of threats to biodiversity. J Appl Ecol https://doi.org/10.1111/1365-2664.13427
Legge S, Woinarski JCZ, Burbidge AA, Palmer R, Ringma J, Radford JQ et al. (2018) Havens for threatened Australian mammals: The contributions of fenced areas and offshore islands to the protection of mammal species susceptible to introduced predators. Wildlife Res https://doi.org/10.1071/WR17172
Gould J (1842) On some new species of Australian mammals.Proceedings of the Zoological Society of London 10, 10–14.
Gray JE (1841) Contributions towards the geographicaldistribution of the Mammalia of Australia, with notes onsome recently discovered species, a letter addressed to the Author. Appendix C. in G Grey (ed.) Journalsof Two Expeditions of Discovery in north-west and WesternAustralia, During the Years 1837, 38, and 39, Under theAuthority of Her Majesty’s Government. Describing manynewly discovered, important, and fertile districts, withobservations on the moral and physical condition of theaboriginal inhabitants, &c. T. & W. Voone, London. Volume 2 pp. 397–414.
Gruber B, Unmack PJ, Berry OF, Georges A (2018) dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resources https://doi.org/10.1111/1755-0998.12745
Haig SM, Miller MP, Bellinger R, Draheim HM, Mercer DM, Mullins TD (2016) The conservation genetics juggling act: Integrating genetics and ecology, science and policy. Evolut Appl https://doi.org/10.1111/eva.12337
Hanson JO, Schuster R, Morrell N, Strimas-Mackey M, Edwards B, Watts ME et al. (2022) prioritizr: Systematic Conservation Prioritization in R (R package version 7.2.2). https://CRAN.R-project.org/package=prioritizr
Harrisson KA, Pavlova A, Gonçalves da Silva A, Rose R, Bull JK, Lancaster ML, et al. (2016) Scope for genetic rescue of an endangered subspecies though re-establishing natural gene flow with another subspecies. Mol Ecol https://doi.org/10.1111/mec.13547
Hoban S, Bruford M, D’Urban Jackson J, Lopes-Fernandes M, Heuertz M, Hohenlohe PA et al. (2020) Genetic diversity targets and indicators in the CBD post-2020 Global Biodiversity Framework must be improved. Biolog Conserv https://doi.org/10.1016/j.biocon.2020.108654
Hoban S, Bruford MW, Funk WC, Galbusera P, Griffith MP, Grueber CE et al. (2021) Global commitments to conserving and monitoring genetic diversity are now necessary and feasible. BioScience https://doi.org/10.1093/biosci/biab054
Hoffmann A, Griffin P, Dillon S, Catullo R, Rane R, Byrne M et al. (2015) A framework for incorporating evolutionary genomics into biodiversity conservation and management. Clim Change Responses https://doi.org/10.1186/s40665-014-0009-x
Hoffmann A, Miller AD, Weeks AR (2021) Genetic mixing for population management: From genetic rescue to provenancing. Evolut Appl https://doi.org/10.1111/eva.13154
Jackson S, Groves C (2015) Taxonomy of Australian Mammals. CSIRO Publishing.
Jombart T (2008) adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genetics https://doi.org/10.1186/1471-2156-11-94
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ https://doi.org/10.7717/peerj.281
Kearney SG, Carwardine J, Reside AE, Fisher DO, Maron M, Doherty TS et al. (2019) Corrigendum to: The threats to Australia’s imperilled species and implications for a national conservation response. Pac Conserv Biol https://doi.org/10.1071/pc18024_co
Kershaw F, Bruford MW, Funk WC, Grueber CE, Hoban S, Hunter ME et al. (2022) The Coalition for Conservation Genetics: Working across organizations to build capacity and achieve change in policy and practice. Conserv Sci Pract https://doi.org/10.1111/csp2.12635
Korunes KL, Samuk K (2021) pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Mol Ecol Resources https://doi.org/10.1111/1755-0998.13326
Leigh DM, Hendry AP, Vázquez-Domínguez E, Friesen VL (2019) Estimated six per cent loss of genetic variation in wild populations since the industrial revolution. Evolut Appl https://doi.org/10.1111/eva.12810
Liddell E, Sunnucks P, Cook CN (2021) To mix or not to mix gene pools for threatened species management? Few studies use genetic data to examine the risks of both actions, but failing to do so leads disproportionately to recommendations for separate management. Biolog Conservat https://doi.org/10.1016/j.biocon.2021.109072
Lohr CA, Nilsson K, Sims C, Dunlop J, Lohr MT (2021) Habitat selection by vulnerable golden bandicoots in the arid zone. Ecol Evolut https://doi.org/10.1002/ece3.7875
Lohr JN, Haag CR (2015) Genetic load, inbreeding depression, and hybrid vigor covary with population size: An empirical evaluation of theoretical predictions. Evolution https://doi.org/10.1111/evo.12802
López-Cortegano E, Pérez-Figueroa A, Caballero A (2019) metapop2: Re-implementation of software for the analysis and management of subdivided populations using gene and allelic diversity. Mol Ecol Resources https://doi.org/10.1111/1755-0998.13015
Lyne AG, Mort PA (1981) A comparison of skull morphology inthe marsupial bandicoot genus Isoodon: its taxonomicimplications and notes on a new species, Isoodonarnhemensis. Australian Mammalogy 4, 107–133.
McDonald JA, Carwardine J, Joseph LN, Klein CJ, Rout TM, Watson JEM et al. (2015) Improving policy efficiency and effectiveness to save more species: A case study of the megadiverse country Australia. Biol Conserv https://doi.org/10.1016/j.biocon.2014.11.030
Milanesi M, Capomaccio S, Vajana E, Bomba L, Garcia JF, Ajmone-Marsan P et al. (2017) BITE: An R package for biodiversity analyses bioRxiv https://doi.org/10.1101/181610
Moritz C (1994) Defining ‘Evolutionarily Significant Units’ for conservation. Trends Ecol Evolut https://doi.org/10.1016/0169-5347(94)90057-4
Muhlfeld CC, Kalinowski ST, McMahon TE, Taper ML, Painter S, Leary RF et al. (2009) Hybridization rapidly reduces fitness of a native trout in the wild. Biol Lett https://doi.org/10.1098/rsbl.2009.0033
Ottewell K, Byrne M (2022) Conservation genetics for management of threatened plant and animal species. Diversity https://doi.org/10.3390/d14040251
Ottewell K, Dunlop J, Thomas N, Morris K, Coates D, Byrne M (2014) Evaluating success of translocations in maintaining genetic diversity in a threatened mammal. Biol Conserv https://doi.org/10.1016/j.biocon.2014.01.012
Pacioni C, Atkinson A, Trocini, S, Rafferty C, Morley K, Spencer PBS (2020) Is supplementation an efficient management action to increase genetic diversity in translocated populations? Ecol Manag Restoration https://doi.org/10.1111/emr.12411
Paris JR., Stevens JR, Catchen JM (2017) Lost in parameter space: a road map forstacks Methods in Ecology and Evolution https://doi.org/10.1111/2041-210X.12775
Peakall R, Smouse PE (2006) GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics https://doi.org/10.1093/bioinformatics/bts460
Pickrell J, Pritchard J (2012) Inference of population splits and mixtures from genome-wide allele frequency data. Nat Precedings https://doi.org/10.1038/npre.2012.6956.1
Raffard A, Santoul F, Cucherousset J, Blanchet S (2019) The community and ecosystem consequences of intraspecific diversity: A meta-analysis. Biol Rev https://doi.org/10.1111/brv.12472
Ralls K, Ballou JD, Dudash MR, Eldridge MDB, Fenster CB, Lacy RC et al. (2018) Call for a paradigm shift in the genetic management of fragmented populations. Conserv Lett https://doi.org/10.1111/conl.12412
Ralphs TK, Güzelsoy M (2005) The Symphony Callable Library for Mixed Integer Programming. In B. Golden, S. Raghavan, E. Wasil (Eds.), The Next Wave in Computing, Optimization, and Decision Technologies (pp. 61–76). Springer US. https://doi.org/10.1007/0-387-23529-9_5
Ramsay EP (1887) Description of three new species ofmammals from north west Australia. Abstract of Proceedingsof the Linnean Society of New South Wales 1887, vi.
Reynolds LK, McGlathery KJ, Waycott M (2012) Genetic diversity enhances restoration success by augmenting ecosystem services. PLOS ONE https://doi.org/10.1371/journal.pone.0038397
Rick K, Ottewell K, Lohr C, Thavornkanlapachai R, Byrne M, Kennington WJ (2019) Population Genomics of Bettongia lesueur: Admixing Increases Genetic Diversity with no Evidence of Outbreeding Depression. Genes https://doi.org/10.3390/genes10110851
Robertson JM, Langin KM, Sillett TS, Morrison SA, Ghalambor CK, Funk WC (2014) Identifying evolutionarily significant units and prioritizing populations for management on islands. Monographs of the Western North American Naturalist https://doi.org/10.3398/042.007.0130
Robinson NM, Rhoades C, Pierson J, Lindenmayer DB, Banks SC (2021) Prioritising source populations for supplementing genetic diversity of reintroduced southern brown bandicoots Isoodon obesulus obesulus. Conserv Genet https://doi.org/10.1007/s10592-021-01341-6
Rossetto M, Yap JYS, Lemmon J, Bain D, Bragg J, Hogbin P et al. (2021) A conservation genomics workflow to guide practical management actions. Global Ecol Conserv https://doi.org/10.1016/j.gecco.2021.e01492
Rousset F (1997) Genetic Differentiation and Estimation of Gene Flow fromF-Statistics Under Isolation by Distance. Genetics https://doi.org/10.1093/genetics/145.4.1219
Roycroft E, MacDonald AJ, Moritz C, Moussalli A, Portela Miguez R, Rowe KC (2021) Museum genomics reveals the rapid decline and extinction of Australian rodents since European settlement. Proc Natl Acad Sci https://doi.org/10.1073/pnas.2021390118
Sayers T, Cameron SF, Wauchope M, Pierson J, Joseph L, Kanowski J (2022) Charnley River—Artesian Range Wildlife Sanctuary Ecohealth Report for 2021. Australian Wildlife Conservancy. https://www.australianwildlife.org/wp-content/uploads/2022/08/CRAR2021_EHReport-PDF.pdf
Schmidt TL, Jasper ME, Weeks AR, Hoffmann AA (2021) Unbiased population heterozygosity estimates from genome-wide sequence data. Methods Ecol Evolut https://doi.org/10.1111/2041-210X.13659
Seaborn T, Andrews KR, Applestein CV, Breech TM, Garrett MJ, Zaiats A et al. (2021) Integrating genomics in population models to forecast translocation success. Restorat Ecol https://doi.org/10.1111/rec.13395
Smouse PE, Banks SC, Peakall R (2017) Converting quadratic entropy to diversity: Both animals and alleles are diverse, but some are more diverse than others. PLOS ONE https://doi.org/10.1371/journal.pone.0185499
Sonderegger DL, Wang H, Clements WH, Noon BR (2009) Using SiZer to detect thresholds in ecological data. Front Ecol Environ https://doi.org/10.1890/070179
von Takach B, Jolly CJ, Dixon KM, Penton CE, Doherty TS, Banks SC (2022a) Long-unburnt habitat is critical for the conservation of threatened vertebrates across Australia. Landscape Ecol https://doi.org/10.1007/s10980-022-01427-7
von Takach B, Penton CE, Murphy BP, Radford IJ, Davies HF, Hill BM et al. (2021) Population genomics and conservation management of a declining tropical rodent. Heredity https://doi.org/10.1038/s41437-021-00418-9
von Takach B, Ranjard L, Burridge CP, Cameron SF, Cremona T, Eldridge MDB et al. (2022b) Population genomics of a predatory mammal reveals patterns of decline and impacts of exposure to toxic toads. Mol Ecol https://doi.org/10.1111/mec.16680
von Takach B, Sargent H, Penton CE, Rick K, Murphy BP, Neave G et al. (2023) Population genomics and conservation management of the threatened black-footed tree-rat (Mesembriomys gouldii) in northern Australia. Heredity https://doi.org/10.1038/s41437-023-00601-0
von Takach B, Scheele BC, Moore H, Murphy BP, Banks SC (2020) Patterns of niche contraction identify vital refuge areas for declining mammals. Diversity Distributions https://doi.org/10.1111/ddi.13145
Taylor SA, Larson EL (2019) Insights from genomes into the evolutionary importance and prevalence of hybridization in nature. Nat Ecol Evolut https://doi.org/10.1038/s41559-018-0777-y
Thavornkanlapachai R, Levy E, Li Y, Cooper SJB, Byrne M, Ottewell K (2021) Disentangling the genetic relationships of three closely related bandicoot species across Southern and Western Australia. Diversity https://doi.org/10.3390/d13010002
Thavornkanlapachai R, Mills HR, Ottewell K, Dunlop J, Sims C, Morris K, et al. (2019) Mixing genetically and morphologically distinct populations in translocations: asymmetrical introgression in A newly established population of the boodie (Bettongia lesueur). Genes https://doi.org/10.3390/genes10090729
Thomas O (1901) On some kangaroos and bandicoots fromBarrow Island, N.W. Australia, and the adjoining mainland.Novitates Zoologicae 8, 394–396.
Undin M, Lockhart PJ, Hills SFK, Castro I (2021) Genetic rescue and the plight of ponui hybrids. Front Conserv Sci https://www.frontiersin.org/articles/10.3389/fcosc.2020.622191
Warburton NM, Travouillon KJ (2016) The biology and palaeontology of the Peramelemorphia: A review of current knowledge and future research directions. Aust J Zool https://doi.org/10.1071/ZO16003
Watts ME, Ball IR, Stewart RS, Klein CJ, Wilson K, Steinback C et al. (2009) Marxan with Zones: Software for optimal conservation based land- and sea-use zoning. Environ Model Softw https://doi.org/10.1016/j.envsoft.2009.06.005
Weeks AR, Heinze D, Perrin L, Stoklosa J, Hoffmann AA, van Rooyen A et al. (2017) Genetic rescue increases fitness and aids rapid recovery of an endangered marsupial population. Nat Commun https://doi.org/10.1038/s41467-017-01182-3
Weeks AR, Sgro CM, Young AG, Frankham R, Mitchell NJ, Miller KA et al. (2011). Assessing the benefits and risks of translocations in changing environments: A genetic perspective. Evolutionary Appl https://doi.org/10.1111/j.1752-4571.2011.00192.x
Weeks AR, Stoklosa J, Hoffmann AA (2016) Conservation of genetic uniqueness of populations may increase extinction likelihood of endangered species: The case of Australian mammals. Front Zool https://doi.org/10.1186/s12983-016-0163-z
Westbury MV, Petersen B, Garde E, Heide-Jørgensen MP, Lorenzen ED (2019) Narwhal genome reveals long-term low genetic diversity despite current large abundance size. IScience https://doi.org/10.1016/j.isci.2019.03.023
Westerman M, Kear BP, Aplin K, Meredith RW, Emerling C, Springer MS (2012) Phylogenetic relationships of living and recently extinct bandicoots based on nuclear and mitochondrial DNA sequences. Mol Phylogenetics Evolut https://doi.org/10.1016/j.ympev.2011.09.009
White LC, Thomson VA, West R, Ruykys L, Ottewell K, Kanowski J et al. (2020). Genetic monitoring of the greater stick-nest rat meta-population for strategic supplementation planning. Conserv Genet https://doi.org/10.1007/s10592-020-01299-x
Woinarski J, Burbidge A, Harrison P (2014) The Action Plan for Australian Mammals 2012. CSIRO Publishing
Woinarski JCZ, Braby MF, Burbidge AA, Coates D, Garnett ST, Fensham RJ et al. (2019) Reading the black book: The number, timing, distribution and causes of listed extinctions in Australia. Biol Conserv https://doi.org/10.1016/j.biocon.2019.108261
Woinarski JCZ, Burbidge AA, Harrison PL (2015) Ongoing unraveling of a continental fauna: Decline and extinction of Australian mammals since European settlement. Proc Natl Acad Sci https://doi.org/10.1073/pnas.1417301112
Woinarski JCZ, Garnett ST, Gillespie G, Legge SM, Lintermans M, Rumpff L (2023) Lights at the end of the tunnel: The incidence and characteristics of recovery for Australian threatened animals. Biol Conserv https://doi.org/10.1016/j.biocon.2023.109946
Woinarski JCZ, Legge S, Fitzsimons JA, Traill BJ, Burbidge AA, Fisher A et al. (2011) The disappearing mammal fauna of northern Australia: Context, cause, and response. Conserv Lett https://doi.org/10.1111/j.1755-263X.2011.00164.x
Wolf JBW, Ellegren H (2017) Making sense of genomic islands of differentiation in light of speciation. Nat Rev Genet https://doi.org/10.1038/nrg.2016.133
Wright B, Farquharson KA, McLennan EA, Belov K, Hogg CJ, Grueber CE (2019) From reference genomes to population genomics: comparing three reference-aligned reduced-representation sequencing pipelines in two wildlife species. BMC genomics https://doi.org/10.1186/s12864-019-5806-y
Zecherle LJ, Nichols HJ, Bar-David S, Brown RP, Hipperson H, Horsburgh GJ et al. (2021) Subspecies hybridization as a potential conservation tool in species reintroductions. Evolut Appl https://doi.org/10.1111/eva.13191
Acknowledgements
We acknowledge the Traditional Owners of the lands on which this research was conducted and pay our respects to their Elders past and present. We thank the following people and organisations for providing tissue samples: Lesley Gibson, Kelly Rayner, Carly Bishop, Adrian Wayne, Colleen Sims and Neil Thomas from Biodiversity and Conservation Science, Department of Biodiversity, Conservation and Attractions; Flora and Fauna Division, Department of Environment, Parks and Water Security, Northern Territory Government; Annika Spiridis, Melissa Bruton, Rosie Hohnen, James Smith and John Kanowski from Australian Wildlife Conservancy; Dambimangari Rangers from Dambimangari Aboriginal Corporation, Bruce Pascoe from Alice Springs Desert Park; Peter Spencer, Alison Hillman and Kate Bryant from Murdoch University and Brian Chambers from The University of Western Australia. Samples were collected with approval from various Animal Ethics committees under the following permits: 2019-38 C, 2016/41, 2011/13, 2010/02, 2019-01 A, 2007/10, 2000/19, 2010/35, 2010/01, 2018-02B, A01001 CDU (06 – 09), A07023 CDU (09 – 13), A13026 CDU (13–16). We also thank Shelly McArthur from the Department of Biodiversity, Conservation and Attractions for undertaking DNA extractions. We acknowledge the contribution of the Oz Mammals Genomics Initiative consortium in the generation of data used in this publication. The Initiative is supported by funding from Bioplatforms Australia through the Australian Government National Collaborative Research Infrastructure Strategy (NCRIS). This work was supported by resources provided by the Pawsey Supercomputing Research Centre with funding from the Australian Government and the Government of Western Australia. K.R. was supported by a University of Western Australia Postgraduate Award and Australian Research Council Linkage Grant LP180100315. We thank three anonymous reviewers for their constructive comments which greatly improved the manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
K.R. and K.O. designed the research and C.L., B.H., S.C. and J.D. collected and provided samples. Funding acquisition was led by C.M., N.M., M.B., K.O., K.T. and S.C.. K.R. carried out the analyses and interpretation of results with input from K.O. and B.v.T. K.R. wrote the paper with input, advice, and contributions from all authors with respect to the manuscript framing, structure and content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rick, K., Byrne, M., Cameron, S. et al. Population genomic diversity and structure in the golden bandicoot: a history of isolation, extirpation, and conservation. Heredity 131, 374–386 (2023). https://doi.org/10.1038/s41437-023-00653-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-023-00653-2
- Springer Nature Switzerland AG