
Abstract
TUBB4A gene variants cause dystonia type 4 and hypomyelination with atrophy of the basal ganglia and cerebellum. We report the case of a child with delayed motor development, intellectual disability, and dystonia. Magnetic resonance imaging revealed hypomyelination and progressive cerebellar atrophy without atrophy of the basal ganglia. Whole-exome sequencing revealed a de novo heterozygous variant, c.1088T > C, p.(Met363Thr), in TUBB4A. The present case further supports the vulnerability of the cerebellum in patients with TUBB4A pathogenic variants.
Similar content being viewed by others

Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) is caused by heterozygous variants in TUBB4A1,2. Variants in TUBB4A are known to cause two different clinical conditions: dystonia type 4 (DYT4) and H-ABC1,3. The DYT4 phenotype is characterized by whispering dysphonia, generalized dystonia, and gait ataxia. Magnetic resonance imaging (MRI) typically reveals a normal brain findings3. Patients with H-ABC exhibit clinical onset in early infancy, with developmental delay, extrapyramidal symptoms, progressive spastic tetraplegia, ataxia, dysarthria, cognitive and sensory deficits, and seizures1. Characteristic MRI findings include white matter hypomyelination, the absence or disappearance of the putamen, and cerebellar atrophy1.
There is great diversity in the age of onset, clinical course, and brain MRI findings associated with TUBB4A variants4,5. In particular, there are a series of TUBB4A-associated phenotypes with isolated hypomyelination that do not fit either DYT or H-ABC patterns4,5,6,7,8,9,10,11,12,13,14,15,16,17,18.
We describe the case of a patient with TUBB4A-related hypomyelination caused by a c.1088T > C, p.(Met363Thr) variant. This variant has been reported once previously, but no details of the course of the disorder were available19. The following report presents the case of this patient with TUBB4A-related hypomyelination and cerebellar atrophy without atrophy of the basal ganglia.
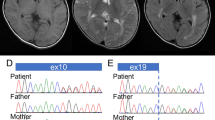
The patient was an 11-year-old boy who presented with a neurodevelopmental disorder. His family and perinatal histories were unremarkable. He first spoke at 12 months of age and began walking independently at 18 months of age. From 3 years of age, he was noted to be clumsy and unsteady and exhibited difficulties with comprehension. He attended both special and regular education classes in elementary school. At the age of 10 years, he was diagnosed with mild intellectual disability (IQ = 52). Physical examination revealed no abnormalities. However, the neurological examination found brisk deep tendon reflexes, a bilateral Babinski sign, ankle clonus, upper limb dystonia, and abnormal tandem gait. MRI at the age of 8 years revealed cerebral white matter hypomyelination but no atrophic findings in the basal ganglia or cerebellum (Fig. 1A). MRI at the age of 11 years revealed hypomyelination and atrophy of the cerebellum, but the basal ganglia size was normal (Fig. 1B, C). Whole-exome sequencing was performed, and the NM_006087.4: c.1088T > C, p.(Met363Thr) variant was detected. Since neither parent had this variant, it was considered a de novo variant. This variant was absent in gnomAD v3.1.2 (accessed January 2022), the ToMMo 14KJPN Allele Frequency Panel (v v20211208) (https://jmorp.megabank.tohoku.ac.jp/202112/), and 218 in-house Japanese exome control datasets. In silico evaluation tools predicted this variant to be deleterious (PROVEAN −3.42, CADD v1.6 phred 23.8, M-CAP 0.869). Based on the American College of Medical Genetics and Genomics standards and guidelines, the c.1088T > C variant was classified as likely pathogenic (PS2, PM2, PP3).

A Axial T2 image at the age of 8 years shows diffuse hypomyelination without atrophy of the basal ganglia. B Axial T2 image at the age of 11 years shows persistent hypomyelination without atrophy of the basal ganglia. C Sagittal T1 image at the age of 11 years shows atrophy of the cerebellum.
TUBB4A variants causing H-ABC were first reported in 2013, with an onset age between 2 months and 4.5 years (median age: 6 months)8. The frequency of atrophy of the basal ganglia in H-ABC has been reported to be 70% in the capsule and 30% in the caudate nucleus within 2 years of onset but progresses to 97% and 53% (2–12 years after onset) and then 100% and 90%, respectively, (>12 years after onset)20. However, some patients with hypomyelination but without atrophy of the basal ganglia have been reported to have isolated hypomyelination4,18. We summarize the variants of TUBB4A-related hypomyelination without atrophy of the basal ganglia (H-without AB) in Table 1. The age at onset of clinical symptoms was between 1 month and 33 years (median age: 15 months), which is later than that for H-ABC. Including the present case, cerebellar atrophy was reported with 12 of the 17 variants (70%) (Table 1).
Several correlations between genotype and phenotypic severity in H-ABC have been suggested. Patients with the common c.745G > A variant have a more benign phenotype than patients with other variants20. Lu et al.8 reported that variants located on the outside of the αβ-tubulin heterodimer, distant from the guanosine triphosphate domain, are likely to result in milder phenotypes without atrophy of the basal ganglia or cerebellum. For the c.900G > T, c.1064A > T, and c.1172G > A variants, there have been two reports of phenotypes characterized by hypomyelination without atrophy of the basal ganglia (Table 1). However, Tonduti et al.10 reported that patients with the same variant showed different disease courses.
To our knowledge, there has been no report on TUBB4A-related hypomyelination with only atrophy of the basal ganglia. Therefore, we speculate that the cerebellum is more vulnerable than the basal ganglia. The highest expression of TUBB4A was in the cerebellum, followed by the putamen and white matter3. There was a twofold difference between the cerebellum and thalamus, which had the lowest expression3. The different TUBB4A expression levels in different brain regions may explain this distinct vulnerability.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3199.
References
van der Knaap, M. S. et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Neuroradiol. 23, 1466–1474 (2002).
Simons, C. et al. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 92, 767–773 (2013).
Hersheson, J. et al. Mutations in the autoregulatory domain of beta-tubulin 4a cause hereditary dystonia. Ann. Neurol. 73, 546–553 (2013).
Curiel, J. et al. TUBB4A mutations result in specific neuronal and oligodendrocytic defects that closely match clinically distinct phenotypes. Hum. Mol. Genet. 26, 4506–4518 (2017).
Purnell, S. M., Bleyl, S. B. & Bonkowsky, J. L. Clinical exome sequencing identifies a novel TUBB4A mutation in a child with static hypomyelinating leukodystrophy. Pediatr. Neurol. 50, 608–611 (2014).
Di Bella, D. E. et al. Hypomyelinating leukodystrophies in adults: Clinical and genetic features. Eur. J. Neurol. 28, 934–944 (2021).
Duncan, I. D. et al. A mutation in the Tubb4a gene leads to microtubule accumulation with hypomyelination and demyelination. Ann. Neurol. 81, 690–702 (2017).
Lu, Y., Ondo, Y., Shimojima, K., Osaka, H. & Yamamoto, T. A novel TUBB4A mutation G96R identified in a patient with hypomyelinating leukodystrophy onset beyond adolescence. Hum. Genome Var. 4, 17035 (2017).
Sagnelli, A. et al. Early-onset progressive spastic paraplegia caused by a novel TUBB4A mutation: brain MRI and FDG-PET findings. J. Neurol. 263, 591–593 (2016).
Tonduti, D. et al. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients. Eur. J. Paediatr. Neurol. 20, 323–330 (2016).
Vanderver, A. et al. Whole exome sequencing in patients with white matter abnormalities. Ann. Neurol. 79, 1031–1037 (2016).
Erro, R. et al. H-ABC syndrome and DYT4: Variable expressivity or pleiotropy of TUBB4 mutations? Mov. Disord. 30, 828–833 (2015).
Isakov, O. et al. Crowdfunding effort identifies the causative mutation in a patient with nystagmus, microcephaly, dystonia and hypomyelination. J. Genet. Genomics 42, 79–81 (2015).
Kancheva, D. et al. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia. Mov. Disord. 30, 854–858 (2015).
Pyle, A. et al. Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain 138, 276–283 (2015).
Ferreira, C., Poretti, A., Cohen, J., Hamosh, A. & Naidu, S. Novel TUBB4A mutations and expansion of the neuroimaging phenotype of hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). Am. J. Med. Genet. A 164A, 1802–1807 (2014).
Miyatake, S. et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 82, 2230–2237 (2014).
Pizzino, A. et al. TUBB4A de novo mutations cause isolated hypomyelination. Neurology 83, 898–902 (2014).
Okubo, M. et al. GGC Repeat Expansion of NOTCH2NLC in Adult Patients with Leukoencephalopathy. Ann. Neurol. 86, 962–968 (2019).
Hamilton, E. M. et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain 137, 1921–1930 (2014).
Acknowledgements
We would like to thank ELCS, AJE and Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hashiguchi, M., Monden, Y., Nozaki, Y. et al. A TUBB4A Met363Thr variant in pediatric hypomyelination without atrophy of the basal ganglia. Hum Genome Var 9, 19 (2022). https://doi.org/10.1038/s41439-022-00198-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-022-00198-6
- Springer Nature Limited