

Abstract
Systemic lupus erythematosus is a heritable autoimmune disease that predominantly affects young women. To improve our understanding of genetic etiology, we conduct multi-ancestry and multi-trait meta-analysis of genome-wide association studies, encompassing 12 systemic lupus erythematosus cohorts from 3 different ancestries and 10 genetically correlated autoimmune diseases, and identify 16 novel loci. We also perform transcriptome-wide association studies, computational drug repurposing analysis, and cell type enrichment analysis. We discover putative drug classes, including a histone deacetylase inhibitor that could be repurposed to treat lupus. We also identify multiple cell types enriched with putative target genes, such as non-classical monocytes and B cells, which may be targeted for future therapeutics. Using this newly assembled result, we further construct polygenic risk score models and demonstrate that integrating polygenic risk score with clinical lab biomarkers improves the diagnostic accuracy of systemic lupus erythematosus using the Vanderbilt BioVU and Michigan Genomics Initiative biobanks.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) is a chronic multi-organ autoimmune disease that predominantly affects young women and individuals of African, Asian, and Hispanic ancestries1,2. The prevalence of SLE worldwide was estimated to range from 37 to 123 cases per 100,000 individuals depending on geographic locations, ancestry groups, study type, and study period3. No cure or targeted treatment exists for SLE. Current medications for treatments are broadly acting (e.g., corticosteroid) and are associated with many side effects4. The diagnosis of SLE is challenging due to the heterogeneity in clinical symptoms and lack of pathognomonic features or accurate lab tests5. The difficulty in the diagnosis of SLE presents great challenges for treatment, as advanced-stage SLE is associated with a worse prognosis and can lead to organ failure and even deaths6. Thus, accurate early detection and intervention are the key to mitigating SLE disease outcomes and improving quality of life7.
Genome-wide association studies (GWAS), functional genomic studies, and integrative analysis have provided unprecedented understanding of the genetic architecture underlying complex diseases8. Such analyses have enabled the identification of candidate causal genes or tissues/cell types and the development of robust biomarkers and targeted drugs9. GWAS have also been successful in SLE10. To date, GWAS have identified >130 loci that reach genome-wide significance (P value <5 × 10−8)11,12,13,14. Yet, these identified loci explain only a small fraction of overall heritability and the genes or tissues/cell types affected by them remain unclear11,15. Previous SLE GWAS results were primarily from individuals of European ancestry16,17,18,19. Fortunately, several recent GWAS efforts have greatly increased the sample size of non-European ancestry including East Asians11,20,21,22,23.
To take advantage of existing datasets and maximize sample sizes and power, we assembled multi-ancestry SLE GWAS datasets from European, Eastern Asian, and Admixed American ancestries. To exploit the shared genetic basis between different autoimmune diseases, we also aggregated GWAS datasets from 10 genetically correlated autoimmune diseases. We conducted multi-ancestry and multi-trait meta-analysis. To gain mechanistic insights, we conducted transcriptome-wide association studies (TWAS), linking regulatory variants to their target genes. We leveraged computational drug repurposing (CDR) and cell type enrichment analyses to identify novel drugs with the potential to treat SLE as well as to identify previously described immune cell types that are dysregulated in SLE. Lastly, we constructed polygenic risk scores (PRS) and validated the models in two independent biobanks: Michigan Genomic Initiative (MGI)24 and Vanderbilt University Biobank (BioVU) and investigated the utility of PRS to improve the diagnosis of SLE when used in conjunction with clinical lab tests, e.g., anti-nuclear antibody (ANA) and anti-double strand DNA (anti-dsDNA).
Results
SLE GWAS meta-analysis
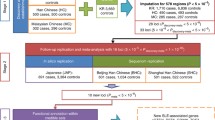
We provide an overview of study design and schematic workflow in Supplementary Fig. 1. Our compiled SLE GWAS dataset contains 12 cohorts from three ancestries [East Asian (EAS), NEAS = 194,435 (5877 cases and 188,558 controls); European (EUR), NEUR = 520,311 (14,355 cases and 505,956 controls); Admixed American (AMR), NAMR = 3720 (1393 cases and 2327 controls)], with a total number of N = 21,625 cases and 696,841 controls. We carefully conducted quality controls to ensure the validity of the analyses, including checking sample overlaps, and manually examining Manhattan and quantile-quantile (QQ) plots for each study. With quality-controlled SLE datasets, we first performed a fixed effect meta-analysis within each ancestry using inverse-variance weighted meta-analysis method.
Next, as SLE has overlapping symptoms and shared genetic basis with other autoimmune diseases25,26,27, we conducted multi-trait association analysis to improve power (Supplementary Data 1 and Supplementary Fig. 2). We first calculated the genetic correlations between SLE and 13 other autoimmune disorders in samples of European ancestry using linkage disequilibrium score regression (LDSC)28,29 (Fig. 1). We identified ten autoimmune disorders (autoimmune thyroid disorder, Crohn’s disease, celiac disease, multiple sclerosis, primary biliary cirrhosis, rheumatoid arthritis, Sjogren’s syndrome, systemic sclerosis, type 1 diabetes, and ulcerative colitis) as significantly genetically-correlated traits with SLE (false discovery rate <0.05) (Fig. 1). We then performed multi-trait analysis across autoimmune disorders of significant genetic correlation with SLE using MTAG30 in each ancestry separately and combined the results across ancestries using fixed effect meta-analysis.

Left panel indicates genetic correlations across 14 autoimmune diseases. Genetic correlation is estimated using cross-trait LDSC. Color intensity and size of square are proportional to strength of genetic correlation (brown = negative correlation, green = positive correlation). Asterisks indicate genetic correlations that are statistically significant at false discovery rate level of 0.05. Red boxes highlight the correlation between SLE and other traits. Right panel illustrates the network of significantly genetically-correlated traits. The colors of the lines represent the magnitude of genetic correlation estimates using LDSC. The widths of the lines represent the statistical significance. Disease name abbreviations: AN = ankylosing spondylitis, ATD = autoimmune thyroid disease, CD = Crohn’s disease, CEL = celiac disease, MS = multiple sclerosis, PBC = primary biliary cirrhosis, PSOA = psoriatic arthritis, RA = rheumatoid arthritis, SJO = Sjogren’s syndrome, SLE = systemic lupus erythematosus, SSC = systemic sclerosis, T1D = type 1 diabetes, UC = ulcerative colitis, VIT = vitiligo.
We show Manhattan plot and Quantile-Quantile (QQ) plot for multi-ancestry and multi-trait meta-analysis (MAMT) for SLE in Fig. 2 and Supplementary Fig. 3, respectively. The meta-analysis results show well calibrated QQ plot with genomic control value 1.03. If we define a locus to be a 1 million basepair window surrounding the sentinel variants, the meta-analysis identified 106 loci that reach genome-wide significance (P value <5 × 10−8). Among the identified loci, 27 are novel that reach genome-wide significance for the first time. Alternatively, we may also define a locus based on the linkage disequilibrium (LD)-based pruning. We created a LD reference panel using a subset of 1000 Genomes Project phase 3 data, with the same ancestry fraction as datasets in the meta-analysis. We performed LD-based pruning using PLINK with the created reference panel and the squared correlation between alleles (r2) cutoff of 0.2. It should be noted that a total of 249 loci, of which 92 were considered novel, were identified when LD pruning was performed (Methods and Supplementary Data 2). For all subsequent presentations, we stick with distance-based loci.

We consider loci reported in the GWAS catalog (as of 12/11/2021) or in the studies used in our meta-analysis as known (brown). We define novel loci (green) as the ones that are 1 million base pairs away from previously reported GWAS loci. Diamonds in the plot represent sentinel variants at novel loci. Diamond with black outline represents novel loci that are replicable according to RATES. In total, we identify 79 known loci and 27 novel loci, of which 74 known loci and 16 novel loci are deemed replicable (PPR > 0.90) by RATES. We also annotated the target genes for top variants at each novel locus using Open Targets Genetics database. Here, y axis is truncated at P value = 1 × 10−18 to visualize the novel loci. The red line represents the genome-wide significance level (5 × 10−8). Two-sided P value associated with each variant is calculated according to the Chi-squared test statistic with 1 degree of freedom.
We further assessed the replicability of each locus using MAMBA31 and its extension RATES (Method), a statistically rigorous model-based method to assess replicability leveraging the strength and consistency of association signals across studies. The term replicability was introduced by McGuire et al. that refers to variants with genuine non-zero effects31. The posterior probability of replicability (PPR) quantifies how likely the signal is genuine and can be replicated in a sufficiently powered replication study, e.g., a large enough study from a matched population31. RATES results confirmed that 74 known loci and 16 novel loci were replicable with PPR > 0.90 (Supplementary Data 3). Across 16 novel and replicable loci, we found that multi-ancestry SLE-only GWAS already yielded genome-wide significant P values in 3 loci and borderline significant P values for the remaining 13 loci (highest P value = 8.19 × 10−5). Importantly, rheumatoid arthritis’s GWAS contributes the most to the identification of novel loci in SLE, having the smallest P values for 8 out of 16 loci in comparison to other autoimmune diseases (Supplementary Table 1). This is likely due to the largest sample size of autoimmune GWAS data and overlapping clinical features between SLE and RA32. Interestingly, all 16 novel loci do not yield significant P values using Cochran’s Q tests for heterogeneities under the Bonferroni threshold of 0.05/106 (Supplementary Fig. 4 and Supplementary Data 4). As fixed effect meta-analysis favors loci with homogeneous effects, we further explored the extent of heterogeneity using additional sub-threshold variants (P < 1.00 × 10−6) and found that 97% of 144 loci do not show evidence of heterogeneity in effect sizes across different SLE studies with P values for Cochran’s Q test ≥0.05/144. We noted that the variances of the effect size estimates from MAMT analysis are 59% smaller than multi-ancestry fixed effect meta-analysis (MA) for SLE, indicating that jointly analyzing multiple traits increases the effective sample sizes by 2.87-fold. 94 of previously known loci did not reach genome-wide significance in our study, yet most of them (91 out of 94 loci) still have association P values <0.05 (Supplementary Data 5). The reduced level of statistical significance for these loci may be due to spurious association, genetic effect heterogeneity between studies, or because multi-trait analysis introduces noise for loci that are uniquely associated with one or few traits and reduces power.
Linking GWAS hits to target genes
95% of identified sentinel variants are non-coding. 79% of variants in the identified loci (as defined by sentinel variants and variants having r2 > 0.8 to sentinel variants) are non-coding. To interpret their functional consequence, we identified potential target genes of each top variant at novel locus using Open Targets Genetics database (Accessed 21 April 2022)33. There is clear biological relevance for the identified target genes (Fig. 2). For example, MKRN2 was previously shown to regulate NF-κB-mediated inflammatory response34. Another target gene CCDC88B is an important regulator of maturation and effector functions of T cell35. Glycosylation changes in T cells by MGAT5 have been shown to impact T cell functions and are implicated in many autoimmune diseases36,37. Moreover, we identified CD83 as another candidate target gene for SLE and studies have found soluble CD83 to be a promising therapeutic to interfere with autoimmunity in SLE38. We provide a description of the sentinel variant and targeted gene at each novel locus in Table 1.
Transcriptome-wide association study implicates additional novel genes
We conducted transcriptome-wide association studies (TWAS). Specifically, we first derived gene expression prediction models using two reference datasets from disease-relevant tissues, i.e., the Genetic European Variation in Disease39 (GEUVADIS; lymphoblastoid cell line (LCL); n = 358) and Depression Gene Network40 (DGN; whole blood; n = 873) datasets. We used a new method PUMICE that integrates 3D genome and epigenetic information to improve prediction accuracy41. To assess the accuracy of prediction models, we calculate Spearman’s correlation coefficients between measured and predicted gene expression using nested cross-validation as described in Khunsriraksakul et al.41 and assess whether Spearman’s correlation is significantly different from zero41. Using PUMICE, we obtained 7028 and 9260 significant genes with Spearman’s correlation coefficients >0.1 and the corresponding P values <0.05 from GEUVADIS and DGN, respectively. Next, we conducted TWAS analysis using TESLA42, which integrates our SLE GWAS summary statistics from multiple participating studies of different ancestries with gene expression prediction models based on samples of European ancestry in DGN or GEUVADIS. Specifically, TESLA uses meta-regression to jointly model the genetic effects across ancestries so that we can borrow strength from shared effects between ancestries to optimize TWAS power (Methods). In total, we identified 99 and 119 significant gene-level associations using GEUVADIS and DGN trained models. We define loci iteratively: we first rank significant genes by their P values. For the most significant gene in the list, we define a locus as a 1 million basepair window surrounding the gene. We then remove all genes in the list that overlap the locus and repeat the process to define the next locus, until we exhaust all significant genes from the list. We define novel loci as the ones >1 million base pairs away from known GWAS sentinel variants. Using this criterion, we found 6 and 17 novel and independent loci from GEUVADIS and DGN analyses, respectively (Fig. 3a, b).

Gene expression prediction models are trained in either lymphoblastoid cell line (GEUVADIS dataset, panel a or whole blood (DGN dataset, panel b. The red horizontal line represents the significance threshold at 2.5 × 10−6 (Bonferroni threshold for testing 20,000 genes). We label the most significant gene at each novel locus outside identified GWAS loci, which we defined as ±1 Mb region surrounding the sentinel variants. Two-sided P value associated with each gene is calculated based on the TWAS Z score for gene-based association test. Panel c illustrates the results for cell type enrichment analyses of TWAS signals from whole blood (DGN) for 15 immune cell types from DICE dataset (left panel) and 5 B cell subsets from SLE patients (right panel). We assess the P value of enrichment using a Gaussian copula-based multiple regression model as described in Chen et al.42. The blue horizontal line represents the significance threshold at 0.05. Bar plots highlighted in green colors represent the significant cell type enrichment (two-sided P value <0.05).
TWAS associations at 106 GWAS loci are reported in Supplementary Data 6 and 7. We further compared the TWAS results with the list of target genes linked to enhancer variants (enVars) via Open Target Genetics43. In total, we were able to link these enVars to 26 unique target genes and subsequently used these genes as a reference to benchmark the accuracy of our TWAS results. We found 22 enVar target genes overlapping TWAS genes with nominal significance levels, and 9 enVar target genes overlapping TWAS hits with significant P values under the Bonferroni threshold for testing multiple genes across the genome.
We identified many novel associations that are supported by biological links to SLE pathogenesis. For example, CD52 mRNA expression is elevated in B cells of SLE patients and may function in B cell homeostasis by inhibiting responses to B cell receptor (BCR) signaling44. Circulating IL8 is also elevated in SLE patients45. IL8 can activate neutrophils, leading to the release of neutrophil extracellular traps (NETs), which are implicated in many immune-mediated conditions46. UBASH3A is a suppressor of T cell receptor (TCR) signaling and is implicated in multiple autoimmune diseases47,48,49,50. Moreover, UBASH3A mRNA expression levels in peripheral blood mononuclear cells are decreased in SLE patients51.
Cell type enrichment analysis in DGN-trained TWAS associations
To identify immune cell types relevant for SLE, we conducted cell type enrichment (CTE) analysis to evaluate if cell type specific genes are enriched with TWAS associations. We provide more detailed description of the method and datasets in Methods.
In the Database of Immune Cell Expression (DICE) dataset, we observed significant enrichment of TWAS hits in various immune cell subsets, including non-classical monocytes, B cells, NK cells, CD8 + T cells, and TH1 cells, whose dysregulation have been implicated in SLE (Fig. 3c). This observation is consistent with previous studies. For example, non-classical monocytes are increased in SLE patients compared to healthy controls. They tend to have inflammatory characteristics with properties of antigen presentation that activate T and B cells52,53. Naïve B cells was observed to be decreased in SLE patients when compared to patients with other autoimmune diseases and healthy controls54. In SLE patients, the chromatin accessibility of naïve B cells shows enrichment for transcription factors that lead to B cell activation (NFKB, AP-1, BATF, IRF4, and PRDM1)55. The role of NK cells remains unclear in SLE, as different subsets have shown to have either pro- or anti- inflammatory activities56,57. Similarly, although the role of naïve CD8+ T cells is unknown in SLE, their effect on the differentiation into activated CD8+ T cells has been studied. Activated CD8+ T cells in the peripheral blood of SLE patients are observed to have decreased effector and cytolytic function which likely contribute to autoimmunity58,59. Lastly, the ratio of TH1/TH2 are observed to be increased in peripheral mononuclear cells of SLE patients60 along with dysregulation of TH1 cytokines, such as IFN-γ61.
We also carried out a second cell-type enrichment analysis, focusing on B cell subtypes collected from SLE patients62. This is because one of the hallmarks of SLE is the production of autoantibodies, which are due to autoreactive B cells. We observed significant enrichment of TWAS signals in cell type specific genes of double negative B cells, transitional 3 B cells, activated naïve B cells, and isotype switched memory B cells, whose dysregulation has been implicated in SLE etiology (Fig. 3c). For example, Scharer et al. observed all B cell subtypes were distinct between healthy controls and SLE62. Double negative B cells are likely to differentiate into antibody secreting cells. Their transcriptomes are more closely related to activated naïve B cells and isotype switched memory B cells in SLE samples than in healthy control cells62. Overall, our analysis highlights dysregulation of specific immune cells subsets that are previously supported to be aberrant in SLE, which could serve as potential targets for drug development and immune therapy.
Computational drug repurposing analysis identified novel therapeutics for SLE
We perform computational drug repurposing (CDR) via Connectivity Map (CMap)63,64 using significant gene targets identified in TWAS associations. Importantly, many clinically informative drug classes were identified, including glucocorticoid receptor agonist, histone deacetylase (HDAC) inhibitor, mTOR inhibitor, and topoisomerase inhibitor (Fig. 4). Some of the identified drug classes, including glucocorticoid receptor agonists, are already being commonly used in the clinic to treat SLE and supports the validity of these results. An HDAC6 inhibitor was previously shown to greatly reduce lupus nephritis in mice65. An mTOR inhibitor was also shown to attenuate SLE by regulating inflammation induced CD11b+ Gr1+ myeloid cells66. Importantly, sirolimus, an mTOR inhibitor, has already been shown to improve SLE disease activity in Phase 1/2 clinical trial67. Irinotecan, a topoisomerase inhibitor, also reverses lupus nephritis and results in prolonged survival in SLE mice68.

We used significant SLE TWAS associations (P value <2.5 × 10−6; two-sided P value associated with each gene is calculated based on the TWAS Z score for gene-based association test) in CMap to identify drugs that could potentially reverse the disease signature. Data are visualized with L1000FWD. Each point represents cell-specific drug-induced signature, and signatures were clustered via k-nearest neighbors. To quantify the correlation between a query signature and reference profile, CMap calculates a τ score. A negative τ score indicates that trait-associated gene expression profile will be normalized by the identified molecule, which we can potentially repurpose to treat the disease. A CMap τ score of −75 indicates that the drug more consistently reverses the expression level of the TWAS significant genes than 75% of all reference gene sets. Diamond shape represents signatures with CMap τ scores less than −75. Signatures are colored by perturbational drug classes. Contour lines and labels represent frequently identified drug class and associated drugs.
Derivation of PRS models
We derived multiple candidate PRS models using a number of methods, including pruning and thresholding (P+T), SBayesR69, SBLUP70, SDPR71, LDpred-inf72, LDpred-funct73, PUMAS74, PRS-CS-auto75, and LASSOSUM76. We only used summary statistics-based PRS methods, which do not require training or replicating cohorts to select tuning hyperparameters. We analyzed three sets of GWAS summary statistics, including 1) SLE GWAS of European ancestry (N = 6748 cases and 11,516 controls)17 (Ref), which we also include as part of MA and MAMT analyses, 2) MA for SLE, and 3) MAMT for SLE. To validate the accuracy of PRS models, we use two independent biobanks that are linked to electronic medical records (EMRs), including MGI (N = 34,702) and BioVU (N = 49,707), which are not part of the GWAS discovery cohorts. Since biobank data may define SLE cases differently from the discovery cohorts, we performed GWAS meta-analysis of BioVU and MGI datasets. We confirmed that the effect size (beta) between the biobank datasets and GWAS discovery cohorts have good concordance with each other (Pearson correlation coefficient of 0.7 and P value = 4.4 × 10−16) (Supplementary Fig. 5).
We calculated the proportion of variance explained on the liability scale (R2) and area under the receiver operating characteristic curve (AUC) for each PRS model in the MGI and BioVU datasets and selected the best PRS model for subsequent downstream analyses (Supplementary Fig. 6). As MGI and BioVU are predominately of European ancestry, we focus on samples of European ancestry in our analyses. We provided other performance metrics, including true negative, true positive, false negative, false positive, sensitivity, specificity, odd ratio (OR) per standard deviation, OR of top 20% vs. bottom 20% and its 95% confidence interval, and Nagelkerke’s R2 and its 95% confidence interval (NKR2 [95% CI]) in Supplementary Data 8.
We found that LASSOSUM on average yielded more accurate models compared to other PRS methods (Supplementary Data 9). LASSOSUM risk scores generated an AUCs of 0.75 and 0.74 in MGI and BioVU, respectively. Moreover, PRS based on GWAS summary statistics derived from MAMT almost always performed better than that of MA or Ref regardless of the PRS methods used. We also provide the number of variants used as predictors in each PRS model in Supplementary Table 2.
Sensitivity analyses of PRS models
Since our external testing datasets are EMR-based biobanks, which are susceptible to incorrect data entry or false positive diagnosis, we conducted sensitivity analyses to evaluate how the PRS models performed using SLE cases defined by three different algorithms. A list of ICD codes used in this study can be found on Supplementary Table 3. Specifically, we utilized algorithms previously evaluated by Barnado et al.77. We considered three different algorithms listed on Supplementary Table 4, including
-
Def1 (least stringent) that only requires at least one count of SLE ICD codes;
-
Def6 (intermediate) that requires at least two separate counts of SLE ICD codes, and excludes patients with systemic sclerosis or dermatomyositis ICD codes, and
-
Def12 (most stringent) that requires at least four separate counts of SLE ICD codes and a recorded ANA positive test result (≥1:160). Moreover, patients with systemic sclerosis or dermatomyositis ICD codes are excluded.
Characteristics of patients identified from different algorithms and biobanks, including sample size, age at first diagnosis, ANA and anti-dsDNA status, and the number of patients with undifferentiated connective tissue disease, can be found on Supplementary Table 5. Importantly, the use of different definitions yielded comparable AUCs of PRS. When defining SLE cases using more stringent criteria (i.e., using Def 12 instead of Def 1), we improved the AUC of PRS from 0.75 to 0.78 in MGI and 0.74 to 0.79 in BioVU (Supplementary Fig. 7).
Integrating PRS with conventional lab tests led to further improvement in diagnostic accuracy
Currently, the diagnosis of SLE follows the 2019 EULAR/ACR classification criteria78. To be considered as SLE, patients must have positive ANA tests (titer ≥1:80) and must tally at least 10 total points from clinical domains (e.g., malar rash) and immunologic domains (e.g., test positive for anti-dsDNA). The biomarkers ANA and anti-dsDNA alone have low accuracy and will only be prescribed when symptoms are already present.
We hypothesized that PRS for SLE can stratify SLE patients, improve the accuracy of conventional lab tests, and facilitate early diagnosis. We first assessed the utility of PRS in stratifying individuals for the risk of developing SLE using two independent biobanks, i.e., BioVU and MGI24. We observed that there were 3.81 times as many cases in the top quintile than in the bottom quintile of the PRS distribution (OR [95% CI]; MGI: 3.46 [2.36, 5.08], BioVU: 4.74 [3.59, 6.25]) (Fig. 5a).

a The odd ratios (OR) and 95% confidence intervals (CIs; bars) of each PRS stratus against individuals in the lowest quintile. We stratified PRS into 5 groups with the bottom quintile used as the reference group. The red dashed line indicates OR = 1. b AUC of PRS (P), ANA (A), anti-dsDNA (D), and when these tests are used together to diagnose SLE. P values are calculated via one-sided Delong’s test. It should be noted that this analysis is done in a subset of patients who have available information on both ANA and anti-dsDNA tests. c Risk gradient curves for patients with positive/negative ANA test results, positive/negative anti-dsDNA test results, and positive ANA and negative anti-dsDNA test results. The lines represent the relationship between PRS quintiles and fractions of SLE cases, and the shaded areas surrounding the lines represent 95% CIs. Here, SLE cases are defined according to Def1 (Supplementary Table 4).
We, then, evaluated the model performance of PRS when used in conjunction with ANA/anti-dsDNA lab results. We found consistent improvement in prediction accuracy when we added PRS into the prediction model. Specifically, in BioVU, the AUC of using PRS + ANA + anti-dsDNA is 0.75, which improves the AUC of using only ANA + anti-dsDNA (0.73) with P value 0.005 (Fig. 5b). Similarly, in MGI, the AUC of using PRS + ANA + anti-dsDNA is 0.75, which improves the AUC of using ANA + anti-dsDNA alone (0.74) with P value of 0.065. When jointly analyzing the MGI and BioVU cohorts, the AUC improves from 0.72 with ANA + anti-dsDNA alone to 0.74 with PRS + ANA + anti-dsDNA (P value = 0.002).
Lastly, we investigated the capability of PRS to stratify patients with certain classical SLE lab results. Among individuals that are ANA positive or anti-dsDNA-negative, the top PRS quintile has approximately 2.36 and 2.34 times more SLE cases than in the bottom PRS quintile, respectively (Fig. 5c). When focusing on patients who had positive ANA and negative anti-dsDNA tests in the MGI and BioVU biobanks (the most common lab test results observed clinically), the top PRS quintile has approximately 2.31 times more SLE cases than in the bottom PRS quintile. Importantly, the risk gradient curve showed a good stratification of positive ANA and negative anti-dsDNA patients, especially, in BioVU database (prevalence of SLE in bottom quintile is 9.91%, middle quintile is 15.09%, and top quintile is 28.88%). The top quintile has 2.91 and 1.91 times more SLE cases than those in the bottom and middle quintiles, respectively. This result shows that PRS can help increase the specificity of ANA test and sensitivity of anti-dsDNA test.
Discussion
In this work, we carried out multi-ancestry and multi-trait GWAS meta-analysis of SLE by aggregating the GWAS results from 12 SLE cohorts, encompassing over 700,000 samples. In total, we identified 106 significant loci, of which 16 loci were deemed as novel and replicable. We further discovered 22 additional novel loci through TWAS and performed computational drug repurposing and cell type enrichment analyses. Our results pointed out a few relevant drug classes, such as glucocorticoid receptor agonist, HDAC inhibitor, and mTOR inhibitor, as well as enriched cell types, such as non-classical monocytes and B cells. The identified drugs are capable of reversing SLE-associated gene expression signatures and are therefore putative candidates for SLE treatment. The roles of these enriched cell types in SLE are also supported by previous experimental evidence.
Based on the meta-analysis results, we applied nine methods to derive PRS models. We assessed the accuracy of the PRS models in two independent EMR-based biobanks, including MGI and BioVU, and evaluated their utility in identifying SLE patients when used in conjunction with conventional lab tests. We found that PRS of SLE was highly effective for identifying patients with an elevated risk of SLE, as there were 3.81 times as many cases in the top PRS quintile than in the bottom PRS quintile. Moreover, we showed that PRS can help increase the specificity of ANA test and the sensitivity of anti-dsDNA test. The results in MGI and BioVU datasets slightly differ, which may be due to factors such as geographic catchment and sampling strategy24. Nonetheless, the results are, in general, concordant with each other and demonstrate the added utility of PRS in diagnosing SLE. Our results demonstrate the potential benefits of incorporating PRS into SLE classification criteria, which can lead to a more accurate early diagnosis for SLE.
Our work helps establish the added benefits of PRS for improved diagnosis when used together with conventional lab tests. Earlier studies have applied PRS in various health outcomes including type 2 diabetes, cardiovascular disease, and others79,80,81,82. However, the added benefits of PRS, when used with other diagnostic criteria and lab tests, remain under-explored, and completely unexplored to our knowledge for SLE. Our work showed that PRS when used in conjunction with conventional lab values such as ANA and anti-dsDNA status, could improve diagnostic accuracy. PRS would not replace conventional lab tests, but as germline DNA usually would not change over a lifetime, the use of PRS could facilitate early diagnosis and risk screening. PRS has the most clinical utility in individuals with extreme PRS values, who will benefit from careful monitoring for progression83. With the improvement in the understanding of the polygenic architecture of complex diseases, the advancement of statistical methodology, and the increase of genetic diversity in genotyped samples, we could expect further improvements in the accuracy of genetic prediction83.
Several future directions in data generation and method development could further enhance the predictive capability of PRS model in diagnosing SLE. First, since SLE is an autoimmune disease with a relatively low prevalence, increasing GWAS sample sizes will further improve marginal effect estimates and in turn, improve the predictive value of PRS. Second, even though SLE is more prevalent in non-European ancestry compared with Caucasians84, 65% of the SLE GWAS samples in our study were from European ancestry. It is imperative to increase the diversity of SLE GWAS in the next phase of studies. Lastly, it remains an open question of how to best combine multi-ancestry genetics data to create a better and more transferable PRS model for different ancestries. It will be of great interest to investigate whether PRS similarly improves the diagnosis for SLE in individuals of non-European ancestry when used together with conventional lab tests. The utility of PRS in samples of European and East Asian populations will help motivate the development and deployment of PRS models in other populations.
In summary, our work represents one of the first attempts of applying PRS and clinical lab tests to enhance the early diagnosis of SLE. We can apply the framework to study other diseases to identify high-risk individuals from the population and facilitate the practice of targeted treatment and precision medicine.
Methods
Quality control of GWAS summary statistics
A list of all GWAS for 14 autoimmune diseases used in this study was listed in Supplementary Data 1. We retrieved GWAS summary statistics from GWAS catalog85, FinnGen website (Release 5 data), Pan-UK Biobank website, and BioBank Japan PheWeb22.
Prior to meta-analysis, we conducted careful quality control including examining the genomic control values, and manually inspecting the QQ plot and Manhattan plot from participating studies. We also harmonized GWAS data from participating studies and calibrated the effect allele to the reference allele. We also ensured that participating studies have little, or no sample overlaps and the effect sizes are homogeneous between studies of similar ancestry, by calculating the λmeta statistic86 (Supplementary Fig. 8). Specifically, for a SNP i with estimated effect size bi,1 and bi,2 and variances \({\sigma }_{i,1}^{2}\) and \({\sigma }_{i,2}^{2}\) in a pair of cohorts 1 and 2, we calculated a statistic \({T}_{i}=\frac{{({b}_{i,1}-{b}_{i,2})}^{2}}{{{\sigma }_{i,1}}^{2}+{{\sigma }_{i,2}}^{2}}\), and denote the vector of statistics for all M SNPs as T=(T1,…,TM). λmeta is then calculated as \(\frac{{median}({{{{{\boldsymbol{T}}}}}})}{{median}({\chi }_{1}^{2})}\). If λmeta is much greater than 1, it suggests that there may be heterogeneity between estimated genetic effects from cohorts 1 and 2. If λmeta is much smaller than 1, it suggests that there may be overlapping samples. We have ensured that all pairs of studies have λmeta values close to 1 (Supplementary Fig. 8). For studies with known sample overlaps, we de-correlated the association statistics to ensure valid meta-analysis results87.
For the situation where one study is a multi-ancestry meta-analysis and included all of the samples from another study (usually occur in multi-ancestry GWAS), we subtracted the GWAS summary statistics of the smaller study from the larger study according to the inverse-variance weighted meta-analysis formula88, so that we can have non-overlapping studies for ancestry-specific analysis.
Previous studies observed that PRS model derived from HapMap3 SNPs have comparable performance to models that uses SNP from 1000 Genomes Project89. As such, we used SNPs in HapMap3 to construct PRS.
Meta-analysis and annotation of the genome-wide significant variants
For MA, we used inverse-variance meta-analysis across 12 SLE cohorts as implemented in the METAL software88. For MAMT, we first analyzed each disease within each ancestry using the inverse-variance weighted fixed effect meta-analysis method in METAL88. Next, we utilized meta-analysis results from European ancestry to calculate genetic correlations among 14 autoimmune diseases using the LDSC software28,29, as samples of European ancestry have the largest sample sizes. We considered traits to be genetically-correlated if genetic correlation P values (two-sided) are significant after controlling false discovery rate (FDR) at 0.05 level. We then performed multi-trait analysis combining SLE and its significantly correlated traits in each ancestry separately using the MTAG software30. Lastly, we combined multiple trait analysis results across ancestries using inverse-variance weighted meta-analysis88. HLA region was excluded from the multi-trait meta-analysis as was done for other autoimmune diseases due to their unusually large effect sizes that violate the model assumptions for MTAG. Cochran’s Q test was performed via METASOFT90 to estimate the heterogeneity of effect sizes across GWAS studies.
Genome-wide significance thresholds were defined as 5 × 10−8. Independent genome-wide significant loci were defined as the ±1 Mb window surrounding the sentinel variant. We deem a locus novel if there were no previously reported significant variants in GWAS catalog or previous studies that fall within ±1 Mb from the sentinel variant. We then annotated potential target genes for sentinel variants according to the Open Target Genetics database, which used functional annotation data (e.g., eQTL, pQTL, and pc-HiC) to link regulatory variants to target genes. Alternatively, we also defined significant loci as the set of variants that are in LD (r2 > 0.2) with the sentinel variants.
Assessing the replicability of association signals
We assessed the replicability of association signals using a model-based approach. The method extends MAMBA31 that uses a mixture model to assess whether the variants identified in a meta-analysis are genuine or spurious based on the strength and consistency of the association between studies. We define “replicable” variants as the ones with genuine association signals, so they can be replicated in a sufficiently powered replication study, e.g., one with large enough sample sizes from a matched population. Replicable signals with genuine non-zero effects tend to have stronger and more consistent signals across studies than spurious association signals. Here, to accommodate the potential heterogeneity of association signals between datasets of different ancestries, we use meta-regression with principal components of genome-wide allele frequencies as covariates and use the residuals as input for MAMBA analysis. RATES will assign a posterior probability of replicability to each signal which rigorously assesses its validity. We consider signals with PPR > 0.90 as strong evidence of replicability. We also visually inspected the Manhattan plot and forest plot for the SNPs deemed as replicable and confirmed the findings.
Imputing gene expression prediction models and TWAS analysis
Gene expression prediction models were constructed via PUMICE41. Briefly, PUMICE utilizes epigenetic information to prioritize essential genetic variants that carry important functional roles. It also uses 3D genomic information to define windows that harbor cis-regulatory variants. Thus, PUMICE can more accurately predict gene expression levels using genotype data as input compared to alternative approaches. For epigenomic data, we utilize four broadly available epigenetic annotation tracks, including H3K27ac mark, H3K4me3 mark, DNase hypersensitive mark, and CTCF mark from ENCODE database91,92. Epigenomic data for whole blood and LCLs were retrieved from the ENCODE database with the following accession codes: ENCFF949VFY and ENCFF028SGJ, respectively. For 3D genomic data, we considered different choices for windows that harbor cis-regulatory variants, including the ones defined by conventional linear windows surrounding gene start and end sites (i.e., ±250 kb and ±1 Mb) as well as by 3D genomic informed regions (i.e., domain93, loop94, pcHiC95, and TAD94). For whole blood (DGN), 3D genomic data from proxy tissue (lung for domain and spleen for loop, pcHiC, and TAD) is used. For LCLs (GEUVADIS), 3D genomic data from matched tissue is available. Model is deemed significant if the cross-validated average Spearman’s correlation coefficient is >0.1 and the P value of the correlation coefficient is <0.05. We applied PUMICE to lymphoblastoid cell line (GEUVADIS) and whole blood (DGN) datasets as these tissues are most relevant to SLE’s pathogenesis and have large sample sizes. For GEUVADIS, we restricted our analysis to European samples (n = 358). For DGN, we only included samples with >90% European ancestry composition as determined from ADMIXTURE software using 1000 Genomes Project Phase 3 Data as reference panel (n = 873). With PUMICE, we obtained 7028 and 9260 gene expression models from GEUVADIS and DGN, respectively, for which the cross-validated average Spearman’s correlation coefficients is >0.1 and the P value of the correlation coefficient is <0.05.
Gene x trait association analysis with TESLA
The PUMICE prediction model is based on samples of European ancestry. We apply TESLA42, a novel method to optimally integrate the prediction model with a multi-ancestry GWAS for TWAS. TESLA exploits shared phenotypic effects between ancestries and accommodates potential effect heterogeneities. TESLA is more powerful than alternative strategies that leverage fixed effect GWAS meta-analysis results to perform TWAS and the methods that only integrates only ancestrally matched GWAS and eQTL datasets.
Cell type enrichment analysis
We retrieved cell type expression from two sources: 1) Database of Immune Cell Expression (DICE)96 and 2) transcriptomic profiles of B cell subsets from SLE subjects62. Specifically, DICE profiled transcriptomic data of 15 immune cell types (2 of which are activated cell types) from 106 samples. For SLE-specific transcriptomic datasets, five B cell subtypes from nine SLE patients were profiled. We processed both datasets uniformly and closely followed the pipeline outlined in the previous study97. First, we normalized RNA-sequencing data in terms of transcript per million (TPM). Next, we computed the average expression for each gene in each cell type. We removed genes not expressed across all cell types. We then rescale gene expression to a total of 1 million TPM for each cell type, to minimize the impact of library size. For each gene, we define the “gene expression specificity score” by dividing the expression of each gene in a given cell type by the total expression of the same gene across all cell types. We define the cell type-specific genes as the ones with gene expression specificity score in the top 5th percentile in each cell type.
To perform cell type enrichment analysis, we defined Y as a vector of squared TWAS Z scores from G significant genes and X as a matrix of G rows and C columns where rows correspond to genes and columns correspond to one of the C cell types. The (g, c) element of X is either 0 or 1, denoting whether gene g is specific to cell type c according to the definition of cell type specific gene above. As the genes may be correlated due to linkage disequilibrium, we calculate the correlation between squared TWAS Z score statistics via bootstrap. For the Z-score statistic from two genes 1 and 2, Z1 and Z2, the correlation between the TWAS Z-statistic can be calculated from LD panel as described in Khunsriraksakul et al.41, which we denote as ρ. To calculate the correlation between squared Z statistics, we simulate bivariate normal distributed statistics \({Z}_{1}^{(b)}\) and \({Z}_{2}^{(b)}\), b= 1,…,B, where
We then calculate the correlation between squared TWAS statistics as
We denote the correlation matrix for the squared Z statistics of G genes as Ρ. We then performed weighted regression to assess whether cell type specific genes of a given cell type are enriched with TWAS hits. Specifically, we use
We estimate the weighted regression coefficients as
We assess the enrichment of each cell type by testing if the corresponding coefficient in \(\hat{{{{{{\boldsymbol{\gamma }}}}}}}\) is significantly different from 0. This approach is similar in principle to MAGMA98 and we adapt it for TWAS analyses.
Computational drug repurposing
We extracted significant TWAS associations (P value <2.5 × 10−6) and used them as a proxy for SLE disease signature. We then applied CMap algorithm to identify drugs capable of reversing disease signature. Specifically, we queried SLE TWAS association signals against the reference profiles in the CMap database (from L1000 assay), which recorded gene expression changes caused by perturbagens as the signature of the drug x gene pair. We only used reference data from touchstone dataset of CMap, which comprised reference signatures across nine cell lines treated with ~3000 well-annotated small molecule drugs. We consider a gene set to have stronger connectivity with the drug if the drug more consistently reverses the expression levels of all genes in the gene set. To quantify the correlation between a query signature and reference profile, CMap calculates a τ score. A negative τ score indicates that trait-associated gene expression profile will be normalized by the identified molecule, which we can potentially repurpose to treat the disease. τ score allows us to compare the strength of connectivity between different gene sets. For example, CMap τ score of −75 indicates that the drug more consistently reverses the expression level of the TWAS significant genes than 75% of all reference gene sets. We consider a more negative τ score as stronger evidence that supports repurposing the drug for treating the disease. Finally, we adapted L1000FWD plot to visualize drug-induced transcriptomic signatures99.
Construction of PRS models
For our PRS model derivation, we applied nine PRS methods that do not require validation datasets for parameter tuning, including P+T, SBayesR69, SBLUP70, SDPR71, LDpred-Inf72, LDpred-funct73, PUMAS74, PRS-CS-auto75, and LASSOSUM76, to three GWAS summary statistics. We used 503 European samples from 1000 Genome Project Phase 3100 as a reference panel for estimating linkage disequilibrium coefficients, per recommendations by previous work89.
For the P+T method, we followed the recommendation from the Global Biobank Meta-Analysis Initiative89. Specifically, we ran PLINK101 with the following flags: -clump-p1 1 -clump-p2 1 -clump-r2 0.1 -clump-kb 250. Next, we applied 13 different P value thresholds, including 5 × 10−8, 5 × 10−7, 1 × 10−6, 5 × 10−6, 5 × 10−5, 5 × 10−4, 5 × 10−3, 0.01, 0.05, 0.10, 0.20, 0.50, 1. For other PRS methods, we used default settings.
Study samples in MGI and BioVU
We first determined ancestry of each sample via ADMIXTURE102 using 1000 Genome Project Phase 3 data as a reference panel. We only included samples with >90% European ancestry composition for subsequent analyses. Genetic data was imputed via Michigan Imputation Server using 1000 Genome phase 3 as the reference panel and only variants with imputation quality metric Rsq > 0.8 were kept for subsequent analyses.
We then performed quality control of the data following the recommendation by Marees et al.103. Specifically, with PLINK, we excluded 1) SNPs with the low genotyping rate (-geno 0.01), 2) individuals who have high rates of genotype missingness (-mind 0.01), 3) SNPs with low minor allele frequency (-maf 0.05), 4) SNPs that deviate from Hardy-Weinberg equilibrium (-hwe 1e-6), 5) individuals with high or low heterozygosity rates, 6) individuals that have a first or second-degree relative in the sample (-rel-cutoff 0.125), and 7) SNPs not within the HapMap3 SNP set.
Next, we extracted phenotypic data from the electronic medical records. Specifically, we used ICD codes to extract disease status (Supplementary Table 3). For ANA test result, we considered a titer of ≥1:80 (e.g., 1:80, 1:160, 1:320, etc.) as positive, and the other values as negative (e.g., negative status, 1:40, 1:20, and etc.). For anti-dsDNA test result, we considered a level of ≥60 IU/mL or positive status as positive, and the other values as negative (e.g., <60 IU/mL or negative status).
Evaluation of prediction performance
We evaluated the prediction performance of constructed PRS in two independent biobank datasets: MGI and BioVU, which are not part of the training data. Specifically, we calculated Nagelkerke’s R2 on the liability scale104 after adjusting for Sex, PC1-10, and HLA alleles105 (HLA-DRB1*03:01, HLA-DRB1*08:01, and HLA-DQA1*01:02) that we imputed using HIBAG106. We assumed population prevalence of SLE to be 0.1%107. Furthermore, we also reported area under the receiver operating characteristic curve (AUC) for full model with the above covariates. We estimated the corresponding 95% confidence intervals from bootstrap with 1000 replicates. Comparison of AUCs between PRS models was calculated with two-sided Delong’s test. Lastly, we divided the target samples into quintiles according to PRS rankings. We calculated odds ratios of developing SLE in each quintile against individuals with PRS in the bottom 20th percentile. Sensitivity and specificity were calculated at the optimal threshold using the Youden’s J statistic.
Software URLs
LDSC software can be found at https://github.com/bulik/ldsc. MTAG software can be found at https://github.com/JonJala/mtag. Open Target Genetics software can be found at https://genetics.opentargets.org. METASOFT software can be found at http://genetics.cs.ucla.edu/meta/. MAMBA software can be found at https://github.com/dan11mcguire/mamba. PUMICE software can be found at https://github.com/ckhunsr1/PUMICE. TESLA software can be found at https://github.com/funfunchen/rareGWAMA. CMap software can be found at https://clue.io/. L1000FWD web application can be found at https://maayanlab.cloud/L1000FWD. SBayesR software can be found at https://github.com/YinLiLin/hibayes. SBLUP software can be found at https://yanglab.westlake.edu.cn/software/gcta. SDPR software can be found at https://github.com/eldronzhou/SDPR. LDpred-Inf software can be found at https://privefl.github.io/bigsnpr/articles/LDpred2.html. LDpred-funct software can be found at https://github.com/carlaml/LDpred-funct. PUMAS software can be found at https://github.com/qlu-lab/PUMAS. PRS-CS-auto software can be found at https://github.com/getian107/PRScs. LASSOSUM software can be found at https://github.com/tshmak/lassosum. PLINK software can be found at https://www.cog-genomics.org/plink. Bedtools software can be found at https://bedtools.readthedocs.io/en/latest. ADMIXTURE software can be found at https://bioinformaticshome.com/tools/descriptions/ADMIXTURE.html. Michigan imputation server can be found at https://imputationserver.sph.umich.edu/index.html. R Project for statistical computing can be found at https://www.r-project.org.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The GWAS summary statistics of the multi-ancestry and multi-trait SLE meta-analysis result have been deposited on the Shiny App [https://liugroupstatgen.shinyapps.io/SLEv] for users to download and interactively explore research results. This meta-analysis result was derived via MTAG30 and METAL88 from the following datasets: SLE11,16,17,18,19,20,21,22,23,108, ATD22, CD109,110, CEL111,112, MS112,113,114, PBC115,116, RA22,117, SJO22,118, SSC119, T1D22,120, and VIT121. We also obtained GWAS data from FinnGen Release 5 website [https://www.finngen.fi/en/access_results], Pan-UK Biobank website [https://pan.ukbb.broadinstitute.org], and BioBank Japan PheWeb [https://pheweb.jp] for traits available in these biobanks. A more detailed information of each study can be found in Supplementary Data 1. For TWAS results, we provided TWAS association statistics from two distinct tissues, including DGN (whole blood) and GEUVADIS (lymphoblastoid cell line). We have also linked GWAS variant to its target gene by labeling the eQTL SNP in the gene expression prediction model with the smallest GWAS P value (“top variant” column). DGN data can be requested at https://www.nimhgenetics.org/request-access/how-to-request-access under “Depression Genes and Networks study (D. Levinson, PI)”. GEUVADIS data can be accessed at https://www.ebi.ac.uk/arrayexpress/experiments/E-GEUV-1. Gene expression prediction models were created using PUMICE41 and TWAS association statistics were calculated by TESLA42. DICE dataset can be requested through dbGaP accession number phs001703.v1.p1. B cell dataset from SLE subjects is available from the NCBI Gene Expression Omnibus under accession number GSE118256.
References
Dall’Era, M. et al. The incidence and prevalence of systemic lupus erythematosus in san francisco county, california: the california lupus surveillance project. Arthritis Rheumatol. 69, 1996–2005 (2017).
Izmirly, P. M. et al. The incidence and prevalence of systemic lupus erythematosus in new york county (manhattan), New York: the manhattan lupus surveillance program. Arthritis Rheumatol. 69, 2006–2017 (2017).
Stojan, G. & Petri, M. Epidemiology of systemic lupus erythematosus: an update. Curr. Opin. Rheumatol. 30, 144–150 (2018).
Fugger, L., Jensen, L. T. & Rossjohn, J. Challenges, progress, and prospects of developing therapies to treat autoimmune diseases. Cell 181, 63–80 (2020).
Felten, R. et al. 10 most important contemporary challenges in the management of SLE. Lupus Sci. Med 6, e000303 (2019).
Kernder, A. et al. Delayed diagnosis adversely affects outcome in systemic lupus erythematosus: cross sectional analysis of the LuLa cohort. Lupus 30, 431–438 (2021).
Carter, E. E., Barr, S. G. & Clarke, A. E. The global burden of SLE: prevalence, health disparities and socioeconomic impact. Nat. Rev. Rheumatol. 12, 605–620 (2016).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet 20, 467–484 (2019).
Cano-Gamez, E. & Trynka, G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front Genet 11, 424 (2020).
Deng, Y. & Tsao, B. P. Genetic susceptibility to systemic lupus erythematosus in the genomic era. Nat. Rev. Rheumatol. 6, 683–692 (2010).
Wang, Y. F. et al. Identification of 38 novel loci for systemic lupus erythematosus and genetic heterogeneity between ancestral groups. Nat. Commun. 12, 772 (2021).
Chen, L., Morris, D. L. & Vyse, T. J. Genetic advances in systemic lupus erythematosus: an update. Curr. Opin. Rheumatol. 29, 423–433 (2017).
Wen, L. et al. Exome-wide association study identifies four novel loci for systemic lupus erythematosus in Han Chinese population. Ann. Rheum. Dis. 77, 417 (2018).
Wang, Y. F. et al. Identification of ST3AGL4, MFHAS1, CSNK2A2 and CD226 as loci associated with systemic lupus erythematosus (SLE) and evaluation of SLE genetics in drug repositioning. Ann. Rheum. Dis. 77, 1078–1084 (2018).
Perez, R. K. et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376, eabf1970 (2022).
Bentham, J. et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464 (2015).
Langefeld, C. D. et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 8, 16021 (2017).
Harley, J. B. et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet 40, 204–210 (2008).
Hom, G. et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med 358, 900–909 (2008).
Song, Q. et al. Genome-wide association study on Northern Chinese identifies KLF2, DOT1L and STAB2 associated with systemic lupus erythematosus. Rheumatol. (Oxf.) 60, 4407–4417 (2021).
Tangtanatakul, P. et al. Meta-analysis of genome-wide association study identifies FBN2 as a novel locus associated with systemic lupus erythematosus in Thai population. Arthritis Res Ther. 22, 185 (2020).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet 53, 1415–1424 (2021).
Alarcón-Riquelme, M. E. et al. Genome-Wide Association Study in an Amerindian Ancestry Population Reveals Novel Systemic Lupus Erythematosus Risk Loci and the Role of European Admixture. Arthritis Rheumatol. 68, 932–943 (2016).
Zawistowski, M. et al. The Michigan Genomics Initiative: A biobank linking genotypes and electronic clinical records in Michigan Medicine patients. Cell Genomics https://doi.org/10.1016/j.xgen.2023.100257 (2023).
Richard-Miceli, C. & Criswell, L. A. Emerging patterns of genetic overlap across autoimmune disorders. Genome Med 4, 6 (2012).
Zhernakova, A., van Diemen, C. C. & Wijmenga, C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nat. Rev. Genet 10, 43–55 (2009).
Shah, S., Chengappa, K. & Negi, V. Systemic lupus erythematosus and overlap: A clinician perspective. Clin. Dermatol. Rev. 3, 12–17 (2019).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
McGuire, D. et al. Model-based assessment of replicability for genome-wide association meta-analysis. Nat. Commun. 12, 1964 (2021).
Icen, M. et al. Systemic lupus erythematosus features in rheumatoid arthritis and their effect on overall mortality. J. Rheumatol. 36, 50–57 (2009).
Ghoussaini, M. et al. Open targets genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–d1320 (2021).
Shin, C. et al. MKRN2 is a novel ubiquitin E3 ligase for the p65 subunit of NF-κB and negatively regulates inflammatory responses. Sci. Rep. 7, 46097 (2017).
Kennedy, J. M. et al. CCDC88B is a novel regulator of maturation and effector functions of T cells during pathological inflammation. J. Exp. Med 211, 2519–2535 (2014).
Alves, I. et al. AB0123 Changes in cellular glycosylation as a key factor in the immunopathogenesis of systemic lupus erythematosus. Ann. Rheum. Dis. 79, 1361–1362 (2020).
Pereira, M. S. et al. Glycans as key checkpoints of T cell activity and function. Front Immunol. 9, 2754 (2018).
Starke, C., Steinkasserer, A., Voll, R. E. & Zinser, E. Soluble human CD83 ameliorates lupus in NZB/W F1 mice. Immunobiology 218, 1411–1415 (2013).
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013).
Battle, A. et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res 24, 14–24 (2014).
Khunsriraksakul, C. et al. Integrating 3D genomic and epigenomic data to enhance target gene discovery and drug repurposing in transcriptome-wide association studies. Nat. Commun. 13, 3258 (2022).
Chen, F. et al. Multi-ancestry transcriptome-wide association analyses yield insights into tobacco use biology and drug repurposing. Nat. Genet. https://doi.org/10.1038/s41588-022-01282-x (2023).
Lu, X. et al. Global discovery of lupus genetic risk variant allelic enhancer activity. Nat. Commun. 12, 1611 (2021).
Bhamidipati, K. et al. CD52 Is Elevated on B cells of SLE Patients and Regulates B Cell Function. Front Immunol. 11, 626820 (2020).
Mao, Y. M. et al. Increased circulating interleukin-8 levels in systemic lupus erythematosus patients: a meta-analysis. Biomark. Med 12, 1291–1302 (2018).
Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147 (2018).
Diaz-Gallo, L. M. et al. Evidence of new risk genetic factor to systemic lupus erythematosus: the UBASH3A gene. PLoS One 8, e60646 (2013).
Frederiksen, B. N. et al. Evidence of stage- and age-related heterogeneity of non-HLA SNPs and risk of islet autoimmunity and type 1 diabetes: the diabetes autoimmunity study in the young. Clin. Dev. Immunol. 2013, 417657 (2013).
Kim, K. et al. High-density genotyping of immune loci in Koreans and Europeans identifies eight new rheumatoid arthritis risk loci. Ann. Rheum. Dis. 74, e13 (2015).
Plagnol, V. et al. Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PLoS Genet 7, e1002216 (2011).
Liu, J. et al. Decreased UBASH3A mRNA expression levels in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Inflammation 38, 1903–1910 (2015).
Mukherjee, R. et al. Non-Classical monocytes display inflammatory features: validation in sepsis and systemic lupus erythematous. Sci. Rep. 5, 13886 (2015).
Zhu, H. et al. CD16(+) monocyte subset was enriched and functionally exacerbated in driving t-cell activation and b-cell response in systemic lupus erythematosus. Front Immunol. 7, 512 (2016).
Odendahl, M. et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J. Immunol. 165, 5970–5979 (2000).
Scharer, C. D. et al. ATAC-seq on biobanked specimens defines a unique chromatin accessibility structure in naïve SLE B cells. Sci. Rep. 6, 27030 (2016).
Liu, M., Liang, S. & Zhang, C. NK cells in autoimmune diseases: protective or pathogenic? Front Immunol. 12, 624687 (2021).
Arazi, A. et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat. Immunol. 20, 902–914 (2019).
Comte, D. et al. Signaling lymphocytic activation molecule family member 7 engagement restores defective effector CD8+ T cell function in systemic lupus erythematosus. Arthritis Rheumatol. 69, 1035–1044 (2017).
Stohl, W. Impaired polyclonal T cell cytolytic activity. A possible risk factor for systemic lupus erythematosus. Arthritis Rheum. 38, 506–516 (1995).
Akahoshi, M. et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum. 42, 1644–1648 (1999).
al-Janadi, M., al-Balla, S., al-Dalaan, A. & Raziuddin, S. Cytokine profile in systemic lupus erythematosus, rheumatoid arthritis, and other rheumatic diseases. J. Clin. Immunol. 13, 58–67 (1993).
Scharer, C. D. et al. Epigenetic programming underpins B cell dysfunction in human SLE. Nat. Immunol. 20, 1071–1082 (2019).
Lamb, J. The connectivity map: a new tool for biomedical research. Nat. Rev. Cancer 7, 54–60 (2007).
Subramanian, A. et al. A next generation connectivity map: l1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452.e17 (2017).
Ren, J. et al. Selective histone deacetylase 6 inhibition normalizes b cell activation and germinal center formation in a model of systemic Lupus Erythematosus. Front Immunol. 10, 2512 (2019).
Shi, G. et al. mTOR inhibitor INK128 attenuates systemic lupus erythematosus by regulating inflammation-induced CD11b(+)Gr1(+) cells. Biochim Biophys. Acta Mol. Basis Dis. 1865, 1–13 (2019).
Lai, Z. W. et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet 391, 1186–1196 (2018).
Frese-Schaper, M., Zbaeren, J., Gugger, M., Monestier, M. & Frese, S. Reversal of established lupus nephritis and prolonged survival of New Zealand black x New Zealand white mice treated with the topoisomerase I inhibitor irinotecan. J. Immunol. 184, 2175–2182 (2010).
Lloyd-Jones, L. R. et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun. 10, 5086 (2019).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Zhou, G. & Zhao, H. A fast and robust Bayesian nonparametric method for prediction of complex traits using summary statistics. PLoS Genet 17, e1009697 (2021).
Privé, F., Arbel, J. & Vilhjálmsson, B. J. LDpred2: better, faster, stronger. Bioinformatics 36, 5424–5431 (2020).
Márquez-Luna, C. et al. Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. Nat. Commun. 12, 6052 (2021).
Zhao, Z. et al. PUMAS: fine-tuning polygenic risk scores with GWAS summary statistics. Genome Biol. 22, 257 (2021).
Ge, T., Chen, C.-Y., Ni, Y., Feng, Y.-C. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Mak, T. S. H., Porsch, R. M., Choi, S. W., Zhou, X. & Sham, P. C. Polygenic scores via penalized regression on summary statistics. Genet Epidemiol. 41, 469–480 (2017).
Barnado, A. et al. Developing electronic health record algorithms that accurately identify patients with systemic lupus erythematosus. Arthritis Care Res. 69, 687–693 (2017).
Aringer, M. et al. 2019 european league against rheumatism/american college of rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 71, 1400–1412 (2019).
Inouye, M. et al. Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J. Am. Coll. Cardiol. 72, 1883–1893 (2018).
Mars, N. et al. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 11, 6383 (2020).
Natarajan, P. et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 135, 2091–2101 (2017).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med 12, 44 (2020).
Drenkard, C. & Lim, S. S. Update on lupus epidemiology: advancing health disparities research through the study of minority populations. Curr. Opin. Rheumatol. 31, 689–696 (2019).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47, D1005–D1012 (2019).
Chen, G. B. et al. Across-cohort QC analyses of GWAS summary statistics from complex traits. Eur. J. Hum. Genet 25, 137–146 (2016).
Han, B. et al. A general framework for meta-analyzing dependent studies with overlapping subjects in association mapping. Hum. Mol. Genet. 25, 1857–1866 (2016).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Wang, Y. et al. Global Biobank analyses provide lessons for developing polygenic risk scores across diverse cohorts. Cell Genomics 3, 100241 (2023).
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet 88, 586–598 (2011).
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
The ENOCDE Project Consortium. A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046 (2011).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Wang, Y. et al. The 3D genome browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151 (2018).
Jung, I. et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet 51, 1442–1449 (2019).
Schmiedel, B. J. et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell 175, 1701–1715.e16 (2018).
Bryois, J. et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet. 52, 482–493 (2020).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 11, e1004219 (2015).
Wang, Z., Lachmann, A., Keenan, A. B. & Ma’ayan, A. L1000FWD: fireworks visualization of drug-induced transcriptomic signatures. Bioinformatics 34, 2150–2152 (2018).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Alexander, D. H. & Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinforma. 12, 246 (2011).
Marees, A. T. et al. A tutorial on conducting genome-wide association studies: quality control and statistical analysis. Int J. Methods Psychiatr. Res. 27, e1608 (2018).
Lee, S. H., Goddard, M. E., Wray, N. R. & Visscher, P. M. A better coefficient of determination for genetic profile analysis. Genet Epidemiol. 36, 214–224 (2012).
Morris, D. L. et al. Unraveling multiple MHC gene associations with systemic lupus erythematosus: model choice indicates a role for HLA alleles and non-HLA genes in Europeans. Am. J. Hum. Genet 91, 778–793 (2012).
Zheng, X. et al. HIBAG-HLA genotype imputation with attribute bagging. Pharmacogenomics J. 14, 192–200 (2014).
Rees, F. et al. The incidence and prevalence of systemic lupus erythematosus in the UK, 1999-2012. Ann. Rheum. Dis. 75, 136–141 (2016).
Julià, A. et al. Genome-wide association study meta-analysis identifies five new loci for systemic lupus erythematosus. Arthritis Res Ther. 20, 100 (2018).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet 47, 979–986 (2015).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet 49, 256–261 (2017).
Dubois, P. C. et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet 42, 295–302 (2010).
Glanville, K. P., Coleman, J. R. I., O’Reilly, P. F., Galloway, J. & Lewis, C. M. Investigating Pleiotropy Between Depression and Autoimmune Diseases Using the UK Biobank. Biol. Psychiatry Glob. Open Sci. 1, 48–58 (2021).
Sawcer, S. et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 (2011).
Beecham, A. H. et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet 45, 1353–1360 (2013).
Gervais, O. et al. Regional heritability mapping identifies several novel loci (STAT4, ULK4, and KCNH5) for primary biliary cholangitis in the Japanese population. Eur. J. Hum. Genet 29, 1282–1291 (2021).
Cordell, H. J. et al. International genome-wide meta-analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat. Commun. 6, 8019 (2015).
Ha, E., Bae, S. C. & Kim, K. Large-scale meta-analysis across East Asian and European populations updated genetic architecture and variant-driven biology of rheumatoid arthritis, identifying 11 novel susceptibility loci. Ann. Rheum. Dis. 80, 558–565 (2021).
Taylor, K. E. et al. Genome-wide association analysis reveals genetic heterogeneity of sjögren’s syndrome according to ancestry. Arthritis Rheumatol. 69, 1294–1305 (2017).
López-Isac, E. et al. GWAS for systemic sclerosis identifies multiple risk loci and highlights fibrotic and vasculopathy pathways. Nat. Commun. 10, 4955 (2019).
Chiou, J. et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402 (2021).
Jin, Y. et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet 48, 1418–1424 (2016).
Acknowledgements
This work was supported by the National Institutes of Health grants R01HG008983, R56HG011035, R01HG011035, R01GM126479, R21AI160138, R03OD032630, R01AI174108, R56HG012358, T32GM118294, T32LM012415, and U01AR071077. This work was also funded in part by the Penn State College of Medicine’s Artificial Intelligence and Biomedical Informatics (AIBI) Program in the Strategic Plan, the Lupus Research Alliance, CURE funds from the Pennsylvania Department of Health, and by generous support from Robert and Sevia Finkelstein. BioVU acknowledgment: The datasets used for part of the PRS analysis were obtained from Vanderbilt University Medical Center’s BioVU, which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10OD017985 and S10RR025141; and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/.
Author information
Authors and Affiliations
Contributions
C.K., L.T., B.J., and D.L. conceived the study. C.K. led the data analysis. C.K., Q.L., H.M., M.P., R.S., D.M., X.W., C.W., L.W., and S.C. conducted analyses. R.S., D.M., G.S., B.L., X.Z., N.O., and L.C. helped with data interpretation. C.K. and D.L. prepared the manuscript. All authors contributed to manuscript editing and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Leah Kottyan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khunsriraksakul, C., Li, Q., Markus, H. et al. Multi-ancestry and multi-trait genome-wide association meta-analyses inform clinical risk prediction for systemic lupus erythematosus. Nat Commun 14, 668 (2023). https://doi.org/10.1038/s41467-023-36306-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36306-5
- Springer Nature Limited
This article is cited by
-
Investigation of immune-related diseases using patient-derived induced pluripotent stem cells
Inflammation and Regeneration (2023)
-
Increased binding of anti-dsDNA antibodies to short oligonucleotides modified with topoisomerase I reveals a potential new enzyme function independent from DNA relaxation
BMC Research Notes (2023)