

Abstract
Dysfunction of pancreatic δ cells contributes to the etiology of diabetes. Despite their important role, human δ cells are scarce, limiting physiological studies and drug discovery targeting δ cells. To date, no directed δ-cell differentiation method has been established. Here, we demonstrate that fibroblast growth factor (FGF) 7 promotes pancreatic endoderm/progenitor differentiation, whereas FGF2 biases cells towards the pancreatic δ-cell lineage via FGF receptor 1. We develop a differentiation method to generate δ cells from human stem cells by combining FGF2 with FGF7, which synergistically directs pancreatic lineage differentiation and modulates the expression of transcription factors and SST activators during endoderm/endocrine precursor induction. These δ cells display mature RNA profiles and fine secretory granules, secrete somatostatin in response to various stimuli, and suppress insulin secretion from in vitro co-cultured β cells and mouse β cells upon transplantation. The generation of human pancreatic δ cells from stem cells in vitro would provide an unprecedented cell source for drug discovery and cell transplantation studies in diabetes.
Similar content being viewed by others
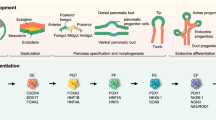


Introduction
The human islet of Langerhans is mainly comprised of three key endocrine cell types that secrete the peptide hormones insulin (β cells), glucagon (α cells), and somatostatin (δ cells). In healthy subjects, these hormone-expressing endocrine cells function within the islets of Langerhans to precisely regulate blood glucose homeostasis and energy metabolism. As blood glucose levels rise, islet β cells secret insulin to let the body make use of glucose. During hypoglycemia, islet α cells secrete glucagon to raise blood glucose levels via increasing glucose output (glycogenolysis and gluconeogenesis) in the liver1. δ cells, despite their scarcity (~ 5% within the islets), can potently suppress insulin and glucagon release via specific somatostatin receptors on β cells and α cells2,3. A recent study has revealed that paracrine signaling by δ cells determines the glycemic set point in vivo4. Therefore, the somatostatin-secreting δ cells can effectively prevent potential over-secretion of insulin and glucagon, maintaining plasma glucose levels within a narrow, physiological range. Given the high potency of somatostatin in suppressing insulin and glucagon release, it is undoubtedly that dysfunction of δ cells and/or disruption of somatostatin signaling contributes to the etiology of diabetes.
There is increasing evidence that δ cells play a role in the etiology of diabetes5,6, although β-cell dysfunction and α-cell defects are the primary contributors. In vitro exposure of islets to free fatty acids resulted in decreased glucose-induced somatostatin and derepression of glucagon secretion7. Moreover, δ-cell death (decreased volume and number) has also been reported in diabetic baboons8. somatostatin release was abolished during high glucose perfusion despite increased somatostatin content9,10. Furthermore, there is evidence that hyperglucagonemia in poorly controlled diabetes can be suppressed by somatostatin11,12. Indeed, somatostatin infusion or use of somatostatin analogues in type 1 and type 2 diabetic patients resulted in improved glycemic control despite lower insulin requirements by reducing glucagon secretion13,14. Therefore, it has been proposed that a long-acting somatostatin agonist may be useful as an adjunct to insulin therapy15. On the contrary, several early findings have shown that inhibition of somatostatin signaling improves glucagon counter-regulation, suggesting that increased somatostatin signaling may be the cause of reduced counterregulatory glucagon secretion during insulin-induced hypoglycemia in diabetic animals16,17. However, the discrepancies in the effects of somatostatin on glycemic control have not been elucidated due to the lack of δ-cell source and appropriate models. In human pancreatic islets, somatostatin receptor-2 (SSTR2) is the dominant receptor in both β cells and α cells, although other isoforms may also contribute to the effects of somatostatin18. Surface expression of SSTR2 has been shown to be reduced in type 2 diabetes, suggesting a mechanism for the observed somatostatin resistance2. In addition, the dynamics of Ca2+ spiking in δ cells are significantly altered in prediabetes when blood glucose is elevated. Similarly, in mice with prediabetes, δ cells have impaired [Ca2+]i dynamics and altered morphology, which may contribute to early stages of β-cell failure and diabetes pathophysiology6. Collectively, these lines of evidence merit growing interest in pancreatic δ cells.
Our current understanding of the pathophysiology of δ cells is limited due to their scarcity; therefore, there is an urgent need to expand the source of human δ cells. In the past decade, the generation of human pancreatic islet α cells and β cells from stem cells has been widely reported as a promising approach to provide sufficient cells for cell replacement therapy studies and drug development19,20,21. Despite no adopted method to efficiently generate pancreatic δ cells by the field, several laboratories have reported a minor proportion of δ cells during the generation of pancreatic β cells19,20,22,23,24. These advances suggest the possibility of generating δ cells from human pluripotent stem cells in a similar route.
It’s known that human δ cells develop in a common way with β cells, sharing the same initiation transcription factors pancreatic and duodenal homeobox 1 (PDX1), basic helix-loop-helix factor neurogenin 3 (NEUROG3), neuronal differentiation 1 (NEUROD1), and additional transcription factors that specify endocrine cell fate including PAX4, NKX2.2 and PAX625,26,27,28,29. δ cells are then divergent from β cells and are characterized by the absence of NKX6.126 and the exclusive presence of hematopoietically expressed homeobox (HHEX), which serves as a δ-cell-specific transcription factor30. In addition, single cell RNA-sequencing has shown that the SST +/HHEX+ cells selectively express FGFR119.
FGF signaling has been reported to regulate the expansion and differentiation of the pancreatic epithelium. In the early rodent pancreas, a number of FGF ligands and receptors have emerged. Among the eighteen FGF members, FGF2 initiates PDX1 expression during the rodent and human pancreas development31,32, while FGF10 influences bud growth and morphogenesis by promoting proliferation of PDX1 positive pancreatic progenitors33. Multiple isoforms of FGFR are present during pancreatic development, and suppression of FGFR signaling decreases PDX1 expression32. In the developing human pancreas, FGFR2 was highly expressed in trunk and ductal cells, and FGF7 and FGF9 were expressed in fibroblasts and mesothelial cells34,35,36. Thus, FGF7 has been shown to increase the number of endocrine progenitor cells but suppress their differentiation into endocrine cells within the pancreatic epithelium via FGFR2b37. In addition, FGFR1 and FGFR4 are transiently expressed during early pancreatic development and are downregulated during adulthood38. Taken together, this offers the promise of generating specific endocrine cell types by modulating FGF signaling.
Here we developed a method for the robust generation of δ cells by applying FGF7 and FGF2 during pancreatic endoderm and endocrine precursors induction stages. We showed that FGF7 helps maintain higher expression levels of PDX1 and CHGA at pancreatic endoderm/endocrine precursors stages. Strikingly, FGF2 specifically promotes the endocrine progenitor cells toward the pancreatic δ-cell lineage by inducing the mRNA and protein expression of SST and HHEX, which is mediated by FGF receptor 1 (FGFR1). Combining FGF2 with FGF7 to modulate transcription factor expression during endocrine precursors induction, we developed a differentiation protocol to generate human δ cells with in vitro and in vivo function. RNA sequencing analyses confirmed that these δ cells are partially mature and express specific δ-cell markers and ion channels. In addition, these δ cells secreted somatostatin in response to various stimuli, suppressed insulin secretion from the co-cultured β cells, and exhibited calcium oscillations in response to high glucose. Furthermore, δ cells survived after transplantation into nude mice and suppressed β-cell insulin secretion and impaired glucose homeostasis. The efficient generation of somatostatin-secreting pancreatic δ cells from human stem cells would provide a stable cell source for mechanistic studies, drug discovery and cell transplantation studies in the field.
Results
FGF7 facilitates pancreatic endoderm lineage induction
To develop a δ-cell differentiation protocol, we first aimed to generate a high proportion of pancreatic endoderm and endocrine progenitors from human pluripotent stem cells. Early ex vivo studies in mouse and human pancreas showed that FGF7 could increase the number of pancreatic endocrine progenitors via FGFR2b35,37. Therefore, the later developed methods to generate pancreatic endocrine cells from human pluripotent stem cells have used FGF7 and/or FGF10 to promote the genesis of endocrine progenitor cells from the definitive endoderm (DE)19,20,39,40. However, the dose and duration of FGF7 treatment varies in these reports. In this context, we first used different doses of FGF7 during stages 2 and 3 (Supplementary Fig. 1a) to test its effect on the induction of pancreatic endoderm and endocrine precursors, using a method optimized from published β-cell protocols19,20 (Supplementary Data 1). Our basic protocol was derived from the seven-step method of the Kieffer laboratory20, where they clearly demonstrated the presence of somatostatin-positive cells and SST gene expression. In addition, inspired by the six-step protocol of the Melton laboratory41, we modified the seven-step protocol by adding or depleting certain factors or adjusting the concentration of some key factors during several steps (e.g., RA, heparin, vitamin C, Y27632).
We first examined the impacts of FGF7 on liver and intestinal lineage directions since FGFs have been widely used to induce definitive endodermal cells during liver, intestine or pancreas development42,43. The results showed that FGF7 had no obvious effects on liver and intestinal lineage differentiation at S2D2 and S3D2, despite slight elevations in PCSK9 and MUC2, which are lowly expressed at these timepoints (Supplementary Fig. 1b, c). When looking at the pancreatic lineage markers (Supplementary Data 2), we found that with concentrations ranging from 0 to 50 ng/ml, FGF7 dose-dependently induced PDX1, NEUROG3 and NEUROD1 expression (Supplementary Fig. 1d). FGF7 at higher concentration than 50 ng/ml, however, slightly decreased the expression of these genes (Supplementary Fig. 1d). In addition, FGF7 dose-dependently induced the expression of specific endocrine cell markers, including NKX6.1, HHEX and ARX, with 100 ng/ml having the strongest effects (Supplementary Fig. 1e). Last, FGF7 showed strong dose-dependent effects on INSULIN (INS) and GLUCAGON (GCG) induction, but saturated at 10 ng/ml on SOMATOSTATIN (SST) induction (Supplementary Fig. 1f). The data suggest that higher concentrations of FGF7 could help induce pancreatic endoderm and endocrine precursors (Supplementary Fig. 1g). Thus, we chose 50 ng/ml FGF at stages 2/3 for the development of δ-cell method thereafter (Fig. 1a), and a small δ-cell fraction amongst the endocrine cell fractions was obtained (Fig. 1b).

a Schematic of the basic differentiation protocol used for planar and 3D suspension differentiations in this study. b Images of clusters at stage 7 day 14 were immuno-stained with SST (purple), INS (green), and GCG (red). Experiment was done with two replicates (differentiations). c, d RT-qPCR of pancreatic δ-cell genes expression (SST and HHEX) across the differentiation period from ES to completion of stage 6. Values are mean ± SD from 6 replicates of two independent differentiations. Mouse islets were used as a reference (purple, n = 4). e Protein expression of the pancreatic δ-cell transcription factor HHEX across the differentiation period from ES to completion of stage 6. Image from one representative experiment was shown. f Schematic of the testing protocol for generating δ cells by prolonged use of 50 ng/ml FGF7 at stages 4 and 5. Timepoints for qPCR determination were indicated by red arrowheads. g–j RT-qPCR of pancreatic endocrine cell genes expression (SST, HHEX, INS and GCG) at the completion of stages 4, 5 and 6. n = 6 independent differentiations for S4D3 and S5D3, n = 5 independent differentiations for S6D5. Individual biological replicates are shown on interleaved box and whisker plots (The central line within the box marks the median. The box represents the 25th to the 75th percentile of the distribution and the line Whiskers above and below the box indicate the 10th and 90th percentiles. The lower bound of the box is the first quartile (Q1), while the upper bound is the third quartile (Q3). The whiskers of the box and whisker plot extend from the box to the minimum and maximum observations within 1.5 times the interquartile range of the lower and upper quartile, respectively). *p < 0.05; **p < 0.01 (unpaired two-tailed Student’s t-test, FGF7 vs control). k Western blotting images showing that protein expression of HHEX was enhanced by FGF7 at the completion of stages 4, 5 and 6. l Western blotting images showing that protein expression of SST was enhanced by FGF7 at completion of stage 6. m, Immunostaining of cell clusters at stage 7 day 7 demonstrating that prolonged treatment with FGF7 promoted δ-cell generation and decreased α-/β-cell production (control cells n = 1104; FGF7 cells n = 999). (k, l, m) Images are one representative experiment of three independent differentiations. Values are mean ± s.e.m. from three independent differentiations. *p < 0.05; **p < 0.01 (unpaired two-tailed Student’s t-test, FGF7 vs control). Scale bars, 50 µm (b, m).
Prolonged FGF7 treatment induces SST and HHEX expression, with resultant increased δ-cell generation
Our next step was to see how the δ-cell marker gene SST is regulated during pancreatic development. This would provide clues as to the stages at which we could modulate δ-cell production. To this end, we examined the mRNA expression of SST throughout the differentiation period. Similar to a previous report20, SST appeared at stage 4 and increased sharply upon entry into stage 6(Fig. 1c). SST gene expression in S6D5 is approximately 10-fold lower than in adult mouse islets. (Fig. 1c). This discrepancy is likely due to the fact that the stem cell-derived δ cells are not yet fully mature, and that the mouse and human islet δ cells differ in terms of their absolute SST expression. Meanwhile, we examined the expression of HHEX, which was previously reported to be essential for the development of DE tissues (liver, intestine, and pancreas)42,43. The HHEX-expressing cells have great capacity to differentiate into derivatives of liver and pancreas44. However, it has been characterized as a gatekeeper for pancreatic lineage specification. It is co-expressed with pancreatic progenitor markers such as PDX1 and NKX6.1 in both hPSC-derived pancreatic islet models and human fetal pancreas45. Deletion of HHEX at gut-tube (stage 3) and pancreatic progenitor 1 (stage 4 and 5) results in impaired pancreatic differentiation and causes ectopic liver differentiation45. Later in the adult or mature islets, HHEX is exclusively expressed in somatostatin-producing δ cells30,45, where it acts as a key transcription factor of δ cells. Its mRNA and protein expression peaked at the end of stage 2 in parallel as the cells entered primitive gut tube stage (Figs. 1d, e). This is consistent with previous findings that mammalian Hhex (hhex) is one of the earliest markers of foregut endoderm giving rise to foregut organs42,43. HHEX mRNA and protein were then declined rapidly during pancreatic progenitor induction (stages 3 to 5) and persisted thereafter, comparable to adult mouse islets. (Figs. 1d, e), which is consistent with a previous report45. This sharp decrease could be due to the restricted expression of HHEX after entering the pancreatic progenitor stage, since HHEX (Hhex) is specifically expressed in δ cells in the adult pancreas, although it may also be present at lower levels in pancreatic ducts and some acinar cells30.
FGF7 has been widely used during stage 4 and/or stage 5 in several β-cell protocols19,20,39,40,46, because of its ability to increase the number of endocrine progenitor cells37. However, it was omitted during these stages in a recently published α-cell protocol21. In this context, it remains to be determined whether FGF7 is also required for δ-cell differentiation is to be clarified. To address this question, we examined SST and HHEX mRNA expression at the completion of stages 4, 5 and 6 (S4D3, S5D3 and S6D5, respectively) with or without 50 ng/ml FGF7 during stage 4 and 5 (Fig. 1f). We found that SST mRNA expression was enhanced at S5D3 and S6D5, and had an increasing trend at S4D3, as compared to control (Ctrl) (Fig. 1g). On the other hand, HHEX mRNA was only significantly increased at S6D5 (Fig. 1h). On the contrary, we found that FGF7 mildly decreased INS expression (Fig. 1i), whereas it remarkably suppressed GCG mRNA expression (Fig. 1j). We further examined the protein expression levels of HHEX and SST at different time points. HHEX protein expression was consistently increased by FGF7 in a dose-dependent manner (ranging from 0 to 50 ng/ml) at all time points examined (Fig. 1k); meanwhile, SST protein was also enhanced by 3.35-fold at S6D5 (0.37 ± 0.09 vs 1.24 ± 0.35, FGF7 vs Ctrl), when it became detectable (Fig. 1l). Consistent with the above mRNA and protein expression studies mentioned above, FGF7 increased the SST positive-cell fraction (17.3 ± 3.4% vs 6.3 ± 1.7%, FGF7 vs Ctrl) and decreased the GCG-positive cell fraction (36.2 ± 5.5% vs 5.1 ± 1.6%, FGF7 vs Ctrl) (Fig. 1m). In addition, FGF7 significantly increased PDX1 expression at S4D3 and S5D3, and CHGA expression at S6D5, while having no effect on NKX6.1 expression (Supplementary Fig. 2a). In line with this, further immunofluorescent and FACS studies showed that FGF7 increased the PDX1 positive fraction and CHGA+ endocrine cells (Supplementary Fig. 2b and S2c, d). Taken together, these data suggest that prolonged use of FGF7 has the potential to maintain endocrine precursors and induce SST and HHEX expression.
A screen identified FGF2, as a more potent SST inducer via activating FGFR1
FGF ligands and receptors, such as FGFR1, FGFR2, and FGFR4, play a crucial role in pancreatic development, specifically in pancreatic lineage specification and endocrine progenitor generation32,38. Mouse embryo studies have demonstrated that the bipotential precursor cell population (DE) defaults to forming the pancreas, and FGFs from the cardiac mesoderm regulate the pancreas-to-liver fate-choice during the early ventral foregut stage (E8.25)47. The proximal endoderm is directed to express liver genes, while the distal endoderm becomes pancreas, thanks to FGF signaling48. However, during E9.5-11.5, FGF10-FGFR2 signaling acts as a liver repressor to suppress hepatic competence49,50. Our analysis of the involvement of FGFs during the development of the human fetal pancreas was based on a recently published scRNA-Seq database of human fetuses34. The network analysis clearly demonstrates the involvement of FGF2, FGF7, and FGF9, along with their receptors, in all cell types during development (Supplementary Fig. 3a). Moreover, FGF2 and FGF7 are expressed in both fibroblast and mesothelial cells, while mesothelial cells exhibit high levels of FGF2, FGF7, FGF9, and FGF10 (Supplementary Fig. 3b). Notably, FGFR1-4 are highly expressed in duct and trunk, with FGFR1 being the most abundant FGF receptor expressed in δ cells (Supplementary Fig. 3c). These data support the use of FGF2, FGF7, and FGF10 to initiate PDX1 expression31,32 and promote PDX1-positive pancreatic progenitor proliferation33 during rodent and human pancreas development34,35. Moreover, FGFs have been commonly utilized to stimulate the expression of HHEX31,43, which is crucial for specifying the pancreatic lineage and suppressing liver-fate during the gut-tube stage. HHEX is co-expressed with PDX1 and NKX6.1 in both hPSC-derived pancreatic islet models and human fetal pancreas (22 wpc)45. Taken together, given that PDX151 and HHEX30 are crucial transcription factors for δ cells by modulating FGF signaling, it is promising to generate pancreatic progenitors and specific endocrine cell types. This motivated us to screen for highly potent FGFs that could induce SST expression specifically.
There are six FGF subfamilies, including FGF1(FGF1/2), FGF4(FGF4/5/6), FGF7(FGF3/7/10/22), FGF8 (FGF8/17/18), FGF9 (FGF9/16/20) and FGF19 (FGF19/21/23). To determine their potency and specificity in inducing δ cells, we selected one from each subfamily and treated differentiating cells with a series of concentrations during stage 4 and 5. The expression of pancreatic endocrine marker genes was examined at S5D3, in comparison to FGF7. FGF2 was found to be the most potent promoter of SST expression, while only mildly inducing PDX1 and CHGA expression (Fig. 2a–c). Similar to FGF7, FGF2 strongly inhibits INS and GCG expression (Fig. 2d, e). The mRNA expression of δ-cell-related genes at S4D3, S5D3, and S6D5 was examined to determine whether the effects of FGF2 are transient or long-lasting, and whether FGF2 treatment ultimately leads to premature induction of δ cells. FGF2 upregulated SST at all time points (Supplementary Fig. 4a), while HHEX was upregulated at S6D5 when the premature δ cells emerged (Supplementary Fig. 4b). Conversely, FGF2 suppressed INS expression at S6D5 (Supplementary Fig. 4c), while significantly suppressing GCG throughout differentiation (Supplementary Fig. 4d). Furthermore, it was found that FGF2 induced SST protein expression in a dose-dependent manner between 0–20 ng/ml (Supplementary Fig. 4e).

a–e RT-qPCR of pancreatic genes expression at the completion of stage 5, under control or serial doses of FGF2, FGF7, FGF8, FGF9, FGF10, FGF21 and FGF4 treatments during stages 4 and 5. Values are mean ± s.e.m. of three independent screens. f–i RT–qPCR of pancreatic endocrine cell genes expression (SST, HHEX, INS and GCG) at completion of stage 5 (FGFR1i, PD-166866, 10 μM; FGFR2i, Alofanib, 5 μM; FGFR3i, BO-264, 1 μM; FGFR4i, Roblitinib, 1 μM). Values are mean ± s.e.m. from 3 independent differentiations. *p < 0.05; **p < 0.01; **p < 0.001 (one way ANOVA followed by Tukey’s multiple comparisons test). j Western blotting images showing that FGF2 induced SST protein expression via FGFR1. Images are one representative experiment from three independent differentiations (shown in Supplementary Fig. 4f). k–m RT–qPCR of FGFR1, SST and HHEX mRNA expression at completion of stage 5 after FGFR1 siRNA or scramble RNA treatment, with or without FGF2 treatments during stages 4 and 5. Values are mean ± SD of one representative experiment from 3 independent experiments. n–p RT–qPCR of PDX1, HHEX and SST mRNA expression at completion of stage 5 in FGFR1 + /- cells (7# and 10# clones). Values are mean ± SD of one representative experiment from 2 independent experiments. k–p *p < 0.05; **p < 0.01; NS nonsignificant (unpaired Student’s T test (two-sided, two-tail), FGF2 vs Ctrl.).
To determine whether the effects are dependent on FGFR1, a receptor favored by FGF2 in humans, we used specific FGFR1-4 inhibitors to block the corresponding FGF receptor during stage 4 and 5 in the presence of FGF2. The results clearly demonstrate that only FGFR1 inhibition (PD-16686652) could abolish the effects of FGF2 on SST and HHEX induction (Fig. 2f, g), as well as on INS and GCG suppression (Fig. 2h, i). The inhibitor significantly blocked the inductive effect of FGF2 on SST protein expression (Fig. 2j and Supplementary Fig. 4f). Knocking down FGFR1 expression using a siRNA strategy at stage 4 and 5 (Supplementary Fig. 5a, b) demonstrated that FGF2 induced FGFR1, SST, and HHEX mRNA expression in scramble siRNA-treated cells but not in the FGFR1 siRNA-treated cells (Fig. 2k–m). In addition, it’s also unable to suppress INS, GCG, and PDX1 expression in the FGFR1 siRNA-treated cells (Supplementary Fig. 5c–e). Using CRISPR/Cas9, we knocked out FGFR1 in H1 stem cells. As FGFR1 is crucial for maintaining stemness in stem cells53, homozygous FGFR1-/- cells lost pluripotency while heterozygous FGFR1+/- cells maintained stemness to some extent (Supplementary Fig. 5f, g). We then compared the efficiency of δ-cell differentiation with and without FGF2 treatment in FGFR1+/- cells (7# and 10#) to that of parent H1 cells. As shown in Supplementary Fig. 4h, heterozygous cell lines (7# and 10#) successfully differentiated into DE and posterior foregut, critical for pancreatic endoderm formation. The application of FGF2 at stage 4 and 5 failed to induce the expression of PDX1, HHEX, and SST (Fig. 2n, p). On the other hand, the suppressive effects of FGF2 on INS, GCG expression were ameliorated in these two cell lines (Supplementary Fig. 5i, j), and the inductive effect on CHGA was also impaired (Supplementary Fig. 5k). These data clearly show that FGF2 induces SST expression by activating FGFR1, leading to the promotion of pancreatic δ-cell differentiation. Given that FGFR1 is predominantly and selectively present in premature SST + /HHEX+ cells19 and in δ cells of the human fetal pancreas (Supplementary Fig. 3), it is plausible to efficiently direct the generation of pancreatic δ cells by activating FGFR1.
Addition of FGF2 to FGF7 further promotes SST expression
We have shown that FGF7 promotes PDX1 and CHGA expression, thereby maintaining a high proportion of endocrine precursors (Fig.1 and Supplementary Fig. 2), and modestly induces SST and HHEX expression (Fig.1g, h, k, l). Meanwhile, activation of FGFR1 by FGF2 has been shown to facilitate δ-cell differentiation (Fig. 2). Therefore, we propose that building on the high expression levels of PDX1/CHGA achieved by FGF7, activation of FGFR1 by FGF2 could synergistically promote δ-cell generation.
To confirm our hypothesis, we investigated whether the addition of a specific FGFR1 ligand to FGF7 could effectively enhance SST/HHEX expression (Fig. 3a). We conducted a comparison of the effects of FGF2 and FGF7 alone with the combination of FGF2 and FGF7 (FGF2/7) on the expression of SST and HHEX mRNA expression. Our results showed that FGF2 significantly induced SST mRNA expression at S4D3 and S6D5 compared to FGF7, whereas FGF2/7 induced SST mRNA expression at all time points (Fig. 3b). The combination demonstrated a significantly greater ability to induce SST mRNA expression at S5D3 compared to FGF2 alone (Fig. 3b). Furthermore, both the FGF2 and combination groups exhibited a significant increase in SST protein expression compared to FGF7 (Fig. 3c). Third, FGF2 significantly increased HHEX mRNA expression only at S6D5 compared to FGF7, while the combination group increased HHEX mRNA expression at S5D3 and S6D5 (Fig. 3d). However, the protein expression pattern shown in Fig. 3e indicates that HHEX protein was drastically increased by FGF2 and FGF2/7 at all time-points. Fourth, as shown in Fig. 3f, g, FGF2 and FGF2/7 demonstrated a higher potential to suppress INS and GCG expression compared to FGF7 alone. Additionally, the combination of FGF2/7 tended to suppress both genes more effectively than FGF2 alone.

a Schematic of the testing protocol in this study. b RT-qPCR of SST at the completion of stages 4, 5 and 6. c Western blotting images showing that protein expression of SST was enhanced by FGF7, FGF2 and further by FGF2/7 at the completion of stage 6. Images are one representative experiment from three independent differentiations. d RT-qPCR of HHEX at the completion of stages 4, 5 and 6. e Western blotting images showing that protein expression of HHEX was enhanced by FGF7, FGF2 and further by FGF2/7 at completion of stages 4, 5 and 6. Images are one representative experiment from three independent differentiations. f, g RT-qPCR of pancreatic endocrine cell genes expression (INS and GCG) at the completion of stages 4, 5 and 6. (b, d, f, and g) Values are mean ± s.e.m.. n = 6 independent differentiations for S4D3 and S5D3, n = 5 independent differentiations for S6D5. * p < 0.05 (one way ANOVA followed by Tukey’s multiple comparisons test). In the interleaved box and whisker plots (b, d, f, g), the central line within the box marks the median. The box represents the 25th to the 75th percentile of the distribution and the line Whiskers above and below the box indicate the 10th and 90th percentiles. The lower bound of the box is the first quartile (Q1), while the upper bound is the third quartile (Q3). The whiskers of the box and whisker plot extend from the box to the minimum and maximum observations within 1.5 times the interquartile range of the lower and upper quartile, respectively). h Representative flow cytometry results of cells at stage 6 day 5, with FGF7, FGF2 or FGF2/7 treatments demonstrating that addition of FGF2 greatly increased the SST positive fraction. Values are mean±s.e.m. from 3 independent differentiations. *p < 0.05; **p < 0.01; ****p < 0.0001 (one way ANOVA followed by Tukey’s multiple comparison test). i Immunostaining of cell clusters at stage 7 day 7 with SST (purple), INS (green), and GCG (red). Results show that adding FGF2 to FGF7 greatly promoted δ-cell generation and decreased α-/β-cell production (n = 3 independent differentiations; control cells n = 729, FGF7 cells n = 779, FGF2 cells n = 708, FGF2/7 cells n = 734). Scale bars, 50 µm. j–m RT-qPCR of LEPR, HER4, FGFR1 and FGFR2 gene expression levels at the completion of stage 6. Values are mean ± SD from one representative experiment. *p < 0.05; **p < 0.01 (one way ANOVA followed by Tukey’s multiple comparisons test).
To confirm the requirement of FGF2/7, we conducted a comparative analysis of HHEX, PDX1, and SST mRNA expression at S5D3 and S6D5 between the treatment of stage 4 plus 5 and the treatments of stage 4 or stage 5 alone. Addition of FGF2/7 in either stage 4 or stage 5 resulted in the induction of HHEX, PDX1, and SST mRNA expression compared to the control group. However, the effects were significantly enhanced when FGF2/7 were used in both stages (Supplementary Fig. 6a). In addition, the combination treatment showed similar effects on PDX1, NKX6.1 and CHGA expression as compared to FGF2 or FGF7 alone (Supplementary Fig. 6b, c). These gene expression profiles suggest that the addition of FGF2 to FGF7 would benefit δ-cell production as this treatment is expected to result in a larger δ-cell fraction but smaller α- and β-cell fractions.
The FACS analysis results from three independent differentiations demonstrate that all FGF-treatment groups increased the SST+ population significantly (29.5% of FGF2/7, 18.2% of FGF2, and 13.3% of FGF7 vs 6.9% of Ctrl, respectively). Although not significant, FGF2 resulted in a larger SST+ population compared to the FGF7 group (18.2% of FGF2 vs 13.3% of FGF7). Moreover, the combination group exhibited a significant increase in the SST+ fraction in comparison to FGF7 (29.5% of FGF2/7 vs 13.3% of FGF7) or FGF2 alone (29.5% of FGF2/7 vs 18.2% of FGF2) (Fig. 3h). Similarly, the combination group exhibited a higher number of SST-positive cells (27.1% for FGF2/7, 19.3% for FGF2, 15.8% for FGF7, and 7.2% for Ctrl) in accordance with the gene expression, FACS, and immunofluorescent data (Fig. 3i, left). The combination of FGF2 and FGF7 resulted in a significant decrease in GCG-positive cells (9.1%) compared to the control group (32.6%) and the FGF7 (10.1%) and FGF2 (7.3%) groups (Fig. 3i, right). The fractions of INS-positive cells were also significantly decreased by FGF2/7 (FGF2/7, 21.8%; FGF2, 18.9%; FGF7, 19.3%; Ctrl, 29.7%) (Fig. 3i, middle). The expression of typical δ-cell surface marker genes, including leptin receptor (LEPR) and human epidermal growth factor receptor 4 (HER4)54, was also examined. Figures 3j and 3k demonstrate a significant increase in the expression of LEPR and HER4 in FGF2 and FGF2/7 groups (Fig. 3j, k). The combination treatment also induced a significant expression of FGFR1 mRNA (Fig. 3l) and protein (Supplementary Fig. 6d), as well as LEPR and HER4, indicating an enhancement of SST+/HHEX+ pre-δ cells by the combination treatment. On the other hand, FGFR2 mRNA (Fig. 3m) and protein (Supplementary Fig. 6e) expression were consistently down-regulated by FGFs treatment. Given that FGFR2 is highly expressed in non-endocrine cells19, the data suggest that FGF2/7 treatment could limit non-endocrine cell populations, thereby enhancing the production of δ cells. These findings demonstrate that the use of FGF2 and FGF7 during pancreatic progenitor induction works together to efficiently generate SST+ δ cells.
Next, we examined additional δ-cell marker genes including ESE3B, ETV1, GABRG2, POU3F1, PDLIM4, SHARP1, LEDGF, NEC1, and SFRP3 and found that most of them were significantly induced by FGF2/7 compared to the control group (Supplementary Fig. 7a). Since enterochromaffin-like cells (EC-like cells) are a known off-target of β-cell differentiation19, we wondered what happened to this cell type in the presence of FGF2/7 in the present protocol. We examined a panel of EC-like cell markers including TPH1, LMX1A, SLC18A1, TRPA1, and DDC. FGF2/7 significantly downregulated the expression of TPH1, LMX1A, SLC18A1, and DDC at both S5D3 and S6D5, whereas TRPA1 was downregulated only at S6D5 (Supplementary Fig. 7b).
Based on the screening study, we observed that FGF4 and FGF21 could induce PDX1 and CHGA expression and FGF4 also enhanced SST expression (Fig. 2a–c). It is possible that other combinations could also efficiently generate δ cells. In this regard, we compared all the possible combinations with FGF2, FGF4, FGF7 and FGF21 and examined the effects on PDX1, CHGA, SST, NKX6.1, INS and GCG at S4D3 and S5D3. The data showed that FGF2/7 consistently increased PDX1 induction, although FGF7/4 and FGF7/21 also strongly induced PDX1 at S4D3 and S5D3, respectively (Supplementary Fig. 8a). FGF2/4 and FGF7/4 consistently induced CHAG expression at both stages (Supplementary Fig. 8b). However, FGF2/7 had the greatest effect on SST induction at both S4D3 and S5D3, whereas FGF7/4 and FGF4/21 had weaker effects (Supplementary Fig. 8c).
To evaluate the robustness of our protocol in other human pluripotent stem cells, we differentiated H9 embryonic stem cells and UE005 inducible pluripotent stem cells (iPSC) and examined the expression levels of critical genes in both control and FGF2/7 groups. In these two cell lines, FGF2/7 similarly and significantly induced the expression of PDX1, HHEX, SST and NEUROD1 expression (Supplementary Fig. 9a), resulting in higher SST protein expression (Supplementary Fig. 9b) and greater numbers of SST+ cells (Supplementary Fig. 9c). Compared to H1, H9 showed higher levels of HHEX and SST expression at S5D3, while the UE005 iPS cell line showed lower levels of these two genes (Supplementary Fig. 9a, Fig. 3b–d).
FGF2/7 synergically directs pancreatic δ-cell induction
Bulk RNA sequencing was conducted at S5D3 and S7D6 to comprehensively understand the impact of FGFs, specifically FGF2/7, on endocrine cell fate determination. A heat map of the 1000 most differentially expressed genes demonstrates that the combination treatment produced the most distinct profile compared to the control (Figs. 4a, b). Specifically, the combination treatment shifted the gene expression profile of cells toward that of the FGF2-treated cells at S5D3 and S7D6 (Fig. 4a, b). However, it specifically upregulated a number of genes at S7D6 (Fig. 4b and Supplementary Data 3). Among these genes, LMX1B functions downstream of NEUROG3 as a critical transcription factor for human islet endocrine cell generation55,56 and CRABP1/2 functions in retinoic acid signaling57. Next, we used a published single-cell dataset as a predefined, cell type signature gene set58 to examine gene alterations between all groups. A significant positive normalized enrichment score (NES) value indicates that members of the gene set tend to appear at the top of the ranked transcriptome data, and a significant negative NES indicates the opposite. The data showed that the FGF-treated groups were enriched in the endocrine cell transcriptome (identity) but not in the non-endocrine (acinar and ductal) cell transcriptome (Fig. 4c). Based on these data, we conclude that FGFs promote endocrine cell differentiation while dampening non-endocrine cell generation. However, all FGFs treatment cells similarly enriched in the δ-cell gene set (Fig. 4c), suggesting that FGF2/7 did not produce distinct δ cells as compared to FGF2 or FGF7 alone. We further investigated the impacts of FGF treatments on δ-cell differentiation. Most δ-cell markers were expressed at low levels at S5D3, and the FGF treatment groups showed little or no effects. In contrast, at S7D6, FGF-treated groups consistently increased most of the markers (Fig. 4d). FGF2/7 had greater effects on EGR1, HHEX, ADGRL2, ERBB4, CADM1, MS4A8 and NSG1 compared to FGF2 or FGF7 alone (Fig. 4d, with green asterisks). However, no significant differences between FGF2/7 and FGF2 or FGF7 alone for most of the other marker genes were found, possibly due to that these marker genes are also expressed in other cell types58. For instance, PDX1, RBP4, SEC11C and PCP4 are also abundantly expressed in β cells, HHEX is expressed in δ and ductal cells, and UCP2, TPP3 and many others are expressed in α and δ cells. Given that FGF2/7 exerted a significant influence on these cells, the overall gene expression profile derived from bulk RNA sequencing may not align with the observed change in δ-cell number. In addition to δ cells, FGF treatments suppressed acinar and ductal cell differentiation (Supplementary Fig. 10a–f) and promoted maturation of other endocrine cell types. FGF treatments decreased several α/ε-cell-specific markers, including GCG, CD26, BHMT, and ASGR1. Conversely, it was found to globally increase β/γ-cell markers, with FGF2/7 exhibiting the most significant effects on some specific marker genes (Supplementary Fig. 10a–d). FGF2/7 treatment also significantly decreased the cholesterol pathway at both S5D3 and S7D6, as demonstrated by further KEGG analysis (Supplementary Fig. 10h–g).

a, b H1 cells differentiated to stage 4 were untreated, treated with FGF7, FGF2 or FGF2/7 throughout stage 4 and stage 5. Bulk RNA sequencing at the completion of stage 5 (S5D3, n = 5) and 6 days into stage 7 (S7D6, n = 4) were used to generate a heat map of the 1000 most differentially expressed genes between the FGF7 group and the control group. A set of genes that were specifically upregulated by FGF2/7 at S7D6 were marked and listed in Supplementary Data 3. c Gene set enrichment analysis from bulk RNA sequencing of select gene sets from multiple pancreatic cell types. d A heat map from bulk RNA sequencing of select genes from reported δ-cell transcription, top markers, surface markers and others. Volcano plots from bulk RNA sequencing data showing expression differences of select genes between FGF2/7-treated cells and cells treated with FGF2 or FGF7 alone at S5D3 (e, f) and S7D6 (g, h) (log2FC cutoff >0.5 or <−0.5; P value < 0.05).
To visualize the preferred cell fate choice during endocrine progenitor induction and cell maturation with FGF2/7 treatment, we generated volcano plots of marker genes for multiple cell types that may emerge from the protocol. The volcano plots clearly showed that S5D3 cells of the FGF2/7 group had lower levels of GCG, INS, CDX2, AFP and ALB, similar levels of ARX, PRSS1 and KLF5, and higher levels of FGFR1, HHEX, SST (not significant) and NEUROG3 as compared to that of FGF7 alone (Fig. 4e). Compared to FGF2, the FGF2/7 group had lower levels of INS, AFP, PNP, GCG and CDX2, similar levels of ARX, FGFR1 and KLF5, and higher levels of PDX1, HHEX, SST and NEUROG3 (Fig. 4f). At S7D6, the FGF2/7-treated cells had higher expression levels of HHES and SST (not significant) but lower levels of GCG, KRT19 and AFP, despite comparable levels of INS, ARX, FGFR1 and PDX1 as compared to that of FGF7 alone (Fig. 4g). Compared to FGF2, FGF2/7 slightly decreased GCG, KRT19, PRSS1 and CDX2, mildly increased HHEX, FGFR1 and SST, and significantly increased HHEX (Fig. 4h). The data showed that FGF2/7 suppressed α-/β-cell, but induced δ-cell differentiation, and that it directed the differentiation toward pancreas rather than liver or intestinal cell fate. In conclusion, our research suggests that FGF2/7 plays a crucial role in promoting δ cell differentiation probably through three distinct mechanisms: directing differentiation towards pancreatic cell fate; controlling the differentiation of cells into pancreatic endocrine precursors and suppressing non-endocrine cell differentiation; promoting δ-cell progenitors while suppressing α- and β-cell differentiation.
To uncover the potential mechanisms by which FGF2/7 promotes the differentiation of δ cells, we performed a regulon analysis using the pySCENIC package to infer the gene regulatory networks (GRN)59. By assuming that the different cell types maintain the same regulatory mechanisms, we would be able to use this method to predict the regulons at the sample level60. This would help to identify the master regulators that determine cell fate at multiple stages. First, we compared all FGF treatment groups to the control group and identified the significantly altered regulons. The top regulons at both time points are shown in Fig. 5a. Interestingly we found that about 40 regulons were more active at S7D6 while 32 regulons were more active in at S5D3 (Fig. 5a). Next, we extracted the top regulons higher than 2.0 (fold change FGF treatment vs. control) and plotted them in a heat map (Fig. 5b, c). We found that FGF2/7 significantly induced δ-cell transcription factors including MESI1, ETV1, BHLHE41 and EHF61 at both stages in comparison to the control group. It also significantly induced MESI1 and BHLHE41 at S5D3 in comparison to FGF2, and significantly induced all these genes at both stages in comparison to FGF7 (Fig. 5d). In addition, FGF2/7 significantly induced the somatostatin upstream enhancer-binding factors CREB1 and PBX162,63 at S4D3 and S5D3 compared to the control group. In comparison to FGF7, the combination group exhibited a significant increase in the expression of these two genes. However, the induction of PBX1 expression was only observed at S5D3 in the combination group, in comparison to FGF2 (Fig. 5e). Together with the data that PDX1 and HHEX were upregulated by FGF2/7, these results suggest that FGF2/7 may induce δ-cell differentiation by upregulating multiple transcription factors and enhancer activators, although the mechanism is unclear.

a Regulon analysis for the cells treated with control or FGFs at S5D3 and S7D6. Top regulons were listed in the heatmap. b, c Heatmaps showing the changes of posterior foregut lineage-related regulons and pancreatic lineage-related regulons under FGFs treatments. d RT-PCR of δ-cell transcription factor gene expression (MEIS1, ETV1, BHLHE41 and EHF) at the completion of stage 5. Values are mean ± s.e.m. from 5 independent differentiations. *p < 0.05; **p < 0.01 (one way ANOVA followed by Tukey’s multiple comparisons test). e RT-PCR of δ-cell SST gene activator expression (CREB1, and PBX1) at the completion of stage 5. Values are mean±s.e.m. from 5 independent differentiations. *p < 0.05; **p < 0.01 (one way ANOVA followed by Tukey’s multiple comparisons test). f, g Heatmaps showing the changes of posterior foregut lineage-related regulons and pancreatic lineage-related regulons under FGFs treatments. h A schematic diagram showing how FGF2 and FGF7 facilitate the differentiation of pancreatic δ cells from human pluripotent stem cells (PFG posterior foregut, PE pancreatic endoderm, EP endocrine precursors;—indicates suppression while + indicates induction; blue color for FGF7 and red color for FGF2; double—or + indicates stronger effects).
In addition, several cell fate-related regulons, in particular NR5A2, TBX3, CDX1, CDX2 and HNF4A, were consistently downregulated by FGF2/7 treatment at both stages (Fig. 5f, g). At S5D3, TBX3 and its regulatory genes HNF4A and CDX2, were significantly decreased by FGF2/7 treatment (Supplementary Fig. 11a). HNF4A, as a master transcription factor for the liver and endocrine pancreas during organ development, has been found to inhibit differentiation of the liver lineage, while not affecting pancreatic progenitors in human stem cell modeling strategies64,65. These data may partially explain why FGF2/7 suppressed the intestinal cells (FABP1, GASTRIN, REG1A, DMBT1, LGR5, MUC2) and liver cells (ALBUMIN, LDR, PSK9, KRT7) (Supplementary Fig. 11b, c). Taken together, these data indicate that FGF2/7 promotes pancreatic endoderm/progenitor formation while inhibiting intestinal and liver differentiation. These data also partially explain the reason for PDX1 induction (Supplementary Fig. 2a, b, and 6b), as loss of TBX3 enhances pancreatic progenitor generation from PSCs via de-repression of PDX1 and other pancreatic progenitor marker genes66.
Furthermore, FGF2/7 significantly downregulated NR5A2 and its target genes PRSS1, KRT19 and GATA4 (Supplementary Fig. 11d). NR5A2 is a master gene that controls multiple stages of pancreatic development, including the maintenance of the exocrine phenotype and the formation of bipotent cells with ductal and islet endocrine potential67. The pancreas deficient in NR5A2 exhibited defects in acinar and ductal tissue, as well as a slight decrease in α-/β-cells. FGF2/7 directs cell differentiation towards pancreatic endocrine progenitors instead of non-endocrine cells, according to the data.
The treatment resulted in significant alterations to key regulons related to endocrine cells. Specifically, at S4D3 and S5D3, PAX4 and SOX9 were upregulated, while NKX2.2, PAX6, and MAFB were downregulated (Fig. 5a, f). Additionally, at S7D6, NEUROD1, SOX9, and NKX2.2 were upregulated (Fig. 5a, g). It is worth noting that SOX9, a downstream regulator of FGF signaling68, controls adoption of an endocrine fate69 and maintains pancreatic ductal identity70. During pancreatic development, SOX9 functions as a multipotency maintenance gene to maintain the pool of pancreatic progenitor cells71. Pancreatic endocrine cells are primarily derived from Sox9+ bipotent progenitors in the stem epithelium71. Sox9 triggers endocrine differentiation by inducing Neurog3 expression, which in turn induces Neurod1 expression71,72. In light of this, we compared SOX9 mRNA expression levels at both stages with or without FGF2/7 treatment. We found that 1) SOX9 expression was enhanced by FGF2/7 treatment at S4D3 and S5D3 (Fig. 5f and Supplementary Fig. 11e), and 2) SOX9 had a pronounced decrease at S7D6 compared to that at S5D3 (Fig. 5a, g). This is consistent with a previous report that SOX9 is restricted to a subset of PDX1-positive progenitors and is absent from committed endocrine progenitors or differentiated cells71.
The loss of SOX9 leads to hepatic cell fate choice49, while its cooperation with PDX1 suppresses genes that encode intestinal cell fate regulators73. This, in turn, enhances the effect of FGF2/7 treatment in restricting intestinal differentiation (Supplementary Fig. 11b). As anticipated, FGF2/7 treatment at S5D3 reduced NKX2.2, which is present in pancreatic α and β cells but not in δ cells, indicating defects in α and β cell differentiation. The combination treatment at S5D3 resulted in a significant increase of PAX4 and a corresponding decrease of PAX6 (Supplementary Fig. 11e). These findings strongly support the hypothesis that FGF2/7 actively promotes the differentiation of endocrine progenitors, with a particular emphasis on β and δ cells. PAX4 is a well-known suppressor of ARX and plays a crucial role in the genesis of β and δ cells, while PAX6 is primarily involved in the differentiation of all endocrine cell types, with a predominant effect on α cells through the regulation of proglucagon and MAFB.
In summary, based on the bulk RNA-seq data and subsequent qPCR analyses, we proposed a model depicting how FGF2 and FGF7 synergistically regulate pancreatic δ-cell differentiation (Fig. 5h). In the combination of FGF7 and FGF2, FGF7 increased PDX1 expression and mildly suppressed liver and intestinal differentiation by downregulating the TBX3 and NR5A2 regulons, whereas the addition of FGF2 synergistically inhibited these two regulons, resulting in more pronounced suppression of liver/intestinal and non-endocrine differentiation. In addition, activation of the δ-cell-specific FGFR1 by FGF2 significantly and robustly enhanced pancreatic δ-cell lineage differentiation while suppressing α- and β-cell lineage differentiation. Finally, FGF2 and FGF7 enhanced additional δ-cell transcription factors ETV1, MEIS1, BHLHE41, and EHF, and SST enhancer activators PBX1 and CREB1.
δ-cell characterization and in vitro function
Subsequently, we characterized the SST-positive cells generated by our protocol (Fig. 6a). First, given that NKX6.1 is absent in the adult pancreatic δ-cells, we determined whether these δ cells were indeed premature by examining the presence of NKX6.1 in the SST+ cells at S7D6. Immunostaining results showed that almost all the SST-positive cells were negative for NKX6.1 (Fig. 6b). Second, HHEX has been reported to be a critical transcription factor in adult δ cells30,45. With this in mind, we examined the co-expression of HHEX with SST. HHEX was present in almost all SST-positive cells (92%, Fig. 6c), which is similar to that observed in FGF7 treated clusters (Supplementary Fig. 12, 91%), although some of the HHEX-positive cells were SST-negative (Fig. 6c), which may be due to the presence of pancreatic ductal cells74. Third, we utilized transmission electron microscopy (TEM) with the SST-P2A-mCherry reporter line to examine the morphology of secretory granules of purified δ cells (Supplementary Fig. 13a–f). The δ cells displayed finely granular cores (Fig. 6d), resembling those of human primary δ cells75. Subsequent FACS analysis revealed that the SST+ cells also expressed the δ-cell transcription factor PDX161 (Fig. 6e), and the endocrine marker CHGA (Fig. 6f). Moreover, most of the SST+ cells were negative for GCG (4.6% colocalization) or INS (6.2% colocalization). Additionally, SST+ cells did not colocalize with CK19 or 5-HT (Supplementary Fig. 14a, b), indicating that these cells are not ductal cells76 or intestinal EC/ enteric serotonergic neurons77.

a Schematic of the final δ-cell differentiation protocol. Immunostaining to characterize the generated δ-cells at stage 7 day 6. SST (green) and NKX6.1 (red) in (b), SST (green) and HHEX (red) in (c). The ratio indicates the percentage of SST positive cells that are also HHEX positive (c, n = 1183 cells, two biological replicates). d Transmission electron microscopes (TEM) to examine the morphology of secretory granules of purified δ cells by using the SST-P2A-mCherry reporter line (S7D14). Images are one representative experiment from two independent differentiations (b-d). e–h Flow cytometry to characterize the generated δ-cells at stage 7 day 6. Co-expression of SST with PDX1, CHGA, GCG or INS was analyzed. i δ-cell in vitro function was assessed by measuring somatostatin secretion under high glucose (20 mM, 20 G) or potassium chloride (30 mM, 1 G + KCl). n = 7 biological replicates from 3 independent differentiations. Values are mean ± s.e.m. *p < 0.05; **p < 0.01 (one way ANOVA followed by Tukey’s multiple comparisons test). j δ-cell somatostatin secretion in response to tolbutamide (10 mM, 1 G + Tol), human recombinant UCN3 (100 nM), GCG (10 nM) and L-arginine (20 mM). n = 5 biological replicates from 3 independent differentiations. Values are mean ± s.e.m. *p < 0.05; unpaired Student’s t-test (two-sided, two-tail). k β-cell insulin secretion was suppressed by somatostatin secreted from cocultured δ cells. β+δ cell mixture (1:1) or pure β cells were sequentially treated with 1 mM glucose (1 G), 20 mM glucose (20 G), and 20 mM glucose +200 nM CYN-154806 (20 G + CYN), insulin secretion levels after each treatment were determined. n = 3 independent experiments. Values are mean ± s.e.m. *p < 0.05; **p < 0.01; **p < 0.001 (one way ANOVA followed by Tukey’s multiple comparisons test). l Ca2+ imaging analysis was done in S7D14-21 δ cells (SST-P2A-mCherry reporter lines). Replated δ cells were incubated in S7 medium containing 5 μmol/L Fluo-8 and Ca2+ imaging was performed by recording of Ca2+ dynamics on live images (mCherry-positive) every 2 seconds for 5 min at 2.8 mM glucose (LG) and 10 min at 20 mM glucose (HG). The mean values of ΔF during the first 5 min (F0) were used to normalize ΔF. The Ca2+ signals of δ cells were represented by ΔF/F0.
Another important characteristic of δ cells is their functional maturity. Mature pancreatic δ cells secrete somatostatin (SST) in response to high glucose, potassium chloride and tolbutamide, a KATP-channel inhibitor78. To this end, we measured the somatostatin release in the presence of 1 mM glucose (1 G), 20 mM glucose (20 G) and 30 mM KCl (1 G+KCl). High glucose and KCl both significantly increased somatostatin secretion, with KCl having a greater effect (Fig. 6i). Tolbutamide (Tol) could also stimulate somatostatin release (Fig. 6j, left). Urocortin3 (UCN3), L-arginine, and glucagon (GCG) were also tested for their effects on somatostatin secretion. These findings demonstrate the efficacy of Tolbutamide, UCN3, and glucagon in stimulating somatostatin secretion. The results showed that 100 nM UCN3 induced SST secretion (Fig. 6j, middle), which is consistent with a previous report5. In addition, 10 nM glucagon also promoted SST secretion (Fig. 6j, middle), as reported by Svendsen et al. using perfused mouse pancreas79. Stimulation of SST secretion was observed with 20 mM L-arginine, which is similar to that in isolated mouse islets80 (Fig. 6j, right). On the other hand, somatostatin secreted by pancreatic δ cells suppresses β-cell insulin release mainly through the β-cell receptor SSTR218, so as to cooperatively control blood glucose levels within a narrow physiological range. Therefore, measuring insulin secretion from β cells that are co-cultured with purified δ cells in the absence or presence of the SSTR inhibitor CYN-154806 (CYN) would allow us to assess the in vitro function of δ cells. To this end, we used reconstructed organoids containing β cells alone or β cells and δ cells to perform a GSIS assay under a series of conditions: low glucose (1 G), high glucose (20 G) and high glucose plus CNY (20 G + CYN). Insulin secretion was approximately 1.5-fold higher under high glucose, and this effect was further enhanced by the addition of CNY (Fig. 6k, left), whereas CYN had no effect on insulin secretion in the control β-cell-only organoids (Fig. 6k, right). These results suggest that blocking SSTR2 alleviates the tonic inhibition by somatostatin.
Finally, pancreatic δ cells secrete somatostatin in a Ca2+-dependent manner involving the influx of extracellular Ca2+ and the mobilization of intracellular Ca2+ oscillations in response to high glucose78,81. Therefore, Ca2+ spiking dynamics and morphology reflect the functional maturity of δ cells6. Therefore, we performed calcium imaging assays on the δ cells (SST-P2A-mCherry reporter line) and found that a fraction of these δ cells exhibited a dynamic calcium oscillation in response to high glucose (20 G) (Fig. 6l, top), although a fraction of the δ cells showed no calcium response (Fig. 6l, bottom). Collectively, all these above results demonstrate that the δ-cells are functionally mature.
Molecular characterization of δ cells
To further characterize the molecular features of the δ cells generated by our protocol, we performed single-cell RNA sequencing. We used the ScType computational platform to unbiasedly and accurately annotate the cell types82. This platform allows cell type identification based solely on our scRNA-Seq data, along with a comprehensive database of primary islet cell markers as background information. The islet organoid cells were identified and clustered as progenitor cells (0), ductal cells (1), α-like cells (2), mesenchymal cells (3), polyhormonal cells (4), EC-like cells (5), δ-like cells (6), β-like cells (7), and acinar cells (8) (Fig. 7a). Next, our analysis of 47 islet-specific genes compared to human islet expression patterns further confirmed the similarity of δ-like cells to primary human δ cells (Fig. 7b). Most of the δ-cell top markers are present in our δ-like cells. Among them, SST, RGS2, and SEC11C are expressed at levels similar to adult δ cells, while others, including HHEX, BCHE, RBP4, FFAR4, PRG4 and SLC38A1 are expressed at lower levels compared to adult δ cells. In addition, δ-like cells expressed higher levels of IAPP and PDX1 compared to adult δ cells, which were also expressed in adult δ cells58,83 (Fig. 7b). However, the expression profile of δ-like cells is quite different from that of EC-like cells.

a UMAPs showing identified cell types from δ cells at stage 7 day 6. b Heat map showing gene expression of markers associated with each cell type in comparison with human adult α, β, and δ cells. Top 13 genes are α-cell specific, middle 24 genes are β-cell specific, and bottom 10 genes are δ-cell specific. c Heat map showing gene expression of pancreatic islet ion channels and cell cycle-related genes in each cell type in comparison with human adult α, β, and δ cells. Top 15 genes are adult islet cell ion channels, and the bottom 2 are cell-cycle marker gene and inhibitor gene. Coloring is based on z-score from 1.5 as high (yellow) to −1.5 as low (purple).
Furthermore, we wondered whether these δ-like cells express ion channels similarly to adult human δ cells by analyzing 13 ion channels in comparison to primary human δ cells. First, the islet alpha/beta enriched channels CNCNA1A/B/C/D are similarly absent in δ-like cells and adult δ cells (Fig. 7c, top). Second, adult δ-cell channels KCNN2, RYR1/381, SCN3A, and especially KCNK16 were present in δ-like cells, at slightly lower levels (Fig. 7c). KCNK16 (TALK-1) is abundant in human δ cells, where it modulates glucose-stimulated changes in cytosolic Ca2+ and somatostatin secretion84. However, some ion channels enriched in adult δ cells, including ANO1, KCNN3, and RYR2, were extremely low in the δ-like cells (Fig. 7c). In addition, the cell cycle-associated genes TOP2A was slightly higher in δ-like cells compared to adult islet δ cells, whereas the cell cycle inhibitor gene CDKN1C was dramatically higher in δ-like cells (Fig. 7c, bottom). Nevertheless, these results suggest that the δ-like cells generated by this protocol are similar to a primary δ-cell signature.
In vivo characterization of δ-cell transplantation
To determine the importance of the production of these cells, we transplanted the sorted δ cells (approximately 3 million per mouse) into kidney capsule of nude mice (Fig. 8a–c). Six weeks after transplantation, most of the δ cells were still alive, as shown by immunostaining with SST and CHGA (Fig. 8d). We monitored blood glucose levels weekly and found that there was no significant difference between the sham control group and the δ cell-transplanted group at all time points, although a smaller blood glucose fluctuation was observed in the δ-cell-transplanted group (Fig. 8e). One of the important physiological functions of pancreatic δ cells is to suppress insulin secretion in response to high glucose54,79,81, and even exogenous somatostatin could suppress insulin secretion85. Therefore, we evaluated the in vivo function of δ cells by assessing glycemic control and insulin secretion in response to glucose challenge. Since the somatostatin ELISA kit used in this study recognizes human and mouse somatostatin (both SST-14 and SST-28), we observed significant increases in somatostatin pools in both groups. In the fasting state, there was an increasing trend in somatostatin in the transplanted group, but 20 min after glucose loading, somatostatin was significantly increased in the transplanted group (Fig. 8f). In support of this, we also observed a reduction in insulin secretion 20 min after glucose loading (Fig. 8g) and higher blood glucose levels at the 15 min and 30 min time points (Fig. 8h) in the δ-cell-transplanted animals. All these data indicate that the δ cells are functional after transplantation.

a-c The procedure of transplantation study of δ cells. S7D7 δ cells were sorted by FACS, aggregated and cultured for 3 days in AggWell plates. A total of 500 clusters (about 3 × 106 cells) were transplanted underneath the kidney capsule (n = 8 animals for each group). d Six-week post-transplantation, the graft was taken, fixed and co-stained with CHGA and SST. e Random blood glucose levels were monitored weekly. Values are mean±S.D., *p < 0.05; unpaired Student’s t- test (two-sided, two-tail). f, g Six-week post-transplantation, mice were fasted for 6 h and then challenged with glucose (2 g/kg body weight). Serum was separated from tail vein whole blood, total somatostatin and mouse insulin levels were determined immediately (n = 6 for each group). *p < 0.05; NS nonsignificant (Two-way ANOVA followed by Sidak’s multiple comparisons test). h Glucose tolerance test in control mice (n = 6 animals) and δ-cell-transplanted mice (n = 8 animals). Area under the curve was calculated by the GraphPad software. All data are presented as mean ± SEM. In the interleaved box and whisker plots, the central line within the box marks the median. The box represents the 25th to the 75th percentile of the distribution and the line Whiskers above and below the box indicate the 10th and 90th percentiles. The lower bound of the box is the first quartile (Q1), while the upper bound is the third quartile (Q3). The whiskers of the box and whisker plot extend from the box to the minimum and maximum observations within 1.5 times the interquartile range of the lower and upper quartile, respectively. *p < 0.05; NS nonsignificant (Unpaired two-sided Student’s T test, transplanted vs sham).
Discussion
The cell replacement approach to diabetes treatment has advanced the field of regenerative medicine by generating and clinically testing two major cell types, the α-cell and the β-cell, that play critical roles in the disease in diabetic patients19,20,21,86. The addition of other physiological cell types would further advance the treatment of diabetes by enhancing glycemic control, which is coordinated by the interplay of multiple islet cell types.
Blood glucose is increased by glucagon from α-cells, and decreased by insulin from β cells. It is well-established that δ cells play a crucial role in regulating blood glucose by balancing insulin and glucagon secretion, as demonstrated by numerous studies3,18,78,81,87,88. Although the α-cells establish a setpoint for insulin secretion through paracrine signaling, δ cells are important modulators to regulate insulin secretion by β cells6,18, and to influence α-cell activity by coupling with β cells89. On the other hand, the mechanism by which glucose regulates somatostatin secretion has been attributed to both intrinsic and paracrine effects exerted by α- and β-cells. Furthermore, the intra-islet regulatory network for δ cells is altered in diabetes. First, the surface expression of SSTR2 is reduced in type 2 diabetes, leading to potential somatostatin resistance2. Second, Ca2+ spiking dynamics and morphology in δ cells are significantly altered in prediabetes6. However, the detail counter-regulatory network of these three cells types and the alterations of δ cells during the development of diabetes remain elusive. Therefore, the generation of human δ cells is of paramount interest to both basic research and regenerative medicine.
The application of FGF7 helps to promote PDX1 expression, resulting in a larger PDX1+ fraction. On the other hand, activation of FGFR1 by its selective ligand FGF2 induces δ-cell generation, since FGFR1 is predominantly and specifically expressed in pancreatic δ cells18. Therefore, the application of FGF2 and FGF7 directs the cells to differentiate into pancreatic δ cells. We observed synergistic effects of FGF2/7 on δ-cell differentiation, likely because FGF7 induces SST through mechanisms other than FGFR1 activation, and because FGF2 and FGF7 both suppress INS and GCG expression. Subsequent bulk RNA-seq analysis revealed that FGF2/7 suppresses liver and intestinal organogenesis, probably by upregulating HHEX, a newly discovered gatekeeper of pancreatic lineage specification45, during the transition from DE to PP (pancreatic progenitors). In addition, FGF2/7 suppressed non-endocrine differentiation by downregulating NR5A2, which was underscored by decreased FGFR2 expression19,90. Moreover, the combined use of FGF2 and FGF7 promoted several δ-cell transcription factors and somatostatin enhancer activators (Fig. 5g). These δ cells are positive for HHEX and PDX1, and resemble adult human pancreatic δ cells by expressing key identity markers and ion channels, as demonstrated by single-cell RNA sequencing. Finally, somatostatin secretion from these δ cells responded to high glucose, potassium chloride, tolbutamide, UCN3, arginine and glucagon, and suppressed insulin secretion of β cells. More importantly, these δ cells are able to function in vivo to modulate glucose homeostasis.
In the present study, we have established a method to efficiently and directly generate pancreatic δ cells from human stem cells. Although several β-cell protocols can generate a small fraction of δ cells as a byproduct (typically less than 10%)19,91,92, they are neither direct nor efficient. More importantly, our study identified a mechanism involving FGFR1, by which δ cells can be specifically induced. This will allow further optimization of δ-cell protocols by modulating FGFR1 signaling or mechanisms other than FGFR1 activation. Finally, by using single cell RNA sequencing, we demonstrated that these δ cells are transcriptionally identical to primary islet δ cells and have in vivo and in vitro functions.
With FGF2 and FGF7 treatment, HHEX protein was dramatically increased during the transition of posterior foregut to pancreatic progenitors. HHEX has recently been discovered as a gatekeeper of pancreatic lineage specification and a regulator that restricts liver and duodenum differentiation programs by inhibiting CDX2 and HNF4A45. Whether the inhibitory effects of FGF2/7 on liver and intestine are due to the upregulation of HHEX, the relationship between HHEX and TBX3 should be further investigated. In addition, we found that at S6D5, 5 days after withdrawal of FGFs, HHEX mRNA and protein levels were still elevated in the treatment groups, although HHEX protein was largely reduced compared to that at stages 4/5. Since HHEX is restricted to δ-cell fate at late stage45, it would be of great interest to investigate how HHEX exerts its role as a δ-cell transcription factor and whether it is a δ-cell identity determinant. In addition to HHEX, we also found that FGFs induced several other transcription factors that are also identified in δ cells, including PDX1, ETV1, MEIS1, BHLHE41, and EHF61, and the SST enhancer activators PBX1 and CREB1. These data may partially explain why the combination of FGF2 and FGF7 additively or synergistically induces SST+ cell generation and why the pattern of HHEX induction by FGF2/7 treatment is not always consistent with that of SST induction.
In the present study, we demonstrated that FGF2 potentiates δ-cell generation by promoting SST and HHEX expression. In parallel, it also suppressed INS and GCG expression, resulting in smaller β-and α-cell fractions. However, the current bulk RNA sequencing data are insufficient to clearly demonstrate the activating and repressive effects of FGF2 and FGF7 on δ, α, and β cells. This necessitates the use of single-cell RNA sequencing approaches to profile and compare the gene expression of individual cell types. Our bulk RNA sequencing study showed that this might be due to strong suppression of PAX6. However, it is still unclear why SST and INS are differentially regulated by FGFR1 signaling, given that β and δ cells share a close developmental lineage. One possible explanation is that HHEX is specifically activated after entry into the pancreatic progenitor stage. This may require further ChIP-seq experiments to identify the cis-regulatory motifs of HHEX that are bound and activated by FGFR1 signaling components. Another important question is the origin of the increased δ cells by FGF treatments. Do they originate from an early pool of multiple destination endocrine progenitors or from the cells that are already destined to become β- and/or α-cell? FGF7 decreased more than 30% of GCG+ cells but only increased SST+ cells by 10%. What are the new 20% of endocrine/islet cells? Single cell RNA-Seq study would help answer these questions by sequencing the cells at stages 4, 5 and 6 to draw a pseudo-time developmental trajectory for these cells. Nevertheless, our current results add to the knowledge of FGFs in regulating organ development and provide clues for the treatment of diabetes with FGFs or their analogues. However, future work will be needed to address how FGFR1 regulates δ-cell development and to decipher the important effectors downstream of FGFR1.
Based on our in vitro and in vivo characterizations, these stem cell-derived δ cells do not fully resemble adult pancreatic δ cells. First, although the δ cells have fine secretory granules (Fig. 6d) similar to those of human primary δ cells75 and secrete somatostatin in response to various stimuli, they show heterogeneity in terms of calcium oscillations as a fraction of SST+ cells were observed with no calcium oscillation (Fig. 6l, bottom). Second, our single-cell RNA-seq analysis identified a fraction of polyhormonal cells located between the alpha-like cell cluster and the δ-cell cluster (Fig. 7a). These polyhormonal cells co-expressing SST and GCG/INS may represent the non-responsive cells and this heterogeneity may explain the discrepancies in somatostatin secretion assays, where some batches showed lower somatostatin secretion. In addition, these δ cells lack some ion channels that are abundant in primary pancreatic δ cells, such as ANO1, KCNN3, and RYR2, although they express comparable or slightly lower levels of KCNN2, RYR1/3, SCN3A, and especially KCNK16 (Fig. 7c). Therefore, future work will need to address the heterogeneity of the stem cell-derived δ cells and seek for approaches to improve the maturation of these cells.
Τhe δ-cell characteristics and in vitro and in vivo function have been partially investigated in this study. However, the physiological function and in vivo maturation were not fully investigated. First, although we have shown that these δ cells survive after transplantation and suppress insulin secretion of mouse β cells, thereby modulating glucose homeostasis, the details of their physiological function have not been elucidated. Since δ cells suppress both α cells and β cells79, it would be important to monitor glucagon and insulin secretion in parallel. In addition, evaluation of the interstitial glucose concentrations in animals during fasting and refeeding periods would help us understand how these cells modulate the hypoglycemia and hyperglycemia21. Second, there is lack of mature δ-cell markers and a lack of animal models suitable for assessing the in vivo function of δ cells. The δ-cell reporter approach used in this study to purify δ cells could not eliminate the polyhormonal GCG+/SST+ or INS + /SST+ cells. In this regard, future work will be the identification of mature δ-cell surface markers based on the SST reporter line. Transplantation of purified mature δ cells into animal models of hyper-insulinemic hypoglycemia would be excellent to explore the physiological effects of these somatostatin-secreting δ cells. In addition, SST ablation animal models would be better suited to clearly evaluate the contribution of transplanted δ cells, since somatostatin derived from pancreatic δ cells accounts for only 5% of circulating somatostatin pool81. The present study did not include primary human islet δ cells due to the limited access to human islet samples, which precluded a more comprehensive comparison. Nevertheless, the method established in this study allows stable and efficient generation of a substantial number of pancreatic δ cells from human pluripotent stem cells. The addition of another important pancreatic endocrine cell type would advance the understanding of the intra-islet regulatory network, islet engineering, small molecule drug screening and cell replacement therapy for diabetes.
Methods
Cell culture
Undifferentiated human embryonic stem cell lines, H1 and H9, and inducible pluripotent cells UE005, were cultured using mTeSR1(Stem cell, Cat# 85850) in Matrigel-coated (Corning, Cat# 354277) 6-well plates in a humidified 5% CO2, 37 °C incubator. Cells were passaged every 6-7 days when the cells reached approximately 70-80% confluence, using gentle cell dissociation reagent (Stem cell, Cat# 7174) with 10 μM Y27632 (Stemcell, Cat# 72307). The Institutional Review Board of Guangzhou National Laboratory reviewed and approved all work involving human pluripotent stem cells carried out in this manuscript.
Directed differentiation
Undifferentiated cells were treated with TrypLE Express (Thermo Fisher, Cat# 12605028) for 3–5 min at 37 °C when they reached approximately 70–80% confluence. Released single cells were rinsed with mTeSR1, and spun at 300 g for 5 min. The resulting cell pellet was resuspended in mTeSR1 medium supplemented with 10 μM Y-27632 (StemCell Technologies, Cat# 72307) and the single cell suspension was seeded at ~1.25 × 105 cells/cm2 on Matrigel-coated 24-well plates. Cultures were fed with mTeSR1 medium daily and differentiation was initiated 48 h after seeding when cells reached ~ 90% confluence. Directed differentiation was performed using the following 7-stage protocol (see also Supplementary Data 1).
Stage 1 (S1, 3 days): Day1 S1 medium + 100 ng/ml Activin A (StemCell Technologies, Cat# 78001) + 3 μM Chir99021 (Stemgent, Cat# 04-0004-10); Day2 S1 medium + 100 ng/ml Activin A + 0.3 μM Chir99021; Day3 S1 medium + 100 ng/ml Activin A
Stage 2 (S2, 2 days): S2 medium + 50 ng/ml FGF7 (StemCell, Cat# 78046)
Stage 3 (S3, 2 days): S3 medium + 50 ng/ml FGF7 + 1 μM retinoic acid (Sigma, Cat# R2625) + 0.25 μM SANT-1 (Sigma, Cat# S4572) + 100 nM LDN193189 (Reprocell, Cat# 40074) + 500 nM PdBU (Millipore, Cat# 524390) + 10 μM Y27632
Stage 4 (S4, 3 days): S4 medium + 50 ng/ml FGF7 (for δ cells) + 20 ng/ml FGF2 (Med Chem Express, Cat# HY-P7004) (for δ cells) + 0.1 μM retinoic acid + 0.25 μM SANT-1 + 100 nM LDN193189 + 100 nM PdBU + 10 μM Y27632 + 10 μM ALK5i II (Cell Guidance Systems, Cat# SM09-50)
Stage 5 (S5, 3 days): S5 medium + 50 ng/ml FGF7 (for δ cells) + 20 ng/ml FGF2 (for δ cells) + 0.05 μM retinoic acid + 0.25 μM SANT-1 + 100 nM LDN193189 + 10 μM zinc sulfate (Sigma, Cat# Z0251) + 10 μM ALK5i II + 1 μM XXI (Millipore, Cat# 595790) + 20 ng/mL Betacellulin (Med Chem Express, Cat# HY-P7005) + 10 μg/ml heparin (Sigma, Cat# H3149-500KU) + 1 μM T3 (Biosciences, Cat# 64245) (depleted for δ cells)
Stage 6 (S6, 5 days): S6 medium + 100 nM LDN193189 + 0.1 μM XXI + 10 μg/ml heparin + 2 μM R428 (Selleck Chem, Cat# S2841) + 10 μM zinc sulfate + 1 mM N-acetyl cysteine + 1 μM T3 (depleted for δ cells)
Stage 7 (S7, 7–14 days): S7 medium + 1 mM N-acetyl cysteine + 10 μM zinc sulfate + 10 μM (±)-α-Tocopherol (Sigma, Cat# T3251) + 10 μg/ml heparin + 1 μM T3 (depleted for δ cells)
Medium
S1 / S2 medium: 500 mL MCDB 131 medium (Thermo Fisher, Cat# 10372-019) supplemented with
0.9 g glucose (Sigma, Cat# G8769), 0.75 g sodium bicarbonate (Sigma, MO, Cat# S6297), 2.5 g bovine serum albumin (BSA) (Proliant, Cat# 68700), 10 μL ITS-X (Invitrogen, Cat# 51500056), 5 mL GlutaMAX (Invitrogen, Cat# 35050079), 22 mg ascorbic acid (Sigma, Cat# A4544), and 5 mL penicillin/streptomycin solution (Thermo Fisher, Cat# 15070063).
S3 /S4 medium: 500 mL MCDB 131 medium supplemented with 0.9 g glucose, 1.25 g sodium bicarbonate, 10 g BSA, 5 ml ITS-X, 5 mL GlutaMAX, 22 mg ascorbic acid, and 5 mL penicillin/streptomycin solution.
S5/S6 medium: 500 mL MCDB 131 medium supplemented with 1.8 g glucose, 0.875 g sodium bicarbonate, 10 g BSA, 5 ml ITS-X, 5 mL GlutaMAX, 22 mg ascorbic acid, and 5 mL penicillin/streptomycin solution.
S7 medium: 500 mL MCDB 131 medium supplemented with 1.8 g glucose, 0.875 g sodium bicarbonate, 10 g BSA, 5 ml ITS-X, 5 mL GlutaMAX, and 5 mL penicillin/streptomycin solution.
Quantitative RT-PCR
RNA was extracted using the RNeasy Mini Kit (Qiagen, Cat# 74106) with DNase treatment (Qiagen, Cat# 79254), and cDNA was synthesized using RT reagent Kit with gDNA Eraser Kit (TAKARA, Cat# RR047A). Real-time qPCR reactions were performed in SYBR Premix Ex TaqII (TAKARA, Cat# RR820A) and analyzed using the ΔΔCt method. All relative expression levels were normalized to the housekeeping gene GAPDH and gene expression levels were expressed as fold induction compared to GAPDH. The primers used in this study are listed in Supplementary Data 4.
Immunohistochemistry
For cryo-sectioning, S5-S7 organoids were fixed with 4% paraformaldehyde (PFA, Sangon Biotech, Cat# E672002) for 1 h at room temperature. After fixation, PFA was removed and organoids were rinsed three times with PBS and incubated overnight at 4 °C in 30% sucrose solution. The samples were then coverslipped with OCT solution (Biosharp, Cat# BL557A), snap frozen in liquid nitrogen, and stored at −80 °C. Sections were cut at 10 μm using a microtome and placed on Superfrost plus slides. For immunostaining of re-plated cells (Supplementary Fig. 2b), clusters were single-cell dispersed using TrypLE Express (Thermo Fisher, Cat# 12604039) and then plated onto Matrigel-coated coverslips and cultured for an additional 2 h. Single cells were fixed with PFA for 30 min at RT.
For immunostaining, sections or replated cells were rinsed with PBS and permeabilized with PBS plus 0.5% Triton X-100 for 20 min, followed by blocking with 5% donkey serum for 30 min at room temperature. Primary antibodies were added at appropriate dilutions overnight at 4 °C. Secondary antibodies were added for 2 h at room temperature, followed by DAPI staining for 15 min. Slides were mounted with VECTASHIELD mounting medium (VECTASHIELD, Cat# H-1000) after washing with PBS. Sections were visualized using a Nikon A1Rsi confocal microscope or a Leica DMI4000 fluorescence microscope. Primary and secondary antibodies (and dilutions) are listed in Supplementary Data 5.
For the quantification of cell ratios, images were analyzed using ImageJ software (with cell counter plug-in). α (GCG + ), β (INS + ) and δ (SST + ) cells were counted and the cell ratio was calculated by dividing the number of individual cells by the total number of DAPI+ cells. 10 clusters for each group (800-1000 total cells per group) were analyzed and three differentiations were evaluated.
Flow cytometry
Cells were dissociated into a single cell suspension with TrypLE and prefixed with a live/dead cell staining reagent (1:500, Fixable Viability Stain 440UV, 566332, BD Biosciences) prior to 4% PFA fixation. For staining, cells were permeabilized with 1×Perm/Wash (BD, Cat# 554723) buffer at RT for 20 min and then incubated with primary antibodies overnight at 4 °C. The primary antibodies were diluted with 1×Perm/Wash buffer. Cells were washed twice in 1×Perm/Wash buffer and incubated with secondary antibodies for 2 h at RT. Cells were then washed twice in 1×Perm/Wash buffer and analyzed using BD LSR-Fortessa. For cell marker analysis, dead cells (Brilliant Violet 421, greater than 1000) were excluded by setting a gate according to the manufacturer’s instructions. For double staining, compensation was performed by using single stained cells to calculate spectral spillover in each channel. FlowJo version 10 software was used for flow cytometry analysis. The antibodies used are listed in Supplementary Data 5.
For δ cell sorting, cell aggregates were dissociated into a single cell suspension with Accutase and suspended in PBS (without Ca2+ and Mg2+) containing 2% BSA, 10 μM Y27632, and 2 mM EDTA). mCherry-positive cells were sorted into 15 ml Falcon tubes containing S7 medium and 10 μM Y27632 using a tube sorting mode (MA900, Sony). The sorted δ cells were aggregated in a 24-well AggreWell plate (AggreWell™400, Stem Cells) and were used for further in vitro and transplantation studies.
Western blotting
2D cultured cells or 3D clusters at different time points were collected in cell lysis buffer RIPA (Beyotime, Cat# P0013) containing protease inhibitor (Beyotime, Cat# P1006) for 30 min on ice. Lysates were centrifuged at 16,000 × g for 15 min at 4 °C, and the resulting supernatants were separated by electrophoresis (4–12% SDS-PAGE gels) with Tris-MOPS-SDS running buffer and transferred to PVDF membranes. Membranes were blocked with blocking buffer (Beyotime, Cat# P0260) for 1 h at room temperature, incubated with primary antibodies overnight at 4 °C, and then incubated with appropriate horseradish peroxidase-conjugated (HRP-conjugated) secondary antibodies for 1 h at room temperature. ECL western blotting detection reagent (Millipore, Cat# WBKLS0500) was used according to the manufacturer’s instructions to expose images, followed by capture on G-Box (SynGene Chemi XR5). Quantification of target protein expression was performed using ImageJ. Values are presented as arbitrary units, obtained by dividing the intensity of the target band by the corresponding housekeeping protein band (alpha-tubulin or beta-actin). The antibodies used are listed in Supplementary Data 5.
Hormone secretion assays
Differentiated islet organoids were washed twice in 6-well plates with low-glucose (1.0 mM) Krebs-Ringer buffer (KRB, 129 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1 mM Na2HPO4, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES, and 0.1% BSA) and fasted for 1 hour at 37 °C in low-glucose KRB. Approximately 15 organoids were transferred to 1.5 ml EP tubes containing 100ul of low glucose KRB and incubated for 1 hour at 37 °C. After incubation, the supernatant was collected and stored at −20 °C until further analysis. For somatostatin secretion assay, organoids were then washed twice with low-glucose KRB and incubated with either low-glucose KRB plus tolbutamide or high-glucose KRB (20 mM) for 1 h at 37 °C followed by low-glucose KRB with 30 mM KCl, the supernatant was collected and stored.
β cells and α cells (generated by our base protocol) were purified using the MACS sorting method with the PE-conjugated CD49a and CD26 antibodies, respectively. Briefly, cell aggregates were dissociated into a single cell suspension as described above. Cells were filtered, counted, and resuspend at a concentration of 1 × 107 cells per 300 µL in sorting buffer. The cells were stained with CD49a-PE (1:50) or CD26-PE (1:50) antibodies at RT for 20 min (covered from light) and washed twice with10 mL of sorting buffer (5 min, 300 g). Subsequently, the cells were resuspended in the initial volume and stained with anti-PE Ultra-Pure beads (40 ul per 1 × 107 cells) in fridge for 15 min, followed by two washes with10 mL of sorting buffer (5 min, 300 g). Cells were counted and positive cells were isolated by using LS columns on a magnetic stand, following the instructions provided by the manufacturer. δ cells were sorted by FACS to select the mCherry-positive cells (for the previous studies before having the SST-mCherry reporter lines, δ cells were collected from the α- and β-cell depleted cell suspension). β cells alone or β cells and δ cells were mixed 1:1 to form clusters in a 24-well AggreWell plate (AggreWell™400, Stem Cells). After 2 days, the clusters were harvested for insulin secretion assays. The insulin secretion assay was performed similarly, but the organoids were sequentially incubated with high glucose KRB and then high glucose KRB with CYN. After the assay, cells were lysed with RIPA buffer and total protein levels were determined using BCA kits (Beyotime, Cat# P0012). The collected supernatants were analyzed by ELISA for human insulin (ALPCO, Cat# 80-INSHUU-E10) and/or somatostatin (Phoenix Pharmaceuticals, Cat# EK-060-03) concentrations and normalized to total cell protein.
Bulk RNA sequencing
Cells generated with the present protocol at S5D3 and S7D6 were harvested and lysed in Trizol reagent and snap frozen in liquid nitrogen. RNA was extracted using the conventional phenol-chloroform RNA extraction method. The library was prepared according to a published protocol115. The library was then sequenced on an MGISEQ-2000RS FAST instrument using MGISEQ-2000RS(PE100) kits.
Bulk RNA sequencing data analysis
Raw reads were aligned to the human GRCh38 genome using HISAT2(version 2.1.0)93. StringTie (version 1.3.4d)94 was used to assemble read alignments into transcripts and quantify gene-level expression. Differential gene expression was performed using the limma-trend method as suggested in the tutorial3. Pairwise comparisons were performed using exactTest and topTags to obtain differentially expressed genes and their respective log fold change (logFC) and adjusted P value (false discovery rate, FDR). Heat maps were generated using the ComplexHeatmap package based on TPM-level expression. Gene set enrichment analysis was used to identify enriched gene sets. Gene sets were curated from MSigDB (v7.5.1, collection 2,5,8) and adapted from the literatures58,95. Gene ontology and KEGG analysis were performed using the Cluster Profiler package. FDR correction was performed using p-adjust. GSVA was used to quantify the expression variation of cell type signature gene sets. pySCENIC package was used to infer the gene regulatory networks (GRN) using GENIE3, cisTarget and AUCell workflow96. Enrichment scores for each regulon were compared using a two-tailed t-test and the resulting p-value was corrected using the FDR method. These values were used to generate volcano plots. Gene set analyses were performed using gene set enrichment analysis58,95. Lineage-specific gene sets, including exocrine cells, ductal cells, pancreatic α, β, δ, γ and PP cells were obtained from the literatures58,95.
Single-cell RNA sequencing
ScRNA-Seq Library Construction and Sequencing. Single-cell libraries were constructed using the Single Cell 3 Library & Gel Bead Kit v.3 according to the manufacturer’s instructions (10X Genomics)120. Briefly, S7D14 cell clusters were dispersed with TrpLE. Single cells with more than 85% viability were adjusted to 1000 cells per μL. Approximately 16,000 cells were added to each channel of a 10X loading chip and then approximately 10,000 cells were captured. The captured cells were divided into Gel Beads in Emulsion (GEMs) and lysed, and the isolated RNA was reverse transcribed, barcoded, and amplified. cDNA libraries were first quantified using Qubit3.0 and further analyzed using Agilent 2100. Libraries were sequenced on an Illumina NovaSeq 6000 platform using a PE150 strategy (Annoroad Gene Technology).
ScRNA-seq data preprocessing. Raw FASTAQ files were processed using the Cell Ranger pipeline (v4.0.0) (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines /latest/advanced/references) with the default options. Reads were mapped to the GRCh38 human genome reference data.
Cell filtering, demultiplexing and clustering. To filter out low quality cells, only cells expressing more than 200 genes (defined as genes detected in at least 3 cells) and less than 20% mitochondrial genes were selected. Datasets were integrated based on ‘anchors’ identified between datasets (nfeatures = 2000, normalization. method = ‘SCT’) before performing linear dimension reduction by principal component (PC) analysis. The top 30 PCs were used in a Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction. To construct the UMAP plot, we selected the number of dimensions mainly according to the ElbowPlot function. We focused the analysis on the identified endocrine cells and EC cells by selecting clusters expressing the markers CHGA, INS, GCG, SST, and TPH1. We then rebuilt the UMAP and clustering on these cells. The annotation of the clusters was done using the scTYPE package82 under the pre-trained model of pancreas. Meanwhile, the expression profile of each cluster was extracted and scaled using the AggregateExpression function from the Seurat packages. Then, the comparison of marker gene expression patterns was performed using the pheatmap packages. To compare the stem cell-derived islet endocrine cells with primary islet cells, healthy primary adult islet cell scRNA-Seq datasets from GSE114297 (donors 1, 2, 3, 4, and 9) were compiled.
FGF network analysis in human fetal pancreas
We used scRNA-seq data from published work in GSA-Human under the accession number HRA00275734. The Seurat objects from the PCW 4–6 and PCW 7-11 samples were simply merged, respectively. The two datasets were normalized by the total counts per cell. The 3000 most variable genes were selected. Cell cycle scores were calculated using the CellCycleScoring function and regressed out together with sequencing depth and batch. Principal component analysis (PCA) was performed with a z score matrix, followed by UMAP and Louvain clustering applied with the top 50 PCs and a resolution of 1. A total of six major cell type classes were identified, including epithelial (EPCAM+ ), mesenchymal (COL3A1+ ), endothelial (PECAM1+ ), neural (ASCL1+ ), immune (PTPRC+ ), and erythroid (HBA1+ ) cells. Cells co-expressing markers of more than one cell class were treated as doublets and removed from the datasets. Then, pancreatic epithelium from different datasets (PCW 4–6 or PCW 7-11) were extracted and merged, followed by repeating the analysis process described above with parameters modified. Briefly, UMAP and Louvain clustering were applied with the top 40 PCs and a resolution of 2.5. We then annotated 13 single-cell type clusters, including dorsal MP (GATA4+ /FOXA2+ /NR2F1+ ), ventral MP (GATA4+ /FOXA2+ /TBX3+ ), early tip/tip/acinar (gradually expressed CPA2+ /RBPJL+ /CTRB2+ ), early trunk (CPA2+ /HES4+ ), trunk (HES4+ /ASCL2+ ), duct (HES4+ /ASCL2-), EP (NEUROG3+ ), alpha (GCG+ ), beta (INS+ ), delta (SST+ ) and epsilon (GHRL+ ) cells.
Cell-cell interaction analysis was performed using the CellChat package as previously described34. Briefly, the CellChatDB.human database containing FGF2, FGF7, FGF9, FGFR1-4 and cell-cell contact ligand-receptor (LR) interactions was used. The Seurat object was imported into the CellChat object and processed according to the guidelines. The chord diagram of the pathway and LR interactions of interest was fulfilled by the netVisual_chord_gene function (p-value < 0.05 were selected).
Small interfering RNA silencing
The siRNA against human FGFR1 (transcript: NM_001174063.1) was purchased from TSINGKE (siRNA sequences are listed in Supplementary Data 4). H1 cells were seeded and differentiated until S3D2 as described above. On the first day of stage 4 (S4D1), cells were detached with TypLE and the single cell suspension (5 × 106 cells) was then transfected with siRNA (a pool of 3 siRNA) using Lipofectamine RNAiMAX (Thermo Fisher, 13778030) according to the manufacturer’s instructions. The cells were then replated onto the precoated plates (1.5 × 106 cells/well of 24-well plates) and differentiation medium was added 2 h later. Differentiation continued until further siRNA transfection on S5D1. Cells were then harvested on S5D3 for qPCR and Western blotting experiments.
Construction of SST-P2A-mCherry reporter cell line
The SST-P2A-mCherry reporter cell line was generated by using the CRISPR/Cas9 knock-in system to insert mCherry at the C-terminus of SST into wildtype H1 cells. The sgRNAs were designed using the CRISPOR web server (http://crispor.tefor.net/). The sequence of sgRNA1 is 5’-GGCTAACTCAAACCCGGCTA-3’, the sequence of sgRNA2 is 5’-GACTAGTTAAGAAAGCTA
AC-3’ (Supplementary Data 4). The donor plasmid (pUC57-SST-P2A-mcherry-NLS-puro) was modified based on the pUC57 backbone. A total of 800 ng sgRNA1-Cas9 plasmids, 800 ng sgRNA2-Cas9 plasmids, and 900 ng donor plasmids were transfected into 1 × 106 H1 cells using Lipofectamine Stem reagent (Thermo Scientific). Twenty-four hours after transfection, single cells were seeded one cell per well in 96-well plates, and the puromycin-resistant cells were selected by 1 μg/ml puromycin. After 5–7 days, the colonies were dissociated and replated into 24-well plates for further expansion and genotyping. The generated heterozygous SST-P2A-mCherry reporter cell line showed normal stem cell morphology with expression levels of pluripotency markers comparable to the parental H1 cells.
Ca2+ imaging and analysis
SST-P2A-mCherry reporter cells were differentiated into δ cells by using the established protocol. S7D14-21 clusters were dispersed into single cells using Accutase (Stemcell, 07920). One day before Ca2+ imaging, 1.0 × 105 cells/cm2 were replated onto Matrigel-coated confocal specialized culture dishes (Nest, 801002) and maintained at 37 °C, 5% CO2. Prior to solution perfusion, the cells were incubated in S7 medium containing 5 μmol/L Fluo-8 (AAT Bioquest, AAT-21082) for 60 minutes at 37 °C. The cells were continuously perfused with KRB buffer (125 mM NaCl, 5.9 mM KCl, 2.56 mM CaCl2, 1.2 mM MgCl2, 1 mM L-glutamine, 25 mM HEPES, 0.1% BSA, pH 7.4) containing 2.8 mM glucose for 60 min at 37 °C in the microfluidic device chamber, while the dishes were held. Imaging was performed using a Carl Zeiss LSM 980 microscope with a 40x oil objective and ZEN software after 1 h of incubation. To locate SST-positive δ cells, we captured a still image of mCherry and GFP, followed by recording of Ca2+ dynamics on live images every 2 seconds for 5 minutes at 2.8 mM glucose and 10 minutes at 20 mM glucose85. All solutions were bathed at 37 °C and perfused to the cells at a speed of 10 rpm/min.
Real-time intensities of GFP fluorescence (ΔF) were exported using ZEN acquisition software. The mean values of ΔF during the first 5 min (F0) were used to normalize ΔF. The Ca2+ signals of δ cells were represented by ΔF/F0. Graphs were generated using Prism software (GraphPad).
Transmission electron microscopy
Ultrastructural analysis of δ cells were performed by transmission electron microscopies using a protocol modified from a previous study41. Briefly, SST-P2A-mCherry cells were differentiated using the present protocol to generate mCherry-positive δ cells. The δ cells (S7D14) were purified by FACS and aggregated by using AggreWell as described above. Cell clusters were fixed with 2.5% glutaraldehyde for 0.5 h at RT, then washed with 0.1 M phosphate buffer (PH 7.4) and postfixed with 1% osmium tetraoxide for 1.5 h at RT. After washing with 0.1 M phosphate buffer (PH 7.4), samples were dehydrated through 50%, 70%, 80%, 90%, 100% ethanol. Propylene oxide and then a 1:1 mixture of propylene oxide and epon were added to the samples for 2–4 h at RT. Next, pure Epon was added to the samples and incubated for 1 h at RT, after which the samples were embedded in Epon overnight. Finally, the samples were kept at 40°C and 60°C for 24 h and 12 h, respectively. Sections were cut and stained with uranyl acetate and lead citrate. A TecnaiG2 Spirit was used to analyze the samples.
Transplantation studies
All animal studies were conducted with approval from Institutional Animal Care and Use Committee of Guangzhou Medical University. Immunodeficient BALB/C nude mice, 8–10 weeks of age, were provided by GemPharmatech (Guangdong, China). Before surgery, animals were housed in groups with unrestricted access to food and water. Ambient temperature was maintained between 18 and 25 °C, humidity 30−70% with 12 h light/dark cycles. Approximately 500 h PSC-derived δ cell clusters (~3.0 × 106 cells per animal) were loaded into a PE50 tube for cell delivery under the kidney capsule of the mice. Control mice was injected with saline into the kidney capsule. Mice were monitored for up to 6 weeks after transplantation.
Random blood glucose levels were measured weekly (Monday morning 9:00–10:00 am) using a glucometer (Accu-Chek, Roche). Six weeks after transplantation, mice were fasted for 6 h (9:00 am to 3:00 pm) and analyzed by performing a glucose challenge (intraperitoneal (IP), 2 g/kg body weight). Blood glucose was measured at 0, 15, 30, 60, and 120 minutes after glucose injection, and serum was collected from the tail vein at 0 and 20 min after glucose injection using microvette tubes (Sarstedt). Mouse serum insulin levels were quantified by the mouse ultrasensitive insulin ELISA (ALPCO, 80-INSMS-E01). Human serum somatostatin levels were determined using somatostatin ELISA kits (Phoenix Pharmaceuticals, Cat# EK-060-03). Kidneys containing the grafts were dissected from the mice, fixed in 4% PFA overnight, cryoprotected in 30% sucrose overnight, and cryo-sectioned for histologic analysis.
Statistics and data reproducibility
Statistical analysis was performed using either one-way ANOVA, two-way ANOVA or unpaired Student’s t-test, as appropriate (GraphPad Prism v.8.0.2). Representative images and Western blotting were performed on at least three biologically independent samples. Hormone secretion assay was performed on samples from three or more independent differentiations. qPCR experiments for the mini-screen were performed on three biologically independent samples, and in other figures, on three or more independent differentiations. All data are presented as mean±s.e.m. unless otherwise noted.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw and processed bulk RNA sequencing data are available in the Sequence Read Archive (SRA) under the accession number PRJNA1126450. Raw data and count matrices of scRNA-seq data are available under the accession number GSE269764. All other data are available from the corresponding authors upon request. Source data are provided with this paper.
Code availability
Code used for analysis of 10X genomics sequencing data can be found at: https://satijalab.org/seurat/ and http://cole-trapnell-lab.github.io/monocle-release/docs/.
References
Jiang, G. & Zhang, B. B. Glucagon and regulation of glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 284, E671–E678 (2003).
Omar-Hmeadi, M., Lund, P.-E., Gandasi, N. R., Tengholm, A. & Barg, S. Paracrine control of α-cell glucagon exocytosis is compromised in human type-2 diabetes. Nat. Commun. 11, 1896–1896 (2020).
Strowski, M. Z., Parmar, R. M., Blake, A. D. & Schaeffer, J. M. Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinol 141, 111–117 (2000).
Huang J. L. et al. Paracrine signalling by pancreatic δ cells determines the glycaemic set point in mice. Nat. Metab. 6, 61–77 (2024).
van der Meulen T. et al. Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med. 21, 769–776 (2015).
Arrojo E. D. R. et al. Structural basis for delta cell paracrine regulation in pancreatic islets. Nat. Commun. 10, 3700 (2019).
Collins, S. C., Salehi, A., Eliasson, L., Olofsson, C. S. & Rorsman, P. Long-term exposure of mouse pancreatic islets to oleate or palmitate results in reduced glucose-induced somatostatin and oversecretion of glucagon. Diabetologia 51, 1689–1693 (2008).
Guardado Mendoza R. et al. Delta cell death in the islet of Langerhans and the progression from normal glucose tolerance to type 2 diabetes in non-human primates (baboon, Papio hamadryas). Diabetologia 58, 1814–1826 (2015).
Abdel-Halim, S. M., Guenifi, A., Efendić, S. & Ostenson, C. G. Both somatostatin and insulin responses to glucose are impaired in the perfused pancreas of the spontaneously noninsulin-dependent diabetic GK (Goto-Kakizaki) rats. Acta Physiol. Scand. 148, 219–226 (1993).
Weir, G. C., Clore, E. T., Zmachinski, C. J. & Bonner-Weir, S. Islet secretion in a new experimental model for non-insulin-dependent diabetes. Diabetes 30, 590–595 (1981).
Gerich J. E. et al. Prevention of human diabetic ketoacidosis by somatostatin. Evidence for an essential role of glucagon. N. Engl. J. Med. 292, 985–989 (1975).
Raskin, P. & Unger, R. H. Hyperglucagonemia and its suppression. Importance in the metabolic control of diabetes. N. Engl. J. Med. 299, 433–436 (1978).
Di Mauro M. et al. Effect of octreotide on insulin requirement, hepatic glucose production, growth hormone, glucagon and c-peptide levels in type 2 diabetic patients with chronic renal failure or normal renal function. Diabetes Res. Clin. Pract. 51, 45–50 (2001).
Gerich, J. E., Schultz, T. A., Lewis, S. B. & Karam, J. H. Clinical evaluation of somatostatin as a potential adjunct to insulin in the management of diabetes mellitus. Diabetologia 13, 537–544 (1977).
Gerich, J. E., Langlois, M., Noacco, C., Karam, J. H. & Forsham, P. H. Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science 182, 171–173 (1973).
Karimian N. et al. Somatostatin receptor type 2 antagonism improves glucagon counterregulation in biobreeding diabetic rats. Diabetes 62, 2968–2977 (2013).
Yue, J. T. Y. et al. Amelioration of hypoglycemia via somatostatin receptor type 2 antagonism in recurrently hypoglycemic diabetic rats. Diabetes 62, 2215–2222 (2013).
Kailey B. et al. SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β- and α-cells. Am. J. Physiol. Endocrinol. Metab. 303, E1107–E1116 (2012).
Veres A. et al. Charting cellular identity during human in vitro β-cell differentiation. Nature 569, 368–373 (2019).
Rezania A. et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 32, 1121–1133 (2014).
Peterson Q. P. et al. A method for the generation of human stem cell-derived alpha cells. Nat. Commun. 11, 2241 (2020).
Hogrebe, N. J., Augsornworawat, P., Maxwell, K. G., Velazco-Cruz, L. & Millman, J. R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 38, 460–470 (2020).
Du Y. et al. Human pluripotent stem-cell-derived islets ameliorate diabetes in non-human primates. Nat. Med. 28, 272–282 (2022).
Ma X. et al. Human expandable pancreatic progenitor-derived β cells ameliorate diabetes. Sci. Adv. 8, eabk1826 (2022).
Chiang, M. K. & Melton, D. A. Single-cell transcript analysis of pancreas development. Dev. Cell 4, 383–393 (2003).
Oster A. et al. Rat endocrine pancreatic development in relation to two homeobox gene products (Pdx-1 and Nkx 6.1). J. Histochem. Cytochem. 46, 707–715 (1998).
Sosa-Pineda, B., Chowdhury, K., Torres, M., Oliver, G. & Gruss, P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature 386, 399–402 (1997).
Afelik, S., Chen, Y. & Pieler, T. Combined ectopic expression of Pdx1 and Ptf1a/p48 results in the stable conversion of posterior endoderm into endocrine and exocrine pancreatic tissue. Genes Dev. 20, 1441–1446 (2006).
Gu, G., Dubauskaite, J. & Melton, D. A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. (Camb., Engl.) 129, 2447–2457 (2002).
Zhang, J., McKenna, L. B., Bogue, C. W. & Kaestner, K. H. The diabetes gene Hhex maintains δ-cell differentiation and islet function. Genes Dev. 28, 829–834 (2014).
Hebrok, M., Kim, S. K. & Melton, D. A. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. 12, 1705–1713 (1998).
Ameri J. et al. FGF2 specifies hESC-derived definitive endoderm into foregut/midgut cell lineages in a concentration-dependent manner. Stem Cells (Dayt., Ohio) 28, 45–56 (2010).
Bhushan A. et al. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Dev. (Camb., Engl.) 128, 5109–5117 (2001).
Ma Z. et al. Deciphering early human pancreas development at the single-cell level. Nat. Commun. 14, 5354 (2023).
Ye, F., Duvillié, B. & Scharfmann, R. Fibroblast growth factors 7 and 10 are expressed in the human embryonic pancreatic mesenchyme and promote the proliferation of embryonic pancreatic epithelial cells. Diabetologia 48, 277–281 (2005).
Gonçalves C. A. et al. A 3D system to model human pancreas development and its reference single-cell transcriptome atlas identify signaling pathways required for progenitor expansion. Nat. Commun. 12, 3144 (2021).
Elghazi, L., Cras-Méneur, C., Czernichow, P. & Scharfmann, R. Role for FGFR2IIIb-mediated signals in controlling pancreatic endocrine progenitor cell proliferation. Proc. Natl. Acad. Sci. USA 99, 3884–3889 (2002).
Hart, A. W., Baeza, N., Apelqvist, A. & Edlund, H. Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature 408, 864–868 (2000).
Xu, X., Browning, V. L. & Odorico, J. S. Activin, BMP and FGF pathways cooperate to promote endoderm and pancreatic lineage cell differentiation from human embryonic stem cells. Mech. Dev. 128, 412–427 (2011).
Zhang D. et al. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 19, 429–438 (2009).
Pagliuca F. W. et al. Generation of functional human pancreatic β cells in vitro. Cell 159, 428–439 (2014).
Shin, D. et al. Bmp and Fgf signaling are essential for liver specification in zebrafish. Dev. (Camb., Engl.) 134, 2041–2050 (2007).
Twaroski K. et al. FGF2 mediates hepatic progenitor cell formation during human pluripotent stem cell differentiation by inducing the WNT antagonist NKD1. Genes Dev. 29, 2463–2474 (2015).
Morrison G. M. et al. Anterior definitive endoderm from ESCs reveals a role for FGF signaling. Cell Stem Cell 3, 402–415 (2008).
Yang D. et al. CRISPR screening uncovers a central requirement for HHEX in pancreatic lineage commitment and plasticity restriction. Nat. Cell Biol. 24, 1064–1076 (2022).
D’Amour K. A. et al. Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 24, 1392–1401 (2006).
Jung, J., Zheng, M., Goldfarb, M. & Zaret, K. S. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science 284, 1998–2003 (1999).
Calmont A. et al. An FGF response pathway that mediates hepatic gene induction in embryonic endoderm cells. Dev. Cell 11, 339–348 (2006).
Seymour P. A. et al. A Sox9/Fgf feed-forward loop maintains pancreatic organ identity. Developement. (Camb., Engl.) 139, 3363–3372 (2012).
Shin, D., Lee, Y., Poss, K. D. & Stainier, D. Y. R. Restriction of hepatic competence by Fgf signaling. Development. (Camb., Engl.) 138, 1339–1348 (2011).
Wang X. et al. Genome-wide analysis of PDX1 target genes in human pancreatic progenitors. Mol. Metab. 9, 57–68 (2018).
Panek R. L. et al. In vitro biological characterization and antiangiogenic effects of PD 166866, a selective inhibitor of the FGF-1. Receptor Tyrosine Knase. J. Pharmacol. Exp. Ther. 286, 569–577 (1998).
Takehara, T., Teramura, T., Onodera, Y., Frampton, J. & Fukuda, K. Cdh2 stabilizes FGFR1 and contributes to primed-state pluripotency in mouse epiblast stem cells. Sci. Rep. 5, 14722 (2015).
Lawlor N. et al. Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 27, 208–222 (2017).
Schreiber V. et al. Extensive NEUROG3 occupancy in the human pancreatic endocrine gene regulatory network. Mol. Metab. 53, 101313 (2021).
Alvarez-Dominguez J. R. et al. Circadian entrainment triggers maturation of human in vitro islets. Cell Stem Cell 26, 108–122.e110 (2020).
Dodge, R., Loomans, C., Sharma, A. & Bonner-Weir, S. Developmental pathways during in vitro progression of human islet neogenesis. Differentiation 77, 135–147 (2009).
Segerstolpe Å. et al. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab. 24, 593–607 (2016).
Aibar S. et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017).
Ma A. et al. IRIS3: integrated cell-type-specific regulon inference server from single-cell RNA-Seq. Nucleic Acids Res. 48, W275–W286 (2020).
van Gurp L. et al. Generation of human islet cell type-specific identity genesets. Nat. Commun. 13, 2020 (2022).
Goudet, G., Delhalle, S., Biemar, F., Martial, J. A. & Peers, B. Functional and cooperative interactions between the homeodomain PDX1, Pbx, and Prep1 factors on the somatostatin promoter. J. Biol. Chem. 274, 4067–4073 (1999).
Vallejo, M., Penchuk, L. & Habener, J. F. Somatostatin gene upstream enhancer element activated by a protein complex consisting of CREB, Isl-1-like, and alpha-CBF-like transcription factors. J. Biol. Chem. 267, 12876–12884 (1992).
Ng N. H. J. et al. HNF4A haploinsufficiency in MODY1 abrogates liver and pancreas differentiation from patient-derived induced pluripotent stem cells. iScience 16, 192–205 (2019).
Vethe H. et al. Probing the missing mature β-cell proteomic landscape in differentiating patient iPSC-derived cells. Sci. Rep. 7, 4780 (2017).
Mukherjee, S., French, D. L. & Gadue, P. Loss of TBX3 enhances pancreatic progenitor generation from human pluripotent stem cells. Stem. Cell Rep. 16, 2617–2627 (2021).
Hale M. A. et al. The nuclear hormone receptor family member NR5A2 controls aspects of multipotent progenitor cell formation and acinar differentiation during pancreatic organogenesis. Dev. (Camb., Engl.) 141, 3123–3133 (2014).
Murakami, S., Kan, M., McKeehan, W. L. & de Crombrugghe, B. Up-regulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA 97, 1113–1118 (2000).
Seymour P. A. et al. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev. Biol. 323, 19–30 (2008).
Seymour P. A. Sox9: a master regulator of the pancreatic program. Rev. Diabet. Stud. 11, 51–83 (2014).
Seymour P. A. et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. USA 104, 1865–1870 (2007).
Bohuslavova R. et al. NEUROD1 reinforces endocrine cell fate acquisition in pancreatic development. Nat. Commun. 14, 5554 (2023).
Shih H. P. et al. A gene regulatory network cooperatively controlled by Pdx1 and Sox9 governs lineage allocation of foregut progenitor cells. Cell Rep. 13, 326–336 (2015).
Villasenor, A., Gauvrit, S., Collins, M. M., Maischein, H.-M. & Stainier, D. Y. R. Hhex regulates the specification and growth of the hepatopancreatic ductal system. Dev. Biol. 458, 228–236 (2020).
de Boer P. et al. Large-scale electron microscopy database for human type 1 diabetes. Nat. Commun. 11, 2475 (2020).
Gribben C. et al. Ductal Ngn3-expressing progenitors contribute to adult β cell neogenesis in the pancreas. Cell Stem Cell 28, 2000–2008.e4 (2021).
Gershon, M. D. & Tack, J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology 132, 397–414 (2007).
Vergari E. et al. Somatostatin secretion by Na+-dependent Ca2+-induced Ca2+ release in pancreatic delta cells. Nat. Metab. 2, 32–40 (2020).
Svendsen, B. & Holst, J. J. Paracrine regulation of somatostatin secretion by insulin and glucagon in mouse pancreatic islets. Diabetologia 64, 142–151 (2021).
Hauge-Evans A. C. et al. Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 58, 403–411 (2009).
Rorsman, P. & Huising, M. O. The somatostatin-secreting pancreatic δ-cell in health and disease. Nat. Rev. Endocrinol. 14, 404–414 (2018).
Ianevski, A., Giri, A. K. & Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat. Commun. 13, 1246 (2022).
DiGruccio M. R. et al. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 5, 449–458 (2016).
Vierra N. C. et al. TALK-1 reduces delta-cell endoplasmic reticulum and cytoplasmic calcium levels limiting somatostatin secretion. Mol. Metab. 9, 84–97 (2018).
Gosak, M., Yan-Do, R., Lin, H., MacDonald, P. E. & Stožer, A. Ca2+ oscillations, waves, and networks in islets from human donors with and without type 2 diabetes. Diabetes 71, 2584–2596 (2022).
Ramzy A. et al. Implanted pluripotent stem-cell-derived pancreatic endoderm cells secrete glucose-responsive C-peptide in patients with type 1 diabetes. Cell Stem Cell 28, 2047–2061.e2045 (2021).
Vergari E. et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 10, 139 (2019).
Ramnanan, C. J., Edgerton, D. S., Kraft, G. & Cherrington, A. D. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes. Metab. 13, 118–125 (2011).
Briant L. J. B. et al. δ-cells and β-cells are electrically coupled and regulate α-cell activity via somatostatin. J. Physiol. 596, 197–215 (2018).
Hiyoshi H. et al. Characterization and reduction of non-endocrine cells accompanying islet-like endocrine cells differentiated from human iPSC. Sci. Rep. 12, 4740 (2022).
Augsornworawat, P., Maxwell, K. G., Velazco-Cruz, L. & Millman, J. R. J. C. R. Single-cell transcriptome profiling reveals β cell maturation in stem cell-derived islets after transplantation. Cell Rep. 32, 108067 (2020).
Velazco-Cruz L. et al. Acquisition of dynamic function in human stem cell-derived β cells. Stem Cell Rep. 12, 351–365 (2019).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Muraro M. J. et al. A single-cell transcriptome atlas of the human pancreas. Cell Syst. 3, 385–394.e383 (2016).
Van de Sande B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276 (2020).
Acknowledgements
We thank Dr. Guangjin Pan for providing the iPS cell line UE005. This work was supported by grants from the National Key Research and Development Program of China (2021YFA1101304, 2020YFA0908200 to H.L.), the Guangdong Basic and Applied Basic Research Foundation (2023A1515010483 to L.C.) and the Young Scientists Program of Guangzhou Laboratory (QNPG23-02 to L.C.).
Author information
Authors and Affiliations
Contributions
H.L. conceived the project and H.L., L.C., and T.X. supervised the research. L.C., N.W., T.Z., J.C., F.Z., H.M., and Z.L. performed the experiments. W.Z. and X.X. performed the single-cell RNA sequencing and bulk RNA sequencing analyses. Z.M. analyzed the single-cell data of human fetal pancreas. L.C., and H.L. analyzed the data and wrote the manuscript, which was further edited by the other co-authors. The authors declare no other competing financial interests.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, L., Wang, N., Zhang, T. et al. Directed differentiation of pancreatic δ cells from human pluripotent stem cells. Nat Commun 15, 6344 (2024). https://doi.org/10.1038/s41467-024-50611-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-50611-7
- Springer Nature Limited