

Abstract
Neutrophil infiltration and subsequent extracellular trap formation (NETosis) is a contributing factor in sterile inflammation. Furthermore, neutrophil extracellular traps (NETs) are prothrombotic, as they provide a scaffold for platelets and red blood cells to attach to. In circulation, neutrophils are constantly exposed to hemodynamic forces such as shear stress, which in turn regulates many of their biological functions such as crawling and NETosis. However, the mechanisms that mediate mechanotransduction in neutrophils are not fully understood. In this study, we demonstrate that shear stress induces NETosis, dependent on the shear stress level, and increases the sensitivity of neutrophils to NETosis-inducing agents such as adenosine triphosphate and lipopolysaccharides. Furthermore, shear stress increases intracellular calcium levels in neutrophils and this process is mediated by the mechanosensitive ion channel Piezo1. Activation of Piezo1 in response to shear stress mediates calpain activity and cytoskeleton remodeling, which consequently induces NETosis. Thus, activation of Piezo1 in response to shear stress leads to a stepwise sequence of cellular events that mediates NETosis and thereby places neutrophils at the centre of localized inflammation and prothrombotic effects.
Similar content being viewed by others
Introduction
Neutrophils are the most abundant type of circulating leukocytes. As a central component of the innate immune system, they represent the first line of defence against various microorganisms such as bacteria, fungi, and protozoa1. In addition to their contribution to host defence, neutrophils have also been recognized as important drivers of chronic inflammatory diseases such as autoimmune disorders, cancers, and cardiovascular diseases1,2,3.
Neutrophils are mechanosensitive, and hemodynamic shear stress regulates their activation e.g., during inflammation. Under static conditions, neutrophils have a spread-out cytoplasmic morphology, can form pseudopods, and migrate on glass slides. Upon exposure to shear stress, they retract their pseudopods and form a rounded morphology4,5. Furthermore, shear stress increases the sensitivity of neutrophils to platelet-activating factor and consequently the shedding of L-selectin into the extracellular space. Additionally, shear stress increases the activation of the integrin αMβ2 on neutrophils and modulates neutrophil morphology6.
One of the most important characteristics of neutrophils is their ability to release their DNA into the extracellular space, forming so-called neutrophil extracellular traps (NETs). NETs are web-like chromatin structures decorated with cytosolic and granule proteins7. NETs trap, neutralize, and kill pathogens but, if dysregulated, can contribute to the pathogenesis of immune-related disorders such as cardiovascular diseases, by contributing to the activation of the coagulation pathway8. For example, NETs have been implicated in the pathogenesis of arterial9,10 and venous thrombosis11,12, sepsis13, and autoimmune vasculitis14.
The NET release process, known as NETosis, occurs via two main pathways. The first is described as the classical pathway, which consists of several steps, including the cessation of actin rearrangement and polymerization, disassembly of the nuclear envelope, and decondensation of nuclear chromatin, leading to the mixing of chromatin with cytoplasm. Following this, the plasma membrane becomes permeable, and DNA content is released into the extracellular space. The classical pathway of NETosis takes 3–8 h upon activation and represents cell death15. The alternative pathway is a non-lytic or vital NETosis, leading to the rapid release of NETs into the extracellular space after exposure to stimuli7. During vital NETosis, cells retain their viability and many of their natural effector functions, such as chemotaxis and phagocytosis16.
Although the initial signalling pathways that trigger NETosis, whether lytic or non-lytic, are not well understood, it is known that NET release is initiated by the activation of membrane receptors that are sensitive to changes in intracellular calcium level, followed by the activation of kinase-signalling cascades and production of reactive oxygen species17,18.
Neutrophils in circulation are constantly exposed to hemodynamic forces and these forces affect their basic biology, including their ability to form NETs within sterile thrombi and the expression of inflammatory cytokines and chemokines19,20.
Hemodynamic forces can trigger the activation of membrane receptors, also known as mechanoreceptors, which can consequently trigger the activation of transduction pathways in neutrophils, leading to changes in functional responses such as retraction of pseudopods, adhesion, and NETosis. However, the identity of the mechanoreceptors that contribute to the sensitivity of neutrophils to hemodynamic forces is not known. Likewise, how hemodynamic forces control the process of NETosis is not yet understood.
Mechanosensitive ion channels are one of the most important and best characterized classes of mechanoreceptors that allow cells to respond to their physical surroundings. Calcium influx via non-selective mechanosensitive cation channels activated by hemodynamic forces is a major contributor to the activation of calcium-dependent biological processes. Among different classes of mechanosensitive ion channels, the Piezo1 channel plays a key role in the sensing of shear stress in different environments, with the ability to directly gate in response to mechanical stress20,21,22,23,24. More importantly, mechanotransduction via Piezo1 is the key regulator linking the external mechanical environment to different cell-signalling pathways and physiological responses24.
This work elucidates the effects of shear stress and mechanosensitive ion channels on NET formation and identifies the signalling pathway that is activated in response to shear stress.
Results
Shear stress triggers NETosis in isolated neutrophils
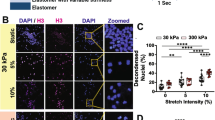
To investigate the effect of shear stress on circulating neutrophils, we used microfluidic channels to expose circulating neutrophils to shear stress levels of 4, 20, and 80 dyne/cm2 for 10 cycles. Following this, we seeded neutrophils onto a chamber slide precoated with Cell-Tak for 3 h to allow them to adhere and form NETs. Next, we measured the percentage of cells with round nuclei as well as the area covered by expelled extracellular DNA or NETs. Under static conditions (no shear stress), the nuclei of the majority of cells were lobulated spatially with heterogeneous DNA staining indicative of regions of varied chromatin condensation and a very low number of NETs. In contrast, exposure of cells to shear stress increased the percentage of cells with swollen and round nuclei, and increased the NET area in a shear stress–dependent manner (Fig. 1a–c).

a Representative confocal images of human neutrophils stained with DAPI and citrullinated histone H3 (H3 citrullination) under static control condition or shear stress of 80 dyne/cm2. Graphs show the: b NET area normalized to the number of neutrophils (n = 35 randomly selected field of view (RSFV) in 0, 20 and 80 dyne/cm2 and n = 32 RSFV in 4 dyne/cm2 from N = 7 independent experiments (IE)); c Percentage of cells with condensed and round nuclei (n = 16 RSFV in 0, 20 and 80 dyne/cm2 and n = 17 RSFV in 4 dyne/cm2 from N = 4 IE) and d Percentage of cells stained positive for H3 citrullination (n = 34 RSFV in 0 and 20 dyne/cm2, n = 33 RSFV in 4 dyne/cm2 and n = 35 RSFV in 80 dyne/cm2 from N = 7 IE). Effect of shear stress on: e NET area ((Static: n = 33 RSFV for Control, LPS and ATP and n = 34 RSFV for PMA) (Shear stress: n = 35 RSFV for control, ATP and PMA and n = 34 RSFV for LPS from N = 7 IE); and f H3 citrullination in response to other NET inducers such as LPS (10 µg/ml), ATP (100 nM), and PMA (3 nM) (n = 35 RSFV from N = 7 IE). b–d are analysed using one-way ANOVA and mixed effects analysis and e, f are analysed using two-way ANOVA and multiple comparisons test. In b–f box plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers.
One of the specific markers of NETosis is citrullinated histone H325. In general, histone citrullination is an epigenetic process that occurs via enzymatic alteration of arginine residue of histone to citrulline. This conversion leads to chromatic relaxation, which plays a central role in the release of chromatin from NETs26. We assessed the effect of shear stress on citrullination of histone H3 by measuring the percentage of cells that were stained positive for citrullinated histone H3. Consistent with the NET area, we found that shear stress increased the percentage of cells with H3 citrullination in a shear stress–dependent manner (Fig. 1d). These results indicate that neutrophils are sensitive to shear stress and that increased shear stress can directly induce NETosis.
Next, we investigated the effect of shear stress on the sensitivity of neutrophils to other NET-inducing agents such as ATP, LPS, and PMA. For this assessment, neutrophils were exposed to 10 cycles of shear stress, followed by incubation inside a chamber slide in the presence or absence of ATP (100 nM), LPS (10 µg/ml), and PMA (3 nM) for 3 h to form NETs. The concentrations of stimuli were based on the minimum amounts required to induce detectable NETosis under static conditions. Using this approach, we found that pretreatment of neutrophils with shear stress increased NETosis induced by LPS and ATP, as evidenced by the increased NET area and degree of H3 citrullination, but did not affect PMA -induced NETosis (Fig. 1e, f).
Neutrophil activation and NETosis are known drivers of thrombosis27. The physical interaction between platelets and neutrophils contributes to NET formation, while NETs provide scaffolds for platelet adhesion and aggregation28. Therefore, here we investigated the effect of shear stress on the interaction of platelets with neutrophils. After shear stress stimulation of neutrophils at 80 dyne/cm2 for 10 cycles (each cycle lasting 1 min), we allowed them to rest for 3 h inside a chamber slide, thereby facilitating the formation of NETs. Subsequently, the chamber slides containing neutrophils, were perfused with platelets for 10 min followed by several washing steps to remove non-adhering cells. We measured the NET area covered by activated platelets using P-selectin staining and assessed changes of the platelet morphology. Exposure of neutrophils to shear stress significantly increased platelet adhesion and average platelet cluster size over the NET area (P < 0.001), compared to the static control group (Fig. 2a–c). Platelet shape change, a prerequisite for platelet aggregation, was confirmed by assessing the platelet morphology after interaction with neutrophils exposed to shear stress or resting neutrophils. Platelets exposed to shear stress-treated neutrophils exhibited a higher percentage of platelets with filopodia compared to those interacting with resting neutrophils (Fig. 2d, e). These data show that shear stress induces NETosis and provides a scaffold for platelets to adhere and aggregates.

a Representative confocal images of neutrophils under static conditions or exposure to shear stress of 80 dyne/cm2 and incubated with platelets. Platelets are stained with P-selectin (red), neutrophils are stained with CD11a (green), and DNA is stained with DAPI (blue). b, c Bar graphs show the average area covered by P-selectin positive platelets normalized to the total neutrophil area (n = 38 RSFV from N = 8 IE) and average P-selectin cluster size (n = 40 RSFV in static and n = 38 RSFV in shear stress from N = 8 IE). d Representative confocal images and e a bar graph showing the effect of shear stress on platelet shape. Red arrows highlight platelets with filopodia, and blue arrows highlight platelets with discoid morphology. (n = 14 RSFV in static and n = 10 RSFV in shear stress groups selected from N = 3 IE). b and c is analysed using two-tailed Student’s T-test and e is analyzed using two-way ANOVA and multiple comparisons test. In b, c and e box plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers.
Shear stress stimulation of neutrophils results in [Ca2+]i elevation
Upregulation of Ca2+ mediated signalling pathways plays an essential role in controlling many of the neutrophil’s defence mechanisms against pathogens29. Therefore, we investigated the effect of shear stress on the intracellular calcium level ([Ca2+]i) of neutrophils using a calcium-sensitive dye, Fluo-4 AM, and confocal microscopy. For this experiment, we immobilized neutrophils inside a microfluidic channel and exposed them to different shear stress levels of 0.2, 2, and 10 dyne/cm2. We found that shear stress increased the [Ca2+]i of neutrophils in a dose-dependent manner (Fig. 3a–c).

a Fluorescent images of firmly adhered neutrophils at different time points in the presence or absence of shear stress at 10 dyne/cm2. b Representative time-course data of the normalized fluorescent intensity of neutrophils (n = 90 cells for 0.2 and 10 dyne/cm2, n = 43 cells for static and n = 89 cells for 2 dyne/cm2 selected from a representative experiment); and c Maximum increase in [Ca2+]i of neutrophils in response to different shear stress levels (n = 118 cells from N = 3 IE). d Time-course response (n = 50 cells for 10 dyne/cm2 and n = 28 cells for static selected from N = 3 IE); and e Maximum increase in [Ca2+]i of neutrophils after exposure to 3 cycles of shear stress at 10 dyne/cm2 (n = 448 cells in static and n = 489 cells in shear stress groups across N = 9 IE). c is analyzed using one-way ANOVA and mixed effects analysis and e is analyzed using two-way ANOVA and multiple comparisons. In c and e box plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers. Error bars in b and d represent standard error. Scale bar is 50 µm.
Next, we assessed the effect of acute exposure to shear stress on neutrophil mechanosensitivity. For this, we exposed neutrophils to three cycles of shear stress at 10 dyne/cm2 with 5 min of static rest between each cycle and measured the [Ca2+]i at each cycle. Using this approach, we found that exposure of neutrophils to multiple cycles of shear stress did not desensitize neutrophils to shear stress (Fig. 3d, e).
Furthermore, removal of calcium from the extracellular space and inhibition of SERCA pumps using thapsigargin both eliminated the effect of shear stress on change in [Ca2+]i, confirming that both calcium influx and calcium release from intracellular stores of Ca2+ contribute to the shear stress sensitivity of neutrophils (Fig. 4a).

a Maximum elevation of [Ca2+]i in response to shear stress (n = 52 cells in static, n = 70 in shear stress, n = 73 in -[Ca2+]ext, n = 74 in Ruthenium Red (RR), n = 75 in -[Ca2+]cyt, n = 47 in GdCl3 and n = 60 in GSMTx4, N = 3 IE). b Normalized NETs (Static: n = 42 RSFV in control, n = 40 in -[Ca2+]ext, n = 11 in RR, n = 15 in GSMTx4 and n = 9 for BAPTA. Shear: n = 30 in control, n = 34 in -[Ca2+]ext, n = 11 in RR, n = 16 in GSMTx4 and n = 8 in BAPTA, N = 3 IE), and c H3 citrullination in response to shear of 80 dyne/cm2 in the presence or absence of EGTA (2 mM), RR (30 µM), GaCl3 (30 µM), GsMTx4 (20 µM), Thapsigargin (1 µM), and BAPTA (10 µM) (Static: n = 27 RSFV in control, n = 41 in -[Ca2+]ext, n = 18 in RR, n = 26 in GSMTx4 and n = 25 in BAPTA. Shear: n = 11 in control, n = 32 in -[Ca2+]ext, n = 9 in RR, n = 9 in GSMTx4 and n = 12 for BAPTA, N = 3 IE). d Immunofluorescence images of neutrophils stained with Piezo1 and DAPI. e Western blot of neutrophils probed for Piezo1 and GAPDH in two different donors. f Representative microscopy images and (g, h) change in [Ca2+]I in response to Yoda1 (g is representative time response of f, n = 30 cells in shear and n = 60 cells in static.) (h n = 79 cells, N = 3 IE). i Normalized NET area (n = 20 RSFV, N = 4 IE) and j percentage of cells with H3 citrullination (n = 20 RSFV, N = 4 IE) and k Net area covered by platelets (n = 23 RSFV in control and n = 24 in Yoda1, N = 6 IE). a and h are analyzed using one-way ANOVA and mixed effects test. b and c are analyzed using two-way ANOVA and multiple comparisons test. i and k are analyzed using two-tailed Student’s T test. In a–c and h–k box plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers. Error bars in g is standard error.
Neutrophils express Piezo1 at a functional level and Piezo1 activation induces NETosis
We further examined the contribution of cation-permeable mechanosensitive ion channels to the shear stress response of neutrophils using ruthenium red, GdCl3, and GSMTx4, which are known blockers of many cationic mechanosensitive ion channels.
Pretreatment of neutrophils with all of the above inhibitors led to a significant decrease in shear-induced calcium influx of neutrophils (Fig. 4a). Furthermore, removal of calcium from the media using EGTA or pretreatment of neutrophils with ruthenium red and GSMTx4 eliminated the shear-induced NETosis and citrullination of H3, confirming that Ca2+ influx via mechanosensitive ion channels controls shear-induced NETosis (Fig. 4b, c).
Piezo1 is a calcium-permeable cation channel that is inherently mechanosensitive with high sensitivity to shear stress and is expressed in several cell types24. To examine whether Piezo1 is expressed in primary human neutrophils, we assessed its expression using immunostaining and Western blotting (Fig. 4d, e) and found that Piezo1 is expressed in human neutrophils. However, it should be noted that protein expression does not provide evidence of the functionality of the receptors, in particular for mechanosensitive ion channels, which are shielded from the external stresses in resting cells and need to be gated to function30.
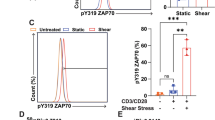
Therefore, we assessed the functional responses of Piezo1 in primary neutrophils by utilizing Yoda1, which is a Piezo1-selective agonist, and measuring [Ca2+]i in the presence or absence of physiological levels of shear stress (10 dyne/cm2). We found that Yoda1 stimulation of neutrophils led to an elevation in [Ca2+]i and shear stress improved the sensitivity of neutrophils to Yoda1 (P < 0.001, Fig. 4f–h). This finding is consistent with what we observed previously in monocytes and endothelial cells31,32,33,34. To determine if activation of Piezo1 can induce NETosis, and consequent increase in platelet adhesion to NETs, we utilized Yoda1. We found that treatment of neutrophils with Yoda1 for 3 h led to a significant increase in NET area (P < 0.001, Fig. 4i), citrullination of H3 (P < 0.001, Fig. 4j) and a consequent increase in platelets adhering to NETs (P < 0001, Fig. 4k). Increased Ca2+ influx after Piezo1 activation is reported to activate calpain in many model systems, essential in downstream signalling events such as cytoskeleton remodelling and NETosis35.
To determine whether calpain activity is modulated by shear stress, we employed a calpain activity assay that utilizes the fluorogenic substrate to assess calpain activity in lyzed cells. Using this approach, we found that exposure of circulating neutrophils to 10 cycles of shear stress at 80 dyne/cm2 modulated calpain activity by 2.4 ± 0.16 fold (Fig. 5a). Furthermore, we found that treatment of neutrophils with 10 µM of Yoda1 increased calpain activity by 2.3 ± 0.2 fold (Fig. 5b). Next, to assess the role of calpain in shear-induced NETosis, we used the calpain inhibitors PD150606 and PD151746, as well as an inactive analogue, PD145305. Consistent with the calpain activity data, pretreatment of neutrophils with both calpain inhibitors blocked the shear-induced NETosis, while the inactive analogue did not have any effect (Fig. 5c), confirming that calpain activity post-shear stress contributes to shear-induced NETosis.

a, b Calpain activity (N = 3 IE); and c Normalized NET area (n = 20 RSFV in control, PD150606 and PD151746 and n = 19 in PD145305, N = 4 IE). a and b are analyzed using one-way ANOVA and mixed effects test. c is analyzed using two-way ANOVA and multiple comparisons test. Box plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers.
Piezo1 siRNA silencing results in reduced shear-induced NETosis
Primary neutrophils are unsuitable for gene knockdown studies due to their short life span36 and mouse neutrophils do not express Piezo137. Therefore, to confirm the contribution of Piezo1 to shear-induced NETosis, we knocked down Piezo1 using an siRNA gene-silencing approach in the HL-60 cell line. This cell line is derived from human promyelocytic leukaemia cells that were originally developed as a model for studying neutrophils38. These cells can be cultured for a long time and differentiated into neutrophils using DMSO treatment. In this study, we differentiated HL-60 cells using 1.25% DMSO for 6 days (dHL-60) and confirmed the expression of the neutrophil markers CD11b and CD16. It should be noted that under this condition, we did not observe a significant decline in cell viability (Supplementary Fig. 1).
First, we confirmed that shear stress induces NETosis in dHL-60 cells. For this, we exposed dHL-60 cells to 10 cycles of shear stress followed by 3 h of static rest to allow them to form NETs. Using this approach, we found that shear stress induces NETosis and increases the H3 citrullination in dHL-60 cells, similar to primary neutrophils. Furthermore, we found that pre-treatment of neutrophils with GsMTx4 blocks shear-induced NETosis and H3 citrullination in dHL-60 cells, confirming a role of mechanosensitive ion channels (Fig. 6a–c). Next, we assessed the expression of Piezo1 in dHL-60 cells and found that these cells expressed Piezo1 (Fig. 6d, e). Next, we used calcium imaging and confirmed that dHL-60 cells were responsive to shear stress and an increase in shear stress improved their sensitivity to Yoda1 (Fig. 6f). These results confirm that dHL-60 cells express Piezo1 at a functional level.

a–c Representative fluorescent images and bar graphs showing the degree of NETosis and H3 citrullination (H3-Cit) in dHL-60 cells under static conditions or after shear stimulation in the presence or absence of GsMTX4. Red arrows in a showing NET release. (b: n = 30 RSFV from N = 6 IE) (c: n = 30 in static and GSMTX4/shear and n = 28 in shear from N = 6 IE). d Confocal images; and e Western blot of dHL-60 cells probed for Piezo1. f Bar graph showing the elevation in [Ca2+]i of dHL-60 cells in response to shear and Yoda1 (control: n = 65 cells in static and n = 79 cells in shear groups, Yoda1: n = 57 cells in static and n = 78 cells in shear group selected from N = 3 IE). g Confocal images and h bar graphs showing the degree of Piezo1 knockdown in dHL-60 cells treated with Piezo1 siRNA compared to the group treated with non-targeting siRNA. (n = 85 cells in Piezo1 siRNA and n = 67 cells in non-targeting siRNA analyzed across N = 3 IE). i qPCR data confirming the knockdown of Piezo1 in HL60 cells treated with Piezo1 compared to non-targeting siRNA. (N = 3 IE in non-targeting and N = 4 IE in Piezo1 siRNA group). b and c are analyzed using one way ANOVA and mixed effects analysis. f is analyzed using two-way ANOVA and multiple comparisons test. h and i are analysed using two-tailed Student’s T test. In b, c, f, h, i plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers.
To confirm the role of Piezo1 in shear-induced NETosis of neutrophils, we compared the NET area in dHL-60 cells transfected with either siRNA-targeting Piezo1 or scrambled siRNA. The Piezo1 expression was reduced by at least 40% in cells treated with siRNA targeted to its transcription compared to the cells treated with scrambled siRNA (Fig. 6g–i).
Furthermore, we found that partial knockdown of Piezo1 in dHL-60 cells blocks the capacity of neutrophils to form NETs and reduces the percentage of cells with H3 citrullination compared to their static control group. While exposure of dHL-60 cells treated with scrambled siRNA to shear stress led to a 1.2 ± 0.1 fold (P < 0.001) increase in NET area and 1.6 ± 0.1 fold (P < 0.001) increase in percentage of cells with H3 citrullination compared to the static control group (Fig. 7a–c).

a Representative confocal images and (b, c) bar graphs showing the degree of NETosis and H3 citrullination in dHL-60 cells treated with Piezo1 or non-targeting siRNA, stained with DAPI (blue) and H3 antibody (green) in response to shear. (b, Static: n = 28 RSFV and Shear: n = 31 RSFV in non-targeting and n = 40 RSFV in Piezo1 siRNA group selected from N = 4 IE) (c, Static: n = 37 RSFV in non-targeting and n = 40 RSFV in Piezo1 siRNA and shear: n = 26 RSFV in non-targeting and n = 40 in Piezo1 siRNA from N = 4 IE). b and c is analyzed using two-way ANOVA and multiple comparisons test. b and c box plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers.
Shear-induced NETosis requires actin cytoskeleton rearrangement
Actin cytoskeleton rearrangement is essential for several cellular functions, including NETosis in response to LPS39, PMA40, Staphylococcus aureus41, and Pseudomonas aeruginosa42. Therefore, we next sought to determine the role of cytoskeleton remodelling in shear-induced NETosis. We initially pretreated the isolated human neutrophils with latrunculin B (that binds to G actin and prevents actin polymerization) or ML-7 (inhibitor of myosin light chain kinase) before shear stress stimulation (80 dyne/cm2) to assess the role of cytoskeleton rearrangement on shear-induced NETosis.
We found that both treatments reduced the NET area and H3 citrullination of neutrophils in response to shear stress (P < 0.001, Fig. 8a, b). Dynamic actin reorganization is essential for multiple cellular functions including cell motility and phagocytosis. To confirm the role of cytoskeleton remodelling, we assessed the effect of shear stress stimulation of circulating neutrophils on their motility once adhered to a collagen-coated substrate by measuring their track length and speed for 10 min. Shear stress stimulation of neutrophils significantly increased their track length (P < 0.001) and speed (P < 0.01, Fig. 8c–e). Thus, to further confirm the effect of shear stress on cytoskeleton remodelling in neutrophils, we subjected them to shear stress (80 dyne/cm2) for 1, 5, and 15 min. We assessed the impact of shear stress exposure time on actin cytoskeleton organization by measuring the intensity and ratio of F-actin to G-actin. During the initial 5 min of shear stress exposure, the intensity of F-actin remains stable but by the 15-min mark, it significantly declined compared to baseline levels (P < 0.05). Concurrently, the intensity of G-actin increased in a time-dependent manner following shear stress stimulation compared to baseline (P < 0.01). Analysis of the F/G-actin ratio revealed a gradual decrease during the first 15 min of shear stress exposure (Fig. 8f–i). Overall, these experiments suggests that shear stress exposure triggers the assembly, disassembly, and remodelling of the actin cytoskeleton.

Graphs show (a) the normalized NET area and (b) H3 citrullination of neutrophils pretreated with ML-7 or Latrunculin B under static conditions or shear stress. (Static: n = 20 RSFV in control and ML-7 and n = 16 RSFV in latrunculin B and shear stress: n = 20 RSFV in control and Latrunculin B and n = 18 RSFV in ML-7 group selected from N = 4 IE). c Representative tracks of neutrophils under static conditions or pretreatment with shear stress and summary graphs showing: d Track length (n = 45 cells that were present during the time course of the experiment analysed from N = 3 IE); and e Speed of neutrophils under static or shear stress conditions. (n = 150 randomly selected cells analyzed across N = 3 IE). f Representative confocal images of neutrophils exposed to shear stress for different duration of time and stained with Alexa Fluor 488 DNase I to label G-actin (green) and Alexa Fluor 568 phalloidin to label F-actin (red). Representative graphs showing the intensity of (g) F- actin, (h) G-actin and (i) F/G actin ratios (n = 39 RSFV from N = 4 IE). j Representative tracks of neutrophils in the presence or absence of Yoda1 and pretreatment with PD150606 and PD151746, and summary graphs showing: k Track length (n = 60 cells in vehicle, n = 75 in Yoda-1 and n = 45 in PD150606 and PD151746 randomly selected cells that were present during the time course of the experiment from N = 3 IE); and l Speed (n = 141 cells randomly selected from N = 3 IE). Data in a and b is analyzed using two-way ANOVA and multiple comparisons test. d and e are analyzed using two-tailed Student’s T-test. g–i, k and l are analyzed using one-way ANOVA and mixed effects analysis. In a, b, d, e, g–i, k and l box plots depict the 25th percentile, the median and the 75th percentile, with minimum to maximum whiskers.
Next, we assessed the contribution of Piezo1 to increased cellular motility after shear stress exposure and found that treatment of neutrophils with Yoda1 increased their track length and speed by 1.4 ± 0.07 (P < 0.001) and 1.6 ± 0.1 (P < 0.001) fold, respectively, compared to the vehicle control group. Moreover, pretreatment of neutrophils with both calpain inhibitors, PD150606 and PD 151746, blocked the effect of Yoda1 in increasing their track length and speed, confirming that calpain activity after Piezo1 activation controls cytoskeleton remodelling and motility of neutrophils (Fig. 8j–l).
Discussion
In the present study, we found that shear stress directly induces NETosis and increases the sensitivity of neutrophils to other NET-inducing agents such as LPS and ATP. We showed that shear-induced NETosis is mediated via calcium influx through the mechanosensitive ion channel Piezo1, calpain activity, and cytoskeleton remodelling (Fig. 9).

Piezo1-mediated calcium influx in response to shear stress activates calpain, leading to cytoskeletal remodelling and the regulation of increased motility and NETosis in neutrophils. (Image created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license).
Neutrophils are a specialized type of immune cell with specific antimicrobial capacity, identified as phagocytosis, degranulation, and NETosis. In general, various stimuli can induce NETosis, including microorganisms, nitric oxide, urate crystals, and inflammatory cytokines, as well as interaction with activated platelets and endothelial cells43. Exaggerated NETosis is proposed to contribute to autoimmune disorders in predisposed individuals14,44,45 as well as providing a scaffold for thrombus formation11,46,47.
Circulating neutrophils, like other leukocytes, constantly receive and respond to hemodynamic forces, namely, shear and tensile stresses. Hemodynamic forces drive the adhesion and migration of inflammatory cells to inflamed tissue. The mechanism that controls these events is of interest to biomedical research. It is not known how transient change in hemodynamics, for example, increase in shear stress as a consequence of stenosis, could affect the basic biology of circulating neutrophils. Many biological processes such as cell-death pathways and inflammatory pathways are mediated by calcium influx via stretch-activated channels expressed in all cell types, including neutrophils48. This raised a question for us as to whether shear stress could be a contributing factor in regulating NETosis. To investigate the direct effect of shear stress on NETosis, we exposed purified neutrophils to various shear stress levels in circulation using microfluidics and allowed the cells to form NETs in a substrate that was precoated with Cell-Tak, in the absence of any integrin-inducing ligand. In terms of timing, we found that neutrophils needed at least 3 h to form detectable NETs after shear stress stimulation, which is consistent with the timing of lethal NETosis. A previous study by Yu et al.19 showed that shear stress could induce rapid NETosis in sterile thrombi.
To assess the effect of shear stress on rapid NETosis, we also performed time-lapse shear stress experiments on neutrophils immobilized in a collagen-coated microfluidic channel and in the presence of various shear stress levels. Our results did not show any NET production within 30 min of shear stress stimulation at 80 dyne/cm2. Therefore, from these experiments we conclude that a high shear stress level can directly induce lethal NETosis and initiate a process of platelet adhesion that can trigger further NETosis along with platelet adhesion and aggregation.
We also assessed the effect of shear stress on neutrophil responses to three other NET-inducing agents, PMA, LPS and ATP. Among these stimuli, ATP is generated abundantly in the cytosol through respiration and glycolysis pathways49. Extracellularly, ATP acts as autocrine and paracrine signalling modulators as well as a potent purinergic signalling modulator. More importantly, activation of purinergic signalling is an important modulator of neutrophils’ effector responses such as NETosis50,51. Here, we found that shear stress enhances H3 citrullination in response to ATP, a marker of neutrophil activation. However, shear stress does not increase the degree of NETosis in response to ATP, and this difference could be attributed to the timing of the experiment. This finding aligns with a previous report that documented an increase in H3 citrullination and PAD4 expression in response to ATP, but not in the context of NETosis52.
On the other hand, LPS, the main component of the outer membrane of gram-negative bacteria, is reported to induce NETosis via different signalling pathways, depending on the strain of bacteria in a dose-dependent manner53,54. Here, we show that shear stress improves the NETosis and H3 citrullination of neutrophil responses to LPS, suggesting that shear stress can regulate the sensitivity of neutrophils to bacterial infection.
The interaction of platelets with neutrophils is one of the strongest inductors of NETosis, mediated by the interaction of cell surfaces and secreted molecules originating from both platelets and neutrophils13,55. In turn, NETs stimulate platelet activation, further adhesion and aggregation, and thrombin generation, consequently accelerating intravascular coagulation56,57. Here, we found that shear-induced NETosis can provide a scaffold for initial platelet adhesion, triggering further NETosis, platelet shape change and aggregation, and activation of downstream pathways.
Ca2+ signals mediated by ion channels via depletion of intracellular Ca2+ stores or influx of Ca2+ from the extracellular environment regulate the majority of neutrophil defence mechanisms against pathogens, including NETosis29,58. One of the primary regulators of calcium entry into neutrophils is IP3-dependent Ca2+ release from the endoplasmic reticulum stores and store-operated calcium entry via the activation of Ca2+ release-activated Ca2+ channels56. The other important triggering mechanism is mediated by ion channels such as TRP channels and P2X receptors, which can mediate Ca2+ influx in response to exogenous or endogenous signals59,60,61,62.
Importantly, we demonstrate that neutrophils are sensitive to shear stress and that exposure of these cells to shear stress leads to increased intracellular Ca2+ directly dependent on the degree of shear stress. Interestingly, upon exposure to multiple cycles of transient elevation in shear stress, neutrophils retain their shear stress sensitivity. First, we confirmed that shear stress induced NETosis is regulated via Ca2+ influx from the extracellular environment, as inhibiting cation channels and mechanosensitive ion channels blocks the shear-induced calcium influx into the cells. Shear stress acting on the membrane can directly or indirectly activate mechanosensitive ion channels expressed on the plasma membrane.
The ion channel Piezo1 is a prime suspect as a mechanoreceptor that mediates sensitivity to shear stress. Its contribution to the activation of mechanotransduction pathways in different classes of cells and its low threshold for shear stress sensitivity are well known. The curve structure of Piezo1 is reported to play an important role in the activation of this ion channel by mechanical stress63,64,65,66. Furthermore, it is reported that membrane curvatures, such as present in microvilli, also regulate the distribution of Piezo1 on the plasma membrane of different cell types67.
To define the contribution of Piezo1 to NETosis, we used dHL-60 cells as an established model to study the mechanism of NETosis in human neutrophil cells68. Here, we confirm that Piezo1 is expressed in both primary human neutrophils as well as dHL-60 cells. Furthermore, knockdown of Piezo1 in dHL-60 cells using siRNA blocked the shear-induced NETosis in these cells. Additionally, we show that stimulation of Piezo1 with Yoda1 triggers NETosis and increases the adhesion of platelets to NETs. The Piezo1 channel can directly gate in response to shear, leading to calcium influx and integrin activation. Our finding provides evidence of the mechanism by which shear stress and activation of Piezo1 may contribute to NETosis and platelet aggregation in the NET area.
Calpain is a protease expressed in eukaryotic cells that can be activated by high concentration of Ca2+ 69. We hypothesized that calpain is involved in signalling of Ca2+-induced NETosis in neutrophils. Piezo1 activity increased calpain activity in neutrophils and inhibition of calpain blocked shear-induced NETosis, indicating that calpain is acting downstream of Piezo1. Calpain activity contributes to chromatin decondensation and NETosis. Furthermore, calpain regulates actin remodelling and cell spreading. We also found that shear stress increases cell motility, consistent with actin remodelling in response to shear stress. This is consistent with earlier reports showing that functional actin cytoskeleton is required for early stage of NETosis. For example, PMA treatment of neutrophils rapidly increases the actin polymerisation, where it reaches a peak at 60 min and then gradually decreases to the baseline at 180 min68. Furthermore, another study has revealed that neutrophil elastase degrades F-actin 30 min post exposure of neutrophils to Candida albicans70. These previous data combined with our data presented here, the changes in the intensity and ratio of G to F actin and increase in cell motility as an independent marker of cytoskeleton remodelling, suggests that a functional actin cytoskeleton exists at the beginning of NETosis process.
Furthermore, inhibition of calpain reduces cellular motility and inhibition of actin polymerization is sufficient to block NETosis. These data together indicate that cytoskeleton remodelling as a result of calpain activity regulates NETosis and improves cell motility.
In conclusion, we demonstrat that Ca2+ influx via Piezo1 regulates NETosis in neutrophils subjected to shear stress. Furthermore, we conclude that shear stress-induced NETosis is part of the classical pathway of NETosis, as this process take at least 3–4 h, and is accompanied by nuclear and cell membrane rupture, which consequently leads to cell death. NETs themselves provide a scaffold for trapping platelets that results in platelet activation and further prothrombotic effects and can also accelerate NET formation further. We identify calpain activity and cytoskeleton remodelling downstream of Piezo1 as mediators of NETosis. Piezo1 activity not only induces NETosis, but also increases the motility of neutrophils, and improves their sensitivity to NET-inducing agents. The central role of Piezo1 in NETosis induced and accelerated by shear stress identifies this ion channel as a pharmacological target for the treatment of conditions that involve excessive NETosis caused by mechanotransduction.
Methods
Our research is in compliance with the guidelines of the RMIT and Alfred Human Research Ethics Committee.
Compounds and buffers
For calcium imaging, standard Hanks’ balanced salt solution (HBSS) supplemented with 10 mM HEPES (imaging buffer) was used34. For calcium-free experiments, HBSS CaCl2 was replaced with 2 mM ethylene glycol tetraacetic acid.
Chemical agents including GSK690693 (Akt inhibitor), ruthenium red (non-selective calcium channel antagonist), Ly294002 and wortmannin (PI3K inhibitors), thapsigargin (non-competitive inhibitor of sarco/endoplasmic reticulum Ca2+ ATPase [SERCA]), lipopolysaccharides (LPS) from Escherichia coli, adenosine triphosphate (ATP), and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich, Australia. Integrin-linked kinase inhibitor (Cpd22) was purchased from Calbiochem, USA. Yoda1 (selective activator of mouse and human Piezo1) and GsMTx4 (inhibitor of stretch-activated ion channels) were obtained from Tocris Bioscience, UK. Lists of antibodies and dyes used in this study are provided in Supplementary Tables 1 and 2.
Cell culturing and differentiation
HL-60 cells were obtained from American Type Culture Collection (ATCC) (Cat#CCL-240) and cultured at 37 °C and 5% CO2 in RPMI plus l-glutamine supplemented with 25 mM HEPES, 10% heat-inactivated foetal bovine serum, and 1% penicillin and streptomycin. Differentiation of HL-60 cells into neutrophil-like cells was achieved by the addition of 1.25% dimethyl sulfoxide (DMSO) to their growth media for 6 days. The differentiation rate of HL-60 cells was assessed by flow cytometry. Cells were washed in iced cold FACS buffer then stained with anti-human CD11b-PE (Cat# 101208, BioLegend, USA) and anti-human CD16-Alexa 647 (Cat# 302023, BioLegend, USA). The viability of differentiated cells was assessed using fixable viability dye eFluor 780 (Thermo Fisher Scientific, Australia) staining. Data were collected using the LSR Fortessa X-20 flow cytometer (BD Biosciences, USA) and were analysed using FlowJo software.
Peripheral human neutrophil isolation
All protocols involving collection of peripheral blood were approved by RMIT and Alfred Human Research Ethics Committee. Blood collection was performed with informed consent in accordance with guidelines from the RMIT and Alfred Human Research Ethics Committee. Peripheral blood was collected from healthy volunteers using sterile vacuum heparin tubes (Becton Dickinson, Australia). Following this, human neutrophils were isolated from the peripheral blood using a Histopaque double-density gradient created by layering 3 mL of Histopaque-1077 solution (Sigma-Aldrich, Australia) above Histopaque-1119 solution (Sigma-Aldrich, Australia). The cell number and purity of neutrophils were determined using an automated haematology analyser (KX-21N, Sysmex, Japan) and only samples with more than 90% purity were used for experiments. After isolation, neutrophils were rested for 20 min and followed by processing for further experiments.
Preparation of wash platelets
Wash platelets were prepared by treating blood with acid citrate dextrose and 0.02 units/mL apyrase, followed by centrifugation at 180 g for 15 min. Following this, the upper platelet-rich plasma was collected and treated with 0.02 U/mL of apyrase, followed by centrifugation at 1500 g for 7 min. Then the supernatant was discarded and platelets were resuspended in Tyrode’s buffer (1.8 mM CaCl2 with 0.5% bovine serum albumin [BSA]) containing 0.01 U/mL apyrase.
In vitro generation of shear stress to assess NETosis and [Ca2+]i
Shear stress stimulation of neutrophils for NET measurement was achieved by resuspending neutrophils in complete RPMI with 10% FBS and recirculation of cells through the µ-Slide 0.1 (ibidi, Australia) for 10 min at flow rates of 50, 250, and 1000 µL/min to induce shear stress levels of 4, 20, and 80 dyne/cm2, respectively. The 10 min exposure time is based on our preliminary experiments. Exposure for less than 10 min did not reliably yield a significant response. Conversely, doubling the shear stress exposure time results in the loss of many activated cells due to their adherence to the tubing. The static control group was rested inside a chamber slide for the duration of the shear stress stimulation, under otherwise the same condition. To assess the role of extracellular Ca2+ in shear induced NETosis, neutrophils were exposed to shear stress in the presence or absence of 2 mM EGTA in the cell culture media.
Followed by that, cells were collected and incubated inside the chamber slide coated with Cell-Tak™ (Cat# CLS354240, Corning, USA) for 3 h to allow them to adhere and form NETs.
Shear stress stimulation for the measurement of changes in intracellular calcium levels ([Ca2+]i) in neutrophils was performed using calcium imaging and confocal microscopy. Isolated neutrophils were seeded inside the µ-Slide 0.1 (ibidi, Australia) precoated with Cell-Tak™ (Cat# CLS354240, Corning, USA) for 15 min to ensure cell adhesion. The neutrophils were then loaded with 2.5 µM Fluo-4 AM (Thermo Fisher Scientific, Australia) at 37 °C in HBSS buffer for 30 min. The µ-Slide channels were subsequently transferred to the stage of a Nikon A1 confocal microscope (Nikon, Japan) and connected to a syringe pump (SDR Scientific, USA) using a calibrated polyvinyl chloride tube (1.02 mm ID, Gilson, Germany) to facilitate precise fluid flow. Measurement of changes in [Ca2+]i was conducted using a 100× objective lens and excitation with a 488 nm laser. Individual regions of interest (ROIs) around neutrophils were defined using NIS Elements software, and changes in fluorescence intensity were monitored throughout the experiment. The maximum fluorescence intensity value for each ROI was recorded to evaluate the cellular response to shear stress. ImageJ software was used for quantitative assessment, including total cell count, NET area measurement, percentage of H3 positive cells, and the extent of nuclear decondensation.
Assessment of platelet adhesion to NETs
To assess the adhesion of wash platelets to neutrophils, neutrophils were seeded inside the 8-well chamber slide at a density of 2 × 105 cells/well precoated with Cell-Tak™ (Corning, USA) for 3 h. Next, neutrophils were incubated with platelets at a density of 1 × 107 cells per chamber for 10 min at 37 °C. This was followed by washing the non-adhered platelets with PBS, fixing the sample with 4% paraformaldehyde, and finally immunostaining the platelets with anti-human CD41/CD61-Alexa Fluor 647 (#362805, BioLegend, USA), the neutrophils with anti-human CD11a-Alexa Fluor 488 (#301216, BioLegend, USA), and the DNA with DAPI, for 3 h at 37 °C.
Gene silencing
HL-60 cells were cultured at a density of 106 cells per mL in the differentiation media for 72 h. Then cells were transfected with either the ON-TARGETplus human Piezo1 siRNA-SMARTpool (Dharmacon, USA) or the nonspecific control pool (non-targeting siRNA, Dharmacon, USA) using Lipofectamine™ 3000 transfection reagent (Thermo Fisher Scientific, Australia) following suppliers’ instructions. After 4 h of incubation with siRNA/Lipofectamine™ complex in serum-free RPMI media, the condition media was topped up with the differentiation media and cells were assessed after 72 h of transfection for Piezo1 expression and functional responses. The knockdown of Piezo1 was confirmed using qPCR approach.
Western blot analysis
For extraction of whole cellular protein, primary neutrophils or HL-60 cells were rinsed with cold PBS and lysed with RIPA buffer, on ice for 30 min. The protein densitometry analysis was performed using Pierce BCA protein assay kit following the supplier’s instructions. Western blot experiments were performed using standard protocols33. Rabbit anti-Piezo1 antibody (Novus biological, NBP1-78537) or mouse anti-GAPDH antibody (Santa Cruz, sc-166574) were used as primary and mouse anti-rabbit HRP (Santa Cruz biotechnology, #sc-2357) or mouse IgG kappa binding protein HRP (Santa Cruz biotechnology, #sc-516102) were used as secondary antibodies. Membranes were imaged using Odyssey Fc (LI-COR, USA).
Quantitative polymerase chain reaction
To assess Piezo1 expression, RNA was extracted using the RNeasy Mini Kit (QIAGEN). The purity of RNA was then assessed using a NanoDrop (Thermo Fisher Scientific), and complementary DNA (cDNA) was synthesized with the SuperScript IV VILO Master Mix kit (Invitrogen). Subsequently, qPCR was conducted using the TaqMan Master Mix kit (QIAGEN) and the following primers: Hs00207230_m1 for PIEZO1 and Hs00609296_g1 for HMBS (Thermo Fisher Scientific), serving as the internal control. The QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems) was employed for qPCR, and all assays were performed according to the suppliers’ instructions.
Neutrophil live cell tracking assay
Isolated neutrophils were labelled with Hoechst 33342 (Abcam, Australia) and Fluo-4 AM (Thermo Fisher Scientific, Australia) in imaging buffer inside a humidified incubator for 30 min. Next, cells were stimulated with shear stress or Yoda1 as described and added to collagen type I–coated (50 µg/mL) wells of an 8-well chambered cover glass (ibidi, Australia). Neutrophils were allowed to adhere for 10 min in imaging buffer supplemented with 10% FBS on the stage of a Nikon A1 confocal microscope (Nikon, Japan) that was maintained at 37 °C. Cells were imaged using a 60× objective lens (Nikon, numerical aperture 1.2) for a duration of 10 min. Cells were individually tracked, and their distance and velocity were calculated using Imaris software (Bitplane, UK).
Calpain activity assay
Primary neutrophils were stimulated with 10 cycles of shear stress or treated with Yoda1 for 10 min followed by Ca2+-activated cysteine protease (calpain) activity assessment using a Calpain Activity Assay Kit (ab65308, Abcam, Australia) following the supplier’s instructions. Plates were read on Clariostar plus (BMG Labtech, Germany).
Immunofluorescence staining and confocal microscopy
For immune staining, neutrophils were permeabilized with 0.1% Triton X-100 for 15 min and blocked with 1% BSA for 1 h at 37 °C to prevent nonspecific antibody binding. Next, samples were stained with goat anti-human myeloperoxidase (R&D Systems, Australia) and rabbit anti-human histone H3 (Abcam or Sapphire Bioscience, Australia) antibodies at a concentration of 10 µg/mL overnight at 4 °C. Secondary antibody staining was achieved using donkey anti-goat Alexa Fluor 546 (Thermo Fisher Scientific, Australia) and goat anti-rabbit Alexa Fluor 647 (Invitrogen, Australia). DNA staining for assessment of NET formation was achieved using Hoechst 33342 (Abcam, Australia) at 1 µg/mL and SYTOX Green (Invitrogen, Australia) at 10 nM for 1 h at 37 °C.
To assess actin dynamics, following each experiment, neutrophils were immediately fixed with 4% paraformaldehyde in PBS for 1 h on ice. Subsequently, cells were permeabilized with 0.1% Triton X-100 in PBS for 5 min, washed with PBS, and stained with Alexa Fluor 488 DNase I to label G-actin and Alexa Fluor 568 phalloidin to label F-actin. Images were acquired using a Nikon A1 confocal microscope (Nikon, Japan) using a 60× objective lens with oil immersion.
Statistical analyses
Statistical analyses were performed using GraphPad Prism V9 (GraphPad Software). Data are presented and statistical significance calculated as appropriate and as indicated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data supporting the findings of this study are available within the paper, its supplementary information, and the source data file. A list of all antibodies and dyes used in this study is provided in the supplementary file. Additional data associated with the paper and source data file is shared via https://figshare.com/s/4a9fbd34041ee1a0740e. Source data are provided with this paper.
References
Mayadas, T. N., Cullere, X. & Lowell, C. A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 9, 181–218 (2014).
Döring, Y., Soehnlein, O. & Weber, C. Neutrophils Cast NETs in Atherosclerosis. Circ. Res. 114, 931–934 (2014).
Castanheira, F. V. S. & Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 133, 2178–2185 (2019).
Coughlin, M. F. & Schmid-Schönbein, G. W. Pseudopod projection and cell spreading of passive leukocytes in response to fluid shear stress. Biophys. J. 87, 2035–2042 (2004).
Fukuda, S. et al. Mechanisms for regulation of fluid shear stress response in circulating leukocytes. Circ. Res 86, E13–E18 (2000).
Mitchell, M. J., Lin, K. S. & King, M. R. Fluid shear stress increases neutrophil activation via platelet-activating factor. Biophys. J. 106, 2243–2253 (2014).
Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147 (2018).
Noubouossie, D. F. et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 129, 1021–1029 (2017).
Maugeri, N. et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 12, 2074–2088 (2014).
Mangold, A. et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 116, 1182–1192 (2015).
von Brühl, M. L. et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 209, 819–835 (2012).
Stark, K. et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 128, 2435–2449 (2016).
Clark, S. R. et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469 (2007).
Kessenbrock, K. et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15, 623–625 (2009).
Yipp, B. G. & Kubes, P. NETosis: how vital is it? Blood 122, 2784–2794 (2013).
Yipp, B. G. et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med 18, 1386–1393 (2012).
Douda, D. N. et al. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl Acad. Sci. USA 112, 2817–2822 (2015).
Vorobjeva, N. V. & Chernyak, B. V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochem. 85, 1178–1190 (2020).
Yu, X., Tan, J. & Diamond, S. L. Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J. Thromb. Haemost. 16, 316–329 (2018).
Abaricia, J. O., Shah, A. H. & Olivares-Navarrete, R. Substrate stiffness induces neutrophil extracellular trap (NET) formation through focal adhesion kinase activation. Biomaterials 271, 120715 (2021).
Murthy, S. E., Dubin, A. E. & Patapoutian, A. Piezos thrive under pressure: mechanically activated ion channels in health and disease. Nat. Rev. Mol. Cell Biol. 18, 771–783 (2017).
Li, J. et al. Piezo1 integration of vascular architecture with physiological force. Nature 515, 279–282 (2014).
Rode, B. et al. Piezo1 channels sense whole body physical activity to reset cardiovascular homeostasis and enhance performance. Nat. Commun. 8, 350 (2017).
Lai, A. et al. Mechanosensing by Piezo1 and its implications for physiology and various pathologies. Biol. Rev. 97, 604–614 (2022).
Wang, Y. et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 (2009).
Sharda, A., et al. Histone posttranslational modifications: Potential role in diagnosis, prognosis, and therapeutics of cancer. In Prognostic Epigenetics (ed. Sharma, S.) p. 351–373 (Academic Press, 2019)
Perdomo, J. et al. Neutrophil Activation and Netosis Are the Key Drivers of Thrombosis in Heparin-Induced Thrombocytopenia. Blood 132, 378–378 (2018).
Thålin, C. et al. Neutrophil Extracellular Traps. Arteriosclerosis, Thrombosis, Vasc. Biol. 39, 1724–1738 (2019).
Immler, R., Simon, S. I. & Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Investig. 48, e12964–e12964 (2018).
Gottlieb, P. A. & Sachs, F. Piezo1: properties of a cation selective mechanical channel. Channels (Austin) 6, 214–219 (2012).
Lai, A. et al. Analyzing the shear-induced sensitization of mechanosensitive ion channel Piezo-1 in human aortic endothelial cells. J. Cell. Physiol. 236, 2976–2987 (2021).
Baratchi, S. et al. Transcatheter Aortic Valve Implantation Represents an Anti-Inflammatory Therapy Via Reduction of Shear Stress–Induced, Piezo-1–Mediated Monocyte Activation. Circulation 142, 1092–1105 (2020).
Baratchi, S. et al. Shear stress mediates exocytosis of functional TRPV4 channels in endothelial cells. Cell. Mol. Life Sci. 73, 649–666 (2016).
Baratchi, S., et al. The TRPV4 Agonist GSK1016790A Regulates the Membrane Expression of TRPV4 Channels. Front. Pharmacol. 10, 6 (2019)
Gößwein, S. et al. Citrullination Licenses Calpain to Decondense Nuclei in Neutrophil Extracellular Trap Formation. Front Immunol. 10, 2481 (2019).
McCracken, J. M. & Allen, L. A. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell Death 7, 15–23 (2014).
Solis, A. G. et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature 573, 69–74 (2019).
Collins, S. J., Gallo, R. C. & Gallagher, R. E. Continuous growth and differentiation of human myeloid leukaemic cells in suspension culture. Nature 270, 347–349 (1977).
Neeli, I. et al. Regulation of extracellular chromatin release from neutrophils. J. Innate Immun. 1, 194–201 (2009).
Sprenkeler, E. G. G. et al. Formation of neutrophil extracellular traps requires actin cytoskeleton rearrangements. Blood 139, 3166–3180 (2022).
Palmer, L. J. et al. Hypochlorous acid regulates neutrophil extracellular trap release in humans. Clin. Exp. Immunol. 167, 261–268 (2012).
Yoo, D. G. et al. Release of cystic fibrosis airway inflammatory markers from Pseudomonas aeruginosa-stimulated human neutrophils involves NADPH oxidase-dependent extracellular DNA trap formation. J. Immunol. 192, 4728–4738 (2014).
Kaplan, M. J. & Radic, M. Neutrophil extracellular traps: double-edged swords of innate immunity. J. Immunol. 189, 2689–2695 (2012).
Garcia-Romo, G. S. et al. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci. Transl. Med. 3, 73ra20–73ra20 (2011).
Lande, R., et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA–Peptide Complexes in Systemic Lupus Erythematosus. Sci. Transl. Med. 3, 73ra19-73ra19 (2011).
Brill, A. et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thrombosis Haemost. 10, 136–144 (2012).
Fuchs, T. A. et al. Extracellular DNA traps promote thrombosis. Proc. Natl Acad. Sci. 107, 15880–15885 (2010).
Martinac, B. Mechanosensitive ion channels: molecules of mechanotransduction. J. Cell Sci. 117, 2449–2460 (2004).
Dunn, J. & Grider, M. H. Physiology, Adenosine Triphosphate. In StatPearls (StatPearls Publishing, 2022).
Elliott, M. R. et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009).
la Sala, A. et al. Alerting and tuning the immune response by extracellular nucleotides. J. Leukoc. Biol. 73, 339–343 (2003).
Carminita, E. et al. DNAse-dependent, NET-independent pathway of thrombus formation in vivo. Proc. Natl Acad. Sci. 118, e2100561118 (2021).
Pieterse, E. et al. Neutrophils Discriminate between Lipopolysaccharides of Different Bacterial Sources and Selectively Release Neutrophil Extracellular Traps. Front. Immunol. 7, 484 (2016).
Khan, M. A. et al. JNK Activation Turns on LPS- and Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci. Rep. 7, 3409 (2017).
Etulain, J. et al. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 126, 242–246 (2015).
Moschonas, I. C. & Tselepis, A. D. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis 288, 9–16 (2019).
Ling, S. & Xu, J. W. NETosis as a Pathogenic Factor for Heart Failure. Oxid. Med. Cell Longev. 2021, 6687096 (2021).
Hann, J. et al. Calcium signaling and regulation of neutrophil functions: Still a long way to go. J. Leukoc. Biol. 107, 285–297 (2020).
Lecut, C. et al. P2X1 ion channels promote neutrophil chemotaxis through Rho kinase activation. J. Immunol. 183, 2801–2809 (2009).
Lindemann, O. et al. TRPC1 regulates fMLP-stimulated migration and chemotaxis of neutrophil granulocytes. Biochim. Biophys. Acta 1853, 2122–2130 (2015).
Heiner, I. et al. Expression profile of the transient receptor potential (TRP) family in neutrophil granulocytes: evidence for currents through long TRP channel 2 induced by ADP-ribose and NAD. Biochem J. 371, 1045–1053 (2003).
Di Virgilio, F. et al. The P2X7 Receptor in Infection and Inflammation. Immunity 47, 15–31 (2017).
Guo, Y. R. & MacKinnon, R. Structure-based membrane dome mechanism for Piezo mechanosensitivity. eLife 6, e33660 (2017).
Yang, X. et al. Structure deformation and curvature sensing of PIEZO1 in lipid membranes. Nature 604, 377–383 (2022).
Lin, Y.-C. et al. Force-induced conformational changes in PIEZO1. Nature 573, 230–234 (2019).
Haselwandter, C. A. & MacKinnon, R. Piezo’s membrane footprint and its contribution to mechanosensitivity. eLife 7, e41968 (2018).
Yang, S. et al. Membrane curvature governs the distribution of Piezo1 in live cells. Nat. Commun. 13, 7467 (2022).
Thiam, H. R. et al. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc. Natl Acad. Sci. 117, 7326–7337 (2020).
Cong, J. et al. The role of autolysis in activity of the Ca2+-dependent proteinases (mu-calpain and m-calpain). J. Biol. Chem. 264, 10096–10103 (1989).
Metzler, K. D. et al. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 8, 883–896 (2014).
Acknowledgements
The authors would like to acknowledge the Australian Research Council for the Linkage grant (LP190100728) to S.B. and K.K. and Discovery grant (DP200101248) to S.B., and the National Health and Medical Research Council for a L3 Investigator Fellowship support (GNT1174098) to K.P. and an Idea grant (GNT2020197) to S.B, K.K. and K.P.
Author information
Authors and Affiliations
Contributions
S.B. and K.P. conceived and supervised the study. S.B. designed the experiments, developed the methodology, analysed and interpreted data, coordinated and supervised the work, and wrote the manuscript with input from all authors. H.D., C.C., Y.Z., A.H., A.L. and M.K. performed experiments and analysed data. K.K. and K.M.Q. provided technical support and advice on data analysis and interpretation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Rachel Scheraga and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Baratchi, S., Danish, H., Chheang, C. et al. Piezo1 expression in neutrophils regulates shear-induced NETosis. Nat Commun 15, 7023 (2024). https://doi.org/10.1038/s41467-024-51211-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51211-1
- Springer Nature Limited