

Abstract
Vast amounts of pathogen genomic, demographic and spatial data are transforming our understanding of SARS-CoV-2 emergence and spread. We examined the drivers of molecular evolution and spread of 291,791 SARS-CoV-2 genomes from Denmark in 2021. With a sequencing rate consistently exceeding 60%, and up to 80% of PCR-positive samples between March and November, the viral genome set is broadly whole-epidemic representative. We identify a consistent rise in viral diversity over time, with notable spikes upon the importation of novel variants (e.g., Delta and Omicron). By linking genomic data with rich individual-level demographic data from national registers, we find that individuals aged < 15 and > 75 years had a lower contribution to molecular change (i.e., branch lengths) compared to other age groups, but similar molecular evolutionary rates, suggesting a lower likelihood of introducing novel variants. Similarly, we find greater molecular change among vaccinated individuals, suggestive of immune evasion. We also observe evidence of transmission in rural areas to follow predictable diffusion processes. Conversely, urban areas are expectedly more complex due to their high mobility, emphasising the role of population structure in driving virus spread. Our analyses highlight the added value of integrating genomic data with detailed demographic and spatial information, particularly in the absence of structured infection surveys.
Similar content being viewed by others
Introduction
Surveillance of infectious diseases increasingly relies on collecting population-level data from various sources, including demographic, clinical, epidemiological and genomic data1. The value of widespread genomic surveillance of infectious diseases was reinforced during the COVID-19 pandemic, with virus sequencing resulting in tens of millions of samples linked with spatial data2,3,4,5,6,7. Genetic sequencing allows us to identify new variants to assess transmissibility, severity and immune escape of new variants8,9,10,11,12, infer transmission networks4,13,14,15,16 and design novel vaccine targets17,18,19. Yet, the major global responses to pooled data during the COVID-19 pandemic also raised striking inequalities in the data available20,21. Certain countries consistently maintained high sampling and sequencing coverage, both as a proportion of infected individuals and in absolute terms20, while fine-grained demographic and geographic information on samples had restricted availability due to patient privacy. Several countries (e.g. Denmark, United Kingdom, and Singapore) made major investments in testing, sequencing and demographic record-keeping, leading to extensive insights into the viral phylodynamics of SARS-CoV-22,22, importation events13,23,24,25 and recombination26,27. These databases can be powerful tools for decision-making and prevention, taking genomic surveillance far beyond the conventional evolutionary analysis of variant emergence and spread among countries and broad geographic regions8,9,10,11,22.
Critically, extensive efforts in genomic surveillance are costly, and the added value of establishing and maintaining high sampling proportions of the population compared to representative infection surveys remains an open question. Studies of pathogen evolution mostly focus on coarse spatial dynamics of variants, limiting the impact of genomic surveillance to inform spread across smaller clusters of the population, such as across age groups, the vaccinated population and individuals infected with rare variants. One fundamental gap that remains to be filled, before we can maximise the value of genomic surveillance, is linking genomic data with fine-grained spatial and demographic information. Denmark provides a unique opportunity to fill this gap: the centralised nature of Denmark’s registry data allows linking sequences from samples of individuals to their relevant demographic information. In addition, the coverage of PCR testing and sequencing in Denmark was sufficiently high in 2021 to consider the set of whole viral genomes as broadly whole-epidemic representative. During the first half of 2021, sequencing rates among PCR-positive individuals were consistently above 60% due to the intensification of PCR testing28. Coupled with the rapid development of new tools for analysing high-volume viral PCR samples and performing large-scale phylogenetic analysis27,29,30,31, the Danish context is a unique opportunity to explore the fine-grained epidemiology of SARS-CoV-2.
Here, we make phylogenetic inferences that provide detailed information on the timing and evolution of SARS-CoV-2 variants, and the spread across locations and demographic groups during the pandemic in 2021 in Denmark. Specifically, our aims included (i) verifying the expected link between viral genomic diversity and observed trends in the epidemic, such as the arrival of sweeping lineages (ii) comparing the transmission dynamics across different viral lineages and variants, (iii) identifying the role of different demographic groups in transmission and (iv) examining the spatial dynamics of transmission by analysing the correlation between geographic and genomic distances.
Results
Population-level trends
In 2021 in Denmark, 966,094 positive tests (antigen and/or PCR) were reported among individuals with a Danish civil registration number, of which 731,122 were positive PCR tests. From this, a total of 293,287 infection episodes with a high-quality SARS-CoV-2 full-length genome were identified, with an infection episode defined as a 60-day window commencing from an individual’s first positive PCR test; this corresponds to 292,481 unique individuals and 806 with a repeat infection episode in the dataset separated by 60 days in the study period. After removing molecular outliers and sequences with missing metadata, a total of 291,791 SARS-CoV-2 genomes were included in the final dataset for phylogenetic analysis (Fig. 1). In the same period, Statens Serum Institut (SSI) recorded 653,004 cases; we therefore included sequences corresponding to 39.9% of all positive PCR tests in our final dataset, which accounts for 44.6% of all cases identified by SSI.

Data preparation included sequencing, identifying consensus sequences, aligning sequences to the reference sequence, masking sites and analysing nucleotide diversity. Phylogenetic analysis included building a preliminary phylogenetic tree, removing molecular clock outliers, partitioning the tree into sub-clades and re-inferring trees using a Bayesian approach for each sub-clade. Post-processing included inferring the effective reproduction number Re value for each clade, linking tips to registries and conducting phylogeographic analysis.
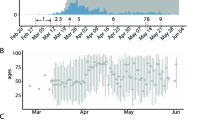
The sampling of full-length genomes showed evidence of ascertainment bias with regard to sex (two-sided χ2 test; χ2 = 1.62 × 101; p = 5.69 × 10−5), age (two-sided χ2 test; χ2 = 1.359 × 103; p < 1 × 10−16) and region (two-sided χ2 = 5.796 × 102, p < 1 × 10−16) (Supplementary Table 1) when compared with individuals with known positive tests (antigen or PCR). Daily collected sequences represented a high proportion of the confirmed cases (Fig. 2a), particularly between March and October, which included the introduction and subsequent sweep of the Delta variant in Denmark. Throughout this period, the proportion of positive PCR tests for which a full-length genome was available was consistently above 60%.

a Number of sequences collected each day by date of testing, separated by lineage, together with the number of confirmed cases published each day by Statens Serum Institut (SSI). b Proportion of sequences collected each day belonging to each major variant. c Infection Ascertainment Rate (IAR) obtained via back-calculation from hospitalisation and mortality data and the proportion of PCR-positive tests taken each day for which we have a WGS. Error bands denote 95% confidence interval. d Proportion of the Danish population that have received a first and second vaccine dose over time. e Nucleotide Diversity calculated for all sequences for each day. f Daily relative growth rate calculated for each major lineage. Error bands denote 98% confidence interval.
For the first half of 2021, before the 1st of June, we observed the co-circulation of several variants, with Alpha being the dominant variant25 (Fig. 2b). Several non-pharmaceutical interventions were implemented and adjusted during this period (Supplementary Table 2), including distancing measures, mask mandates and school closures. The Delta variant quickly dominated all others after the 1st of June, which was preceded by Delta having a much higher observed growth rate compared with other variants throughout May 2021 (Fig. 2f). By August, most restrictions had been lifted (Supplementary Table 2).
By the 1st of September 2021, the central estimate for the infection ascertainment rate (IAR) dropped below 50% and remained consistently below 60% for the rest of the year (Fig. 2c). Although still representing a substantial portion of the estimated infections within the population, this suggests that roughly half of all infections during this period were not captured in the data. This phenomenon coincided with a marked increase in case numbers. Additionally, this time frame witnessed the emergence of two major Omicron lineages (BA.1 and BA.2) which exhibited notably higher estimated growth rates compared to the preceding Delta variant. Consequently, these two variants subsequently dominated, leading to a lineage sweep. We also split the relative growth rates by NUTS 2 (EU Nomenclature of Territorial Units for Statistics) region. The overall pattern was similar across regions; however, there were certain regions where smaller variants did not appear at all, such as the Gamma P.1 variant, possibly due to undersampling, showing that sustained growth of some of these variants was limited to certain geographical regions (Supplementary Fig. 1).
Nucleotide diversity in SARS-CoV-2 genomes from Denmark, a measure of nucleotide-level polymorphism in a population, increased steadily through 2021. Interestingly, large spikes in diversity occurred upon the introduction of new variants (Fig. 2e). This was observed at three time points in our data, with the introduction of the Alpha, Delta and Omicron variants leading to major increases in nucleotide diversity. Once these variants became dominant in the population, the diversity returned to a stable level, after which the nucleotide diversity further increased as mutations accumulated in the viral population. We observed this phenomenon for different diversity metrics, such as average pairwise Hamming and cophenetic distances, as well as the Tajima’s D statistic (Supplementary Fig. 2). Following32, viral diversity was far more stable over time when diversity was considered within-lineage. Our data provide evidence for this phenomenon when considering within-lineage diversity (Supplementary Fig. 2).
To show the impact of novel variant introduction on genetic diversity, we compared the distribution of Hamming distances throughout 2021. When a single major variant was dominant, this distribution was typically unimodal. However, the distribution became bimodal (or, occasionally, multimodal) when new major variants were introduced, coinciding with the timing of new infection waves (Supplementary Fig. 3). This multimodal distribution appears because the pairwise Hamming distances between sequences belonging to the same major variant were lower than between sequences belonging to different major variants. As with nucleotide diversity, spikes in the mean pairwise Hamming distance as well as the appearance of multiple modes in the pairwise Hamming distance distribution coincided with the importation of novel variants and subsequent infection waves. This was because imported variants contained a large number of mutations when compared with existing variants. The phenomenon of the emergence of novel variants that are genetically distinct from existing variants was previously attributed to heterogeneous rates of within-host evolution and the presence of epistasis33.
Clade characterisation and ancestral state reconstruction
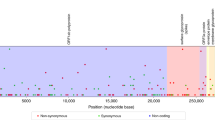
We found that several smaller, lesser-studied variants competed with wild-type and Alpha variants at the beginning of 2021 in Denmark (Fig. 3), including Eta B.1.525, Mu B.1.621, Epsilon (B.1.525 and B.1.429) and Zeta P.2 (Table 1, Supplementary Fig. 4). Larger clades tended to be widely distributed geographically between the five regions of Denmark, albeit with closely related samples tending to be clustered geographically. Some smaller variants were not only highly localised (e.g. Zeta P.2) as one may expect from localised outbreaks but were often broadly regionally distributed (e.g. Beta B.1.351) (Fig. 4). There were also large variations in Re across clades, as inferred from PhyloDeep34 using variant-specific trees, inferred using BEAST35. Newer variants such as Omicron BA.1 and Delta sub-variants tended to have high Re values, yet several smaller sub-clades of these variants had low inferred Re values.

Tip colours represent major variant assignments using pangolin, with yellow tips representing 'others'. Bar plots depict tip distributions across major variants, age groups, and Danish regions. The map delineates the boundaries of Denmark’s five main regions: H (Hovedstaden), M (Midtjylland), N (Nordjylland), SJ (Sjælland) and SY (Syddanmark). In the first quarter of 2021, the population (in millions) in each region according to Statistics Denmark (https://statbank.dk) was: 1.86 (H), 1.33 (M), 0.59 (N), 0.84 (SJ), and 1.22 (SY).

The selected clades are a subset of those shown in Table 1, with several unique clades of the same variant identified during the partitioning of the full tree. Nodes are coloured by their most likely value based on results from ancestral state reconstruction. Heat maps denote the number of directed transitions between regions, z-scored by column such that each column sum = 0. A transition is defined as a node from a given region (source) leading to a subsequent node or tip in the same or different region (target). Map outlines the boundaries of Denmark’s five main regions, with colours corresponding to nodes in the trees: H (Hovedstaden), M (Midtjylland), N (Nordjylland), SJ (Sjælland) and SY (Syddanmark).
Furthermore, upon integrating a parameter for superspreading into the model, we discerned evidence of superspreading within the larger clades (Supplementary Table 5), with variations in their inferred superspreading fractions (i.e. the proportion of superspreaders at equilibrium, fss) and transmission ratios (i.e. the factor by which the transmission rate of super-spreaders exceeds that of typical spreaders, Xss)34,36. For instance, Alpha B.1.1.7 and Omicron BA.1 showed moderate-to-high fss values (0.23 [95% CI: 0.13–0.33] and 0.20 [95% CI: 0.14–0.23], respectively), with corresponding Xss values of 13.98 (95% CI: 8.27–19.4) and 12.80 (95% CI: 8.60–16.73). Conversely, Delta AY.4 and Delta B.1.617 exhibited lower fss values (0.07 and 0.13, respectively), with Xss values of 4.04 (95% CI: 3.21–5.75) and 9.35 (95% CI: 7.05–10.18), respectively. Delta AY.4.2 fell in between, with a fss value of 0.17 (95% CI: 0.11–0.20) and a Xss value of 7.80 (95% CI: 5.95–9.34).
Ancestral state reconstruction analysis across the full tree, as well as for each variant-specific BEAST tree, showed that most transmission events occurred within the same region (61.9% within vs. 38.1% between; two-proportion z-test; χ2 = 64,780; p < 1 × 10−16), as expected under a scenario of geographically constrained spread (Fig. 4 and Supplementary Fig. 5). However, we found substantial transmission between regions, indicated by differences between the inferred region of the origin node and the region of its subsequent target node. While these patterns were consistent for the largest clades, smaller clades showed more variation, with many exhibiting a higher proportion of between-region transmission events. We also found that the time between the first case in a given region to the first date of subsequent cases in another region was highly variable (Supplementary Fig. 6).
In investigating these dynamics across various age demographics, we employed a methodology akin to that utilised for quantifying transitions between regions. Our analysis revealed a predominance of transmissions within homogeneous age cohorts (Supplementary Fig. 7) (69.9% within vs. 30.1% between; two-proportion z-test; χ2 = 182,823; p < 1 × 10−16) when pooling across the calendar year. Upon normalisation of transmission counts within each age cohort, individuals aged between 15 and 30 were most commonly linked with individuals across all other age groups, except for individuals within their age bracket. We also found that, in absolute numbers, all age groups were least likely to be linked with individuals 75+ in age.
Evolutionary rates
To understand how various demographic factors influenced SARS-CoV-2 evolutionary trends, we examined variations in tip lengths and evolutionary rates in the virus across different demographic groups. After adjusting for covariates (sex, age group, region, vaccination status and major variant), we found that molecular rates were marginally faster among individuals in age groups 15–30 and 45–60 compared with those aged 0–15 (two-sample, two-sided t-tests; t15−30 = 2.244; p15−30 = 0.015; t45−60 = 2.539; p45−60 = 0.011), although these differences were negligible in magnitude; no significant disparities were detected among other age groups (Fig. 5). These trends remained when removing zero-length branches (Supplementary Fig. 9), with molecular rates matching those previously estimated for SARS-CoV-237,38,39,40.

Regression coefficients of a model incorporating all four factors to estimate (a) substitution rate using ordinary least squares (OLS) (without interaction between factors) and (b) the number of substitutions using zero-inflated negative binomial regression (without interaction between factors). Groups where the confidence interval does not cross zero (dashed line) indicates significant difference from the reference group. Data are presented as mean ± 95% confidence intervals; n = 289,072 with full metadata for all covariates.
We further investigated these dynamics by examining raw amounts of molecular change (i.e. branch lengths at the tips of the trees), without temporal adjustments (Fig. 5). Notably, individuals aged under 15 and over 75 exhibited a significantly lower contribution to molecular change compared with those between 15 and 75. To test whether this could be due to unsampled individuals along these branches, in which case a terminal branch could represent a whole chain of transmission, we subset the tree to only include samples from April to November, corresponding to the period with the highest infection ascertainment and sequencing rates, finding similar results (Supplementary Fig. 10). The similar molecular rates observed within these age cohorts indicate large amounts of genomic novelty without accelerated rates in the 15–75 age range. In ancestral state reconstruction analyses, we also found individuals in the 15–30 age group to be the most commonly linked to other age groups on the phylogenetic tree, and individuals 75+ in age to be the least likely to be associated with others (Supplementary Fig. 7). Individuals aged 0–15 were most likely to be linked with 30–45 and 45–60 year-olds (and vice-versa), mimicking previously described age-stratified contact patterns corresponding to parent-child contacts41,42,43.
After adjustment for covariates in multivariable regression, the data also revealed marginally slower molecular rates in samples from fully vaccinated individuals (i.e. those who had received two vaccinations) compared with partially and unvaccinated counterparts (Fig. 5; two-sample, two-sided t-tests; tpartial = 2.754; ppartial = 0.006; tunvaccinated = 3.982; punvaccinated = 6.85 × 10−5). Concerning major variants, we found that molecular rates among individuals infected with the Delta and Omicron variants were significantly higher than among those infected with an Alpha variant (Fig. 5; two-sample, two-sided t-tests; tDelta = 7.359; pDelta = 1.86 × 10−13; tOmicron = 24.686; pOmicron = 2.06 × 10−134); this is consistent with previous findings demonstrating higher substitution rates in more recent variants of concern due to incomplete purifying selection, with rates converging over time22,37,44.
Correlation between geographic and genomic distance
Using spatial analyses, we estimated the rate of the geographic spread of SARS-CoV-2 in Denmark in 2021 to be 27,424 km2/year (95% CI: 26,890-27,953), significantly faster than estimates of SARS-CoV-2 diffusion in non-human reservoirs (2050 [95% CI: 233-5470] km2/year)45, Influenza A in avian populations (712 [95% CI: 558-884] km2/year)46 and West Nile Virus in North America (210 [95% CI: 174-25,317] km2/year)47, in line with expectations of comparatively higher rates in human populations due to increased mobility. To place this in the context of known transmission settings, we compared pairwise cophenetic distances (i.e. molecular change) among individuals within and between households. Within each of 1000 unique households including exactly 3 individuals who tested positive in 2021, pairwise cophenetic distances were more similar than distances among individuals in different households, even when adjusting for time (in days) between individuals testing positive (Fig. 6a, b), confirming that genomic data should provide sufficient signal to detect geographically distinct genomic patterns.

a Distribution of mean pairwise cophenetic distances between individuals within the same household (n = 1000 households), normalised to time (i.e. distance divided by time in days between individuals testing positive). b Distribution of mean pairwise cophenetic distances between individuals in different households (n = 1000 households), normalised to time. c Molecular change (i.e. number of nucleotide changes) per 10 km increase in Euclidean distance across various geographic models (national, residential zone, regional, city) (National: n = 20,000 individuals; Urban: n = 18,111; Countryside: n = 1817; Hovedstaden: n = 1000; Midtjylland: n = 1000; Nordjylland: n = 1000; Sjælland: n = 1000; Syddanmark: n = 1000; Copenhagen: n = 3416). Error bars denote 95% confidence intervals. d Molecular change per 10 km increase in car travel distance using OpenStreetMap across different geographic models (national, residential zone, regional, city) (National: n = 20,000 individuals; Urban: n = 18,111; Countryside: n = 1817; Hovedstaden: n = 1000; Midtjylland: n = 1000; Nordjylland: n = 1000; Sjælland: n = 1000; Syddanmark: n = 1000; Copenhagen: n = 3416). Error bars denote 95% confidence intervals. e Mean pairwise cophenetic distance over time, stratified by region (n = 10,000 individuals per region, n = 20,000 for the national subset).
Our analyses of the link between geographic and genomic distance suggest that the spread of SARS-CoV-2 in Denmark in 2021 was complex and unlikely to follow a simple diffusion process. We found no correlation between geographic and genomic distance (Pearson correlation r = −0.008). To explore this relationship in greater detail and to account for temporal delays between individuals testing positive, we investigated the association between geographic and cophenetic distance using multivariable regression; for computational tractability, we randomly sub-sampled 20,000 individuals from the final time tree. We found a weak but negative association (p < 0.001) between travel distance (i.e. shortest distance by car using OpenStreetMap) and cophenetic distance, such that increasing geographic distance between household locations results in smaller cophenetic distances on the national level both when adjusting for time between samples (Fig. 6c, d) and without time-adjustment (Supplementary Fig. 11, Supplementary Table 16, Supplementary Table 17).
Subsequently, we split our 20,000 sub-sample into those living in urban (n = 18,111) and countryside (i.e. rural) (n = 1817) areas, excluding those living in designated summerhouse areas (n = 72) as defined by the Danish Planning Act48. We found that there was a positive association between geographic and cophenetic distance when examining individuals living in rural areas (p < 0.001), but not in urban areas (p < 0.001). We then conducted the same analysis by sampling 10,000 individuals from each of the five regions of Denmark and fitting a model to each independently. We also found that the associations vary significantly between regions. In Midtjylland and Syddanmark, we identified a positive association (p < 0.001) between geographic distance and cophenetic distance, but not in Hovedstaden (p < 0.001), Nordjylland (p < 0.001), or Sjælland (p < 0.001) (Fig. 6c, d). To explore the relationship at finer granularity in urban areas, we identified individuals in the 20,000-individual subset living in the city of Copenhagen (n = 3416, including Frederiksberg municipality), once again finding a strong negative association (p < 0.001) (Fig. 6c, d). Estimates were similar when using Euclidean and travel distances across all geographic models.
Discussion
The significant sampling, PCR testing and sequencing efforts during the COVID-19 pandemic in Denmark in 2021 resulted in a unique dataset of high-coverage and high-volume genomic data that allowed us to understand population-level phylodynamics, even in the absence of structured infection surveys. Using a set of 291,791 sequences from Denmark from 2021, we were able to report the dominant role of novel variants in driving viral genomic diversity and various epidemiological parameters; we revealed the role of working-age adults in mediating viral evolution and transmission; and we showed a strong distinction between urban and rural settings in driving the link between geographic and genomic distance. Overall, our analyses unveil a landscape of links between pooled data types from infectious disease surveillance, only made possible by an integrated data registry and dedicated methods for large-scale analysis of molecular data.
Our findings revealed consistent patterns of slowly increasing nucleotide diversity followed by punctuated spikes with the introduction of new variants. Previous efforts have also reported this saltatory pattern, where notable evolutionary shifts occur in substantial leaps involving multiple mutations rather than through gradual changes, as indicated by a pattern of increasing diversity followed by peaks and subsequent declines in diversity22,33,49. We found consistent signals of diversity across a wide range of metrics, including Tajima’s D, mean pairwise cophenetic distance and mean pairwise Hamming distance, suggesting that these metrics can likely be used interchangeably to monitor population-level evolutionary trends. We also found remarkably consistent rises in within-lineage diversity, which is consistent with stable rates of neutral evolution until new variants are introduced, suggesting that measures of genetic diversity could be used to develop early-warning systems for novel variant emergence.
The evidence of superspreading within larger clades, albeit with variation in estimates between clades and significant estimate-specific uncertainty, also aligned with previous research demonstrating the heterogeneity in SARS-CoV-2 transmission, where the offspring distribution was highly overdispersed50,51,52,53,54. Given the presence of several NPIs during 2021, particularly during the first half of the year, estimates of Xss were likely to be lower than one would expect in periods without NPIs, bounded by the number of effective contacts individuals can have. However, it is worth noting that we only included the first 800 sequences from each of the larger clades when re-inferring the variant-specific BEAST trees; therefore, we cannot rule out that superspreading dynamics may change as specific variants become dominant in the population.
We also found that individuals aged <15 and >75 years had a lower contribution to molecular change (i.e. shorter branch lengths) compared to those between 15 and 75, despite having similar molecular rates. This suggests that mutational novelty in the population is unlikely to be attributable to heightened within-host evolution in any specific age group. Nonetheless, we hypothesise that certain age groups were disproportionately more likely to introduce novel variants from exogenous sources, such as via travel and importation. This would reinforce previous findings which suggest that working-age adults sustained transmission43 and/or initiated rebounds of transmission following lockdowns55. Nevertheless, we cannot dismiss the possibility of significant contributions to transmission by children, especially considering that infections in children were less likely to be detected as they were more likely to experience asymptomatic infections56 and perhaps less likely to be tested in general.
Furthermore, our analysis revealed intriguing trends with regard to the effect of vaccination. Our finding of shorter branch lengths (i.e. less molecular change) among partially vaccinated and unvaccinated individuals compared with fully vaccinated individuals, despite similar rates of molecular evolution, is likely related to increased immune evasion. As within-host immunity increases, the importance of the antigenic novelty of a variant for its reproductive success grows, with immune evasion necessary for subsequent infections57,58. While previous work suggests that mass vaccination could potentially hasten the evolution of SARS-CoV-2, compared to the evolutionary pace seen in natural infections across the population59, we did not find broad evidence that these dynamics are the result of heightened substitution rates in vaccinated individuals. Others have also found that vaccination might influence the diversity of mutations occurring within a host, yet it does not prompt an increase in non-synonymous mutations60. Rather, given the longer branch lengths among vaccinated compared to unvaccinated individuals, the results suggest that evolution and immune escape are necessary for the virus due to the reduced likelihood of infection in vaccinated individuals22,58.
In addition to characterising viral transmission dynamics, our study delved into the spatial and temporal patterns of viral spread. Our findings challenge the assumption of simple diffusion models which assume that geographic distance serves as an accurate proxy for genomic distance47,61,62,63. Our results revealed a weak negative association between geographic and genomic distances on the national level, a trend further strengthened in urban settings, but a weakly positive association in rural settings. This suggests that household location is a poor predictor of genomic dissimilarity, highlighting the importance of including human mobility and social networks to model viral spread, supporting previous work highlighting the importance of effective distance on transmission64, which accounts for the structure of the mobility network. Frameworks such as the gravity or radiation models of mobility65 are alluring due to their simplicity of implementation and interpretation, but these models may only be valid at very broad spatiotemporal scales66. Our analyses highlight the need for access to novel data and realistic models of mobility patterns.
In conclusion, our work demonstrates the major added value of high-coverage sequencing efforts, primarily when pooled with high-resolution demographic and spatial data. Firstly, it allows for the identification of smaller sub-variants, identified through phylogenetic analysis, which would otherwise be undetectable with lower sequencing rates. Secondly, by linking large-scale genomic data with registry data, it is possible to untangle the role of different demographic groups in viral transmission and evolution, particularly when representative sampling strategies are lacking. Such information is crucial from a public health and policy perspective to understand which settings of transmission should be prioritised to break transmission chains. Lastly, understanding the role of vaccination on viral evolution, apart from the mitigating effects on disease and infection rates, can allow us to model the balance between slowing transmission and reducing disease burden through vaccination and the selection pressure this introduces on viruses.
However, while the comprehensive sequencing efforts undertaken in Denmark provide unique insights into the dynamics of SARS-CoV-2 transmission, maintaining high-coverage sequencing efforts is costly and labour-intensive. Systematic sampling surveys can permit inference of population-level trends in disease incidence, such as the Office of National Statistics (ONS) COVID-19 Infection Survey in the United Kingdom67,68, but the design of more parsimonious sampling strategies remains under-explored and is likely to depend on one’s parameter of interest (e.g. novel variant detection, sociodemographic distributions of disease, routes of transmission). Exploring these strategies, taking account of different parameters of interest, is likely to be crucial for future epidemic responses.
It is also worth acknowledging the limitations of the dataset, despite its high coverage. For one, there are ascertainment biases (e.g. age, sex and regional differences in our sample) that may over-attribute infections to certain regions (e.g. the capital region of Denmark) or to certain age groups. Additionally, we assumed that consensus sequences did not vary across an individual’s defined infection episode. While previous research suggests that this is a valid assumption69, with consensus sequences remarkably stable over time even in the context of persistent infections, the role of intra-host variation on inferred phylogenetic relationships between individuals merits further research. Lastly, because we only included sequences from Denmark in our analyses, we may have overlooked the influence of virus exports and subsequent re-introductions. That being said, we expect the effect of this bias on our results to be small. Despite these limitations, our study contributes significantly to a growing understanding of the complex spatial dynamics of virus epidemics, highlighting the need for integrated approaches that consider genomic information, geographic proximity and social connectivity in modelling disease transmission.
Methods
Genomic data and testing strategy
Sequencing SARS-CoV-2 was systematically done as part of Denmark’s COVID-19 response. Denmark implemented a dual testing strategy for SARS-CoV-2, dividing testing into healthcare and community tracks to curb transmission, safeguarding vulnerable populations and preventing the overload of healthcare infrastructure70. The healthcare track initially analysed all samples and later focused on clinical testing, including both in- and out-patients70,71,72. In addition to the healthcare track, Denmark established the community track to offer free, on-demand testing without referrals. During the pandemic, trained professionals collected oropharyngeal swabs for PCR testing at various testing stations established across the country70, while self-administered home tests only became routine by December 202172. The following vaccinations were used as part of the Danish vaccination programme: Comirnaty (Pfizer/BioNTech); Spikevax (Moderna); Vaxzevria (AstraZeneca); and COVID-19 VACCINE Janssen (Johnson & Johnson)72. Individuals were considered fully vaccinated if they had received two vaccinations and partially vaccinated if they had received one vaccination by the date of their infection, with some individuals combining vaccination types. An overview of the timings of different non-pharmaceutical interventions implemented in 2021 can be found in Supplementary Table 2.
The Danish COVID-19 Genome Consortium (DCGC), established in March 2020, conducted the sequencing to monitor the evolution of SARS-CoV-2. The DCGC encompassed all genomic sequences that met the inclusion criteria based on a cycle threshold (Ct) value, with thresholds ranging from 30 to 38 during the study period73. For the first half of 2021, whole genome sequencing was performed mostly at Aalborg University (AAU) with contributions from Statens Serum Institut and different regional clinical microbiology laboratories. Whole genome amplification of SARS-CoV-2 employed a modified version of the ARTIC tiled PCR scheme74 (https://artic.network), targeting 33 overlapping amplicons ranging between 1000 and 1500 base pairs; for barcoding the amplicon libraries, a custom 2-step PCR strategy was utilised. Barcoded libraries underwent normalisation, pooling and preparation for sequencing using the SQK-LSK109 ligation kit from Oxford Nanopore; sequencing was performed on the MinION device using R.9.4.1 flow cells from Oxford Nanopore75. Raw sequencing data underwent base calling with Guppy v.3.6.1 (https://nanoporetech.com) and demultiplexing using a custom cutadapt v.2.10 wrapper76. Consensus sequences were generated using the artic minion function with default settings from the ARTIC network protocol (v.1.1.0), incorporating medaka77 for consensus calling. For the second half of 2021, the majority of whole-genome sequencing was conducted at Statens Serum Institut using the ARTIC v3 amplicon sequencing panel78; this consisted of 98 overlapping amplicons of ~300 nucleotides each, with custom spike-ins to maintain consistent amplicon coverage over time79. Sequencing was performed on either the NextSeq or NovaSeq platforms (Illumina), employing paired read lengths spanning from 51 to 150 nucleotides, with a majority of paired reads of length 74. Reads were trimmed using trim-galore v.0.6.10 (https://github.com/FelixKrueger/TrimGalore) and consensus sequences were generated using either an internally-developed iVar80 (v.1.4.3) implementation or a combination of iVar and a custom BCFtools (v.1.18) command for consensus calling. A PHRED score cut-off of 20 was applied throughout, first at the read trimming step (trimgalore) and then at the primer trimming step (iVar). Furthermore, a fraction of sequences from different regional clinical microbiology labs using either Nanopore or Illumina sequencing were included as part of the sequence database.
Infection episodes were defined by SSI as a 60-day window from the first positive PCR test. The defined consensus sequence for each individual was defined as the best sequence (i.e. the lowest number of ambiguous bases) within an individual’s 60-day window. Only sequences with fewer than 3000 missing (N’s) or ≤ 5 ambiguous base calls, with high yield compared to control and not manually excluded by the quality control (QC) team due to suspicion of plate contamination were considered high-quality and included in the national sequencing database. Sequences were checked against SARS-CoV-2 genome reference models to identify possible errors such as frameshifts using VADR81 before uploading to sequence data repositories. Sequencing metadata contained information about the sampling and sequencing date. We restricted our study sample to the period from January 1, 2021 to December 31, 2021 to analyse the period with the highest infection ascertainment rate. The data included in the final genomic dataset prior to phylogenetic analysis consisted of 293,287 consensus sequences. Among these, 259,106 high-quality annotation-checked genomes were uploaded to GISAID’s EpiCoV database. The remaining sequences were part of SSI’s internal genome collection and were not included in the national sequencing database for not satisfying one of the above-described quality control criteria.
Registry linkage, case counts and infection ascertainment rates
To link sequences with Danish registry data, pseudo-anonymized Danish civil registration numbers (CPR numbers) were used to link sequences to their corresponding registry information stored at Statistics Denmark. Registry information included sex, age, test date, vaccination status and dates, household NUTS 2 region (EU Nomenclature of Territorial Units for Statistics) (Hovedstaden, Midtjylland, Nordjylland, Sjælland and Syddanmark), and household location (i.e. latitude and longitude). To explore selection to sequencing, we compared the distribution of positive individuals (PCR- and/or antigen-positive) with whole-genome sequenced individuals based on sex, age and region (Supplementary Table 1).
Case counts and COVID-specific mortality numbers were sourced from weekly reports by SSI. A semi-mechanistic branching process model, using case counts and reported deaths was used to model the disease transmission dynamics82. A component was added to the model to infer the time-varying weekly Incidence Ascertainment Ratios (IAR) following the approach of Mishra et al. 83. The generation time distribution g was unknown but was approximated with the distribution of the serial interval, which was assumed to be Gamma distributed g ~ Γ(6.5, 0.62)84. This allowed us to link observed cases with infections and infer the IAR. We defined the IAR as a weekly random walk with a link function, specifically a doubly inverse logit function. The parameters of the model were jointly estimated, and the inference was performed in R using Stan85 (v.2.32.5).
Genetic diversity metrics
To characterise the diversity in sequences over time, we calculated nucleotide diversity for the sequences per day as first introduced by Nei and Li86. We choose this measure of diversity as our primary measure since it is the most insensitive to the number of sequences present in the sample per day87. Given a set of N nucleotide sequences of length L, the nucleotide diversity π is given by:
where Ni denotes the number of observations of the allele i at each site, and i ∈ {A, C, G, T}. We calculated this score for all sequences over time throughout 2021, as well as for data separated by each major lineage, using Pangolin version 4.3.1.
In addition to nucleotide diversity, we also calculated the pairwise Hamming distance88 between sequences, which is the raw number of site differences between two given sequences. This allows for a pairwise distance matrix for each day, whose (i, j)th entry is the Hamming distance between sequence i and sequence j. We then considered both the distribution of pairwise Hamming distances between sequences collected each week, as well as the daily Hamming distance of all sequences from the Wuhan WIV04 (MN996528.1) reference sequence89. These provide a complementary measure of the changing diversity of viral lineages in the population.
Finally, we also calculated Tajima’s D statistic for the sequences collected on each day90. Tajima’s D statistic is a measure of the extent to which mutations that arise in a collection of sequences are the result of either purifying selection, an increase in the effective population size, or are consistent with neutral genetic drift91. For a collection of N sequences of length L, we calculate the number of segregating sites S, which is the number of sites at which there is more than one distinct allele present, and subsequently the average proportion θp of nucleotide differences between pairs of sequences in the sample. We then calculate \(a=\mathop{\sum }_{i=1}^{N-1}\frac{1}{i}\) and let θS = S/(La). Tajima’s D statistic for the sample is then given by:
where SE( ⋅ ) is the standard error of the difference of the two statistics. Values of Tajima’s D statistic below D = 2 suggest that the evolution of the virus is not consistent with neutral evolution. We calculated Tajima’s D statistic for 1000 bootstrapped samples of size 100 taken from the sequences collected on each day to obtain confidence intervals for the D statistic.
Relative growth rates
We modelled the proportion over time of observed sequences belonging to each major lineage using a Gaussian Process following32 to calculate the relative growth rate of each lineage compared to all others over time. We used the functions MCMC and NUTS92 from the package numpyro (v.0.13.2) in Python93 to fit a Gaussian Process to the number of sequences collected from each major lineage. This enabled a substantial speedup in the computation, which allowed us to obtain more samples from the Gaussian Process. For estimates of real-time growth rates, we took 5000 samples from a Gaussian Process fitted using a squared exponential kernel in numpyro from a larger sample of 10,000 to reduce sample auto-correlation. The next step was to calculate the growth rate by comparing the results for each lineage with all other lineages sampled at that time. To do this, we took the set of all lineages considered to be \({{\mathcal{L}}}\), then the fitted Gaussian Process has mean Xi(t) for variant \(i\in {{\mathcal{L}}}\) and had mean \({X}_{{{\mathcal{L}}}\backslash \{i\}}(t)\) for all other lineages at time t. The relative growth rate for variant i was then calculated as
Phylogenetic analysis and epidemiological parameter inference
Sequences were aligned to the Wuhan WIV04 (MN996528.1) reference sequence89 using MAFFT (v.7.520)94. Following alignment, problematic sites identified by De Maio et al. 31 were masked using augur mask from the Nextstrain pipeline95 (Augur version v.22.3.0), which included sites at the 5’ (sites 1-55), 3’ ends (site 28804 - end), and several sites along the genome. A full set of masked sites is available in Supplementary Table 6. Masked sites were transformed to ‘N’ and excluded in subsequent analyses. We used MAPLE (MAximum Parsimonious Likelihood Estimation; v.0.2.1)31, a recently published method for inferring large SARS-CoV-2 phylogenies using maximum-likelihood inference, to create a full genetic distance phylogenetic tree including all consensus sequences.
Branch lengths from MAPLE were fit to a gamma distribution, after which we discarded branches longer than the 99th percentile to exclude implausibly divergent sequences. To estimate a time tree for the full dataset, we used Chronumental30 (v.0.0.62), a novel tool to estimate time trees and branch lengths for extremely large phylogenies, which uses stochastic gradient descent to maximise the evidence lower bound under a probabilistic model. To identify molecular clock outliers, we ran a Bayesian regression model using brms96 (v.2.20.3) with sample dates and predicted dates from Chronumental30 as input covariates. After removing several molecular clock outliers as well as sequences with missing date information, the final time tree contained 291,791 tips.
To infer the Pango (Phylogenetic Assignment of Named Global Outbreak Lineages) lineage of each sequence, we used Pangolin (v.4.3.1). We then partitioned the final time tree into smaller clades using a custom Python script from ref. 13 (v.1.0). The script identifies subtrees with a high degree of clustering while accounting for uncertainty in Pango assignments. We segmented the complete tree into 31 distinct clades. To ensure reliable tree inference, we selected 18 representative variant clades and excluded extremely rare clades (<25 samples). Large clades (n > 800) were also characterised in their entirety (Supplementary Table 3).
We then re-inferred the time trees for each clade using Bayesian inference as implemented in BEAST v1.10.435, using a Bayesian Skyline model. To make these analyses computationally tractable, we limited partitions with ≥800 taxa to include only the first 800 sequences chronologically. We ran between two and six Markov chain Monte Carlo (MCMC) chains with between 10 and 300 million states - depending on the size of the clade—using a burn-in of between 10 and 40% (see Supplementary Table 7 for clade-by-clade information). We merged chains and assessed model convergence using the BEAST functions LogCombiner and Tracer, respectively, ensuring convergence for parameters relating to the tree without mandating convergence for population sizes. Tree visualisation was done using ggtree97 (v.3.7.2) and Taxonium98 (v.2.0.110).
To infer epidemiological parameters (i.e. Re, fss, Xss) for clades with >50 tips, we used PhyloDeep34, a recently-developed deep learning-based method to infer birth-death parameters from phylogenetic trees, which outperforms similar methods for diversification rate parameter inference. To avoid issues of non-identifiability for birth-death parameter inference99, we estimated sampling fractions for each clade before running PhyloDeep. We defined each clade-specific period as the time between the first and last samples in each clade. To estimate the sampling fraction across this period, we took the average inferred IAR for each day across the period and multiplied it by the average sequencing rate, defined as the percentage of PCR/antigen-positive individuals that were subsequently sequenced and included in the dataset. Clade-specific sampling fractions ranged from 0.02 to 0.62. To explore the role of the sampling fraction on Re, we repeated analyses by uniformly assuming a sampling fraction of 0.5 (Supplementary Table 11).
For each clade, we inferred the most likely region for each node across each clade-specific tree and across the full tree. This was done by conducting ancestral state reconstruction using maximum likelihood inference100 as implemented using the ace function in ape101 (v.5.7-1). To quantify the connections between different regions, we tallied the occurrences of transitions between each pair of regions. A transition was identified as a movement from one region to another, where a node from the initial region led to a subsequent node or tip in either the same region or a different one, progressing forward in time. We conducted a similar analysis by splitting individuals into different age groupings (0–15, 15–30, 30–45, 45–60, 60–75, 75+) and re-running ancestral state reconstruction across the full tree.
Analysis of evolutionary rates
To explore the relationship between different demographic factors and virus evolutionary rates at the time of sampling, we first calculated the number of molecular changes observed in individual virus samples (the length of phylogenetic tree ’tips’, or terminal branches, hereafter tip length). We calculated the mean and 95% confidence intervals (CIs) of these values across individuals stratified by age, sex, region, month and vaccination status using the full phylogenetic tree. Due to the density of sampling, such that many samples had identical genomic sequences, we also conducted the same analyses excluding samples with tip lengths of zero and while using the number of molecular changes with zero-inflated negative binomial regression. We then fit a multivariable regression model using ordinary least squares to test the five covariates as partial terms predicting molecular rates (i.e. molecular changes divided by time, where time is the corresponding length of branches on the time tree).
Spatial analyses and geographic spread of variants
To explore the spatial relationship between the geographic location of individuals and their genomic distance, we randomly sub-sampled 20,000 people from the final, pruned time tree. Differences by area type were explored by subsetting the 20,000 samples into those living in urban vs. countryside (i.e. rural) areas as defined by the Danish Planning Act48. Additionally, we randomly sub-sampled 10,000 people from each NUTS 2 region in Denmark. Finally, we defined postcodes for the city of Copenhagen and subset the 20,000 samples to include the individuals living in those postcodes. We therefore created a total of 9 unique datasets (one national, two area-type, five regional, and one city dataset).
For each dataset, we calculated pairwise distance matrices for (1) cophenetic distances between virus samples for individuals (from virus phylogeny), (2) geographic distances using household coordinates, (3) driving distances between households, and (4) absolute time (days) between samples. Cophenetic distances were calculated using ape101. Geographic distances were calculated using the haversine formula, implemented in the geosphere package102 (v.1.5-18), assuming a radius of 6,378,137 metres. Driving distances between coordinates were extracted using the Open Source Routing Machine engine103 (v.5.27.1), which leverages OpenStreetMap data to calculate the shortest driving route between two points. Distance in time was calculated by taking the absolute difference (in days) between each person’s PCR test date.
For each distance matrix, a submatrix operation was applied to isolate its lower triangular elements, which were subsequently vectorized. Using cophenetic distance as the dependent variable, a linear model was constructed to investigate the relationship between geographic distance (Euclidean or transportation) and cophenetic distance, while simultaneously adjusting for temporal variations by including the time matrix in the model. Models were constructed for all nine datasets to explore whether national-level trends were consistent with those found on a regional level.
As a confirmatory analysis, we explored whether the relationship between geographic and cophenetic distance was consistent when collapsing infection clusters on a household level. To do this, we selected 1000 random households, all of which had 3 unique individuals testing positive with a full-length genome in 2021. All individuals were registered as living in the given household at the time of infection. As above, we then compared the cophenetic distances among individuals between and within households, with the diagonal of the matrix representing the average pairwise distances between individuals within a household and non-diagonal elements representing the average pairwise distances between individuals in different households.
To estimate the spatial diffusion of SARS-CoV-2 on the national level, we subset the final time tree to include only the tips from the national 20,000-tip sub-sample. We estimated diffusivity under Spherical Brownian Motion (SBM), which provides a model of change in geographic locations through lineages. Parameters were inferred using maximum-likelihood as implemented in the fit_sbm_geobiased_const function of the castorR package104 (v.1.7.11; R v.4.3.2). To address and mitigate geographic sampling biases, this method uses an iterative simulation and fitting procedure until convergence is achieved. Accordingly, we included 1000 bootstrap replicates, 1000 SBM simulations, and a sampling fraction of 0.4 to estimate standard errors and make geographic bias estimates, respectively.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data utilised in this study is accessible under restricted conditions under Danish data protection laws. Researchers can request access to the data from The Danish Health Data Authority and Statens Serum Institut, complying with Danish data protection regulations and any necessary permissions. No data collection or sequencing was conducted specifically for this study. 259,106 high-quality SARS-CoV-2 consensus genomes used in this study are available on GISAID’s EpiCoV database under the EPI-SET accession number EPI_SET_240423qn (https://epicov.org/epi3/epi_set/240423qn). Synthetic data to demonstrate the code functionality is available online at https://github.com/MLGlobalHealth/sars_cov2_290k_denmark105.
Code availability
All code relevant to reproducing the experiments is available online at https://github.com/MLGlobalHealth/sars_cov2_290k_denmark105.
References
Grenfell, B. T. et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303, 327–332 (2004).
Attwood, S. W., Hill, S. C., Aanensen, D. M., Connor, T. R. & Pybus, O. G. Phylogenetic and phylodynamic approaches to understanding and combating the early SARS-CoV-2 pandemic. Nat. Rev. Genet. 23, 547–562 (2022).
Lu, R. et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574 (2020).
Matteson, N. L. et al. Genomic surveillance reveals dynamic shifts in the connectivity of COVID-19 epidemics. Cell 186, 5690–5704.e20 (2023).
Coppée, R. et al. Phylodynamics of SARS-CoV-2 in France, Europe, and the world in 2020. eLife 12, e82538 (2023).
Rich, S. N. et al. Application of phylodynamic tools to inform the public health response to COVID-19: qualitative analysis of expert opinions. JMIR Form. Res. 7, e39409 (2023).
Dellicour, S. et al. Epidemiological hypothesis testing using a phylogeographic and phylodynamic framework. Nat. Commun. 11, 5620 (2020).
McCrone, J. T. et al. Context-specific emergence and growth of the SARS-CoV-2 Delta variant. Nature 610, 154–160 (2022).
Mlcochova, P. et al. SARS-CoV-2 B. Nature 599, 114–119 (2021).
Kraemer, M. U. G. et al. Spatiotemporal invasion dynamics of SARS-CoV-2 lineage B. Science 373, 889–895 (2021).
Tegally, H. et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592, 438–443 (2021).
Rasmussen, M. et al. First cases of SARS-CoV-2 BA.2.86 in Denmark, 2023. Euro Surveill. 28, 2300460 (2023).
Tsui, J. L.-H. et al. Genomic assessment of invasion dynamics of SARS-CoV-2 Omicron BA. Science 381, 336–343 (2023).
Bedford, T. et al. Cryptic transmission of SARS-CoV-2 in Washington state. Science 370, 571–575 (2020).
Truong Nguyen, P. et al. The phylodynamics of SARS-CoV-2 during 2020 in Finland. Commun. Med. 2, 65 (2022).
Chan, W.-M. et al. Phylogenomic analysis of COVID-19 summer and winter outbreaks in Hong Kong: an observational study. Lancet Reg. Health West. Pac. 10, 100130 (2021).
Krammer, F. SARS-CoV-2 vaccines in development. Nature 586, 516–527 (2020).
Corbett, K. S. et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020).
Bambini, S. & Rappuoli, R. The use of genomics in microbial vaccine development. Drug Discov. Today 14, 252–260 (2009).
Brito, A. F. et al. Global disparities in SARS-CoV-2 genomic surveillance. Nat. Commun. 13, 7003 (2022).
Maxmen, A. One million coronavirus sequences: popular genome site hits mega milestone. Nature 593, 21–21 (2021).
Markov, P. V. et al. The evolution of SARS-CoV-2. Nat. Rev. Microbiol. 21, 361–379 (2023).
Du Plessis, L. et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 371, 708–712 (2021).
Hodcroft, E. B. et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 595, 707–712 (2021).
Michaelsen, T. Y. et al. Introduction and transmission of SARS-CoV-2 lineage B. Genome Med. 14, 47 (2022).
Pipek, O. A. et al. Systematic detection of co-infection and intra-host recombination in more than 2 million global SARS-CoV-2 samples. Nat. Commun. 15, 517 (2024).
Turakhia, Y. et al. Pandemic-scale phylogenomics reveals the SARS-CoV-2 recombination landscape. Nature 609, 994–997 (2022).
Kaspersen, K. A. et al. Estimation of SARS-CoV-2 infection fatality rate by age and comorbidity status using antibody screening of blood donors during the COVID-19 epidemic in Denmark. J. Infect. Dis. 225, 219–228 (2022).
Turakhia, Y. et al. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 53, 809–816 (2021).
Sanderson, T. Chronumental: time tree estimation from very large phylogenies. bioRxiv (2024).
De Maio, N. et al. Maximum likelihood pandemic-scale phylogenetics. Nat. Genet. 55, 746–752 (2023).
Lythgoe, K. A. et al. Lineage replacement and evolution captured by 3 years of the United Kingdom Coronavirus (COVID-19) Infection Survey. Proc. R. Soc. B 290, 20231284 (2023).
Nielsen, B. F. et al. Host heterogeneity and epistasis explain punctuated evolution of SARS-CoV-2. PLoS Comput. Biol. 19, e1010896 (2023).
Voznica, J. et al. Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks. Nat. Commun. 13, 1–14 (2022).
Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 1–8 (2007).
Stadler, T. & Bonhoeffer, S. Uncovering epidemiological dynamics in heterogeneous host populations using phylogenetic methods. Philos. Trans. R. Soc. B Biol. Sci. 368, 20120198 (2013).
Tay, J. H., Porter, A. F., Wirth, W. & Duchene, S. The emergence of SARS-CoV-2 variants of concern is driven by acceleration of the substitution rate. Mol. Biol. Evol. 39, msac013 (2022).
Wang, S. et al. Molecular evolutionary characteristics of SARS-CoV-2 emerging in the United States. J. Med. Virol. 94, 310–317 (2022).
Forni, D., Cagliani, R., Pontremoli, C., Clerici, M. & Sironi, M. The substitution spectra of coronavirus genomes. Brief. Bioinform. 23, bbab382 (2022).
Neher, R. A. Contributions of adaptation and purifying selection to SARS-CoV-2 evolution. Virus Evol. 8, veac113 (2022).
Mistry, D. et al. Inferring high-resolution human mixing patterns for disease modeling. Nat. Commun. 12, 323 (2021).
Prem, K., Cook, A. R. & Jit, M. Projecting social contact matrices in 152 countries using contact surveys and demographic data. PLoS Comput. Biol. 13, e1005697 (2017).
Monod, M. et al. Age groups that sustain resurging COVID-19 epidemics in the United States. Science 371, eabe8372 (2021).
Ghafari, M. et al. Purifying selection determines the short-term time dependency of evolutionary rates in SARS-CoV-2 and pH1N1 Influenza. Mol. Biol. Evol. 39, msac009 (2022).
Pekar, J. E. et al. The recency and geographical origins of the bat viruses ancestral to SARS-CoV and SARS-CoV-2. bioRxiv (2023).
Trovão, N. S., Suchard, M. A., Baele, G., Gilbert, M. & Lemey, P. Bayesian inference reveals host-specific contributions to the epidemic expansion of Influenza A H5N1. Mol. Biol. Evol. 32, 3264–3275 (2015).
Pybus, O. G. et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc. Natl Acad. Sci. USA 109, 15066–15071 (2012).
By-, Land- og Kirkeministeriet. Bekendtgørelse af lov om planlægning. https://www.retsinformation.dk/eli/lta/2020/1157 (2020).
Harari, S. et al. Drivers of adaptive evolution during chronic SARS-CoV-2 infections. Nat. Med. 28, 1501–1508 (2022).
Lloyd-Smith, J. O., Schreiber, S. J., Kopp, P. E. & Getz, W. M. Superspreading and the effect of individual variation on disease emergence. Nature 438, 355–359 (2005).
Adam, D. C. et al. Clustering and superspreading potential of SARS-CoV-2 infections in Hong Kong. Nat. Med. 26, 1714–1719 (2020).
Endo, A., Centre for the Mathematical Modelling of Infectious Diseases COVID-19 Working Group, Abbott, S., Kucharski, A. J. & Funk, S. Estimating the overdispersion in COVID-19 transmission using outbreak sizes outside China. Wellcome Open Res. 5, 67 (2020).
Wegehaupt, O., Endo, A. & Vassall, A. Superspreading, overdispersion and their implications in the SARS-CoV-2 (COVID-19) pandemic: a systematic review and meta-analysis of the literature. BMC Public Health 23, 1003 (2023).
Kirkegaard, J. B. & Sneppen, K. Superspreading quantified from bursty epidemic trajectories. Sci. Rep. 11, 24124 (2021).
Tran Kiem, C. et al. SARS-CoV-2 transmission across age groups in France and implications for control. Nat. Commun. 12, 6895 (2021).
Sah, P. et al. Asymptomatic SARS-CoV-2 infection: a systematic review and meta-analysis. Proc. Natl Acad. Sci. USA 118, e2109229118 (2021).
Paton, R. S., Overton, C. E. & Ward, T. The rapid replacement of the SARS-CoV-2 Delta variant by Omicron (B.1.1.529) in England.Sci. Transl. Med. 14, eabo5395 (2022).
Carabelli, A. M. et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat. Rev. Microbiol. 21, 162–177 (2023).
Rouzine, I. M. & Rozhnova, G. Evolutionary implications of SARS-CoV-2 vaccination for the future design of vaccination strategies. Commun. Med. 3, 86 (2023).
Gu, H. et al. Within-host genetic diversity of SARS-CoV-2 lineages in unvaccinated and vaccinated individuals. Nat. Commun. 14, 1793 (2023).
Lemey, P., Rambaut, A., Welch, J. J. & Suchard, M. A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 27, 1877–1885 (2010).
Lemey, P., Rambaut, A., Drummond, A. J. & Suchard, M. A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5, e1000520 (2009).
Faria, N. R., Suchard, M. A., Rambaut, A. & Lemey, P. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 1, 423–429 (2011).
Brockmann, D. & Helbing, D. The hidden geometry of complex, network-driven contagion phenomena. Science 342, 1337–1342 (2013).
Simini, F., Gonzalez, M. C., Maritan, A. & Barabasi, A.-L. A universal model for mobility and migration patterns. Nature 484, 96–100 (2012).
Li, X., Tian, H., Lai, D. & Zhang, Z. Validation of the gravity model in predicting the global spread of influenza. Int. J. Environ. Res. Public Health 8, 3134–3143 (2011).
Walker, A. S. COVID-19 infection survey of the UK general population Institution: ISRCTN. https://doi.org/10.1186/ISRCTN21086382.
Pouwels, K. B. et al. Community prevalence of SARS-CoV-2 in England from April to November, 2020: results from the ONS Coronavirus Infection Survey. Lancet Public Health 6, e30–e38 (2021).
Ghafari, M. et al. Prevalence of persistent SARS-CoV-2 in a large community surveillance study. Nature 626, 1094–1101 (2024).
Gram, M. A. et al. Patterns of testing in the extensive Danish national SARS-CoV-2 test set-up. PLoS ONE 18, e0281972 (2023).
Schønning, K. et al. Electronic reporting of diagnostic laboratory test results from all healthcare sectors is a cornerstone of national preparedness and control of COVID-19 in Denmark. APMIS 129, 438–451 (2021).
Lyngse, F. P. et al. Effect of vaccination on household transmission of SARS-CoV-2 Delta variant of concern. Nat. Commun. 13, 3764 (2022).
Lyngse, F. P. et al. Increased transmissibility of SARS-CoV-2 lineage B. Nat. Commun. 12, 7251 (2021).
Quick, J. nCoV-2019 sequencing protocol v2 (GunIt) v2. https://www.protocols.io/view/ncov-2019-sequencing-protocol-v2-bdp7i5rn (2020).
Sorensen, E. A., Karst, S. M. & Knutsson, S. AAU-nCoV-2019_Tailed_Long_Amplicon_Sequncing V.2 https://www.protocols.io/view/aau-ncov-2019-tailed-long-amplicon-sequncing-bfc3jiyn (2020).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10 (2011).
nanoporetech/medaka original-date: 2017-06-07. https://github.com/nanoporetech/medaka (2024).
Quick, J. nCoV-2019 sequencing protocol v3 (LoCost) v3. https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (2020).
Tang, M.-H. E. et al. Comparative subgenomic -mRNA profiles of SARS-CoV-2 Alpha, Delta and -Omicron BA. eBioMedicine 93, 104669 (2023).
Grubaugh, N. D. et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 20, 8 (2019).
Nawrocki, E. P. Faster SARS-CoV-2 sequence validation and annotation for GenBank using VADR. NAR Genom. Bioinform. 5, lqad002 (2023).
Bhatt, S. et al. Semi-mechanistic Bayesian modelling of COVID-19 with renewal processes. J. R. Stat. Soc. Ser. A Stat. Soc. 186, 601–615 (2023).
Mishra, S. et al. A COVID-19 model for local authorities of the United Kingdom. J. R. Stat. Soc. Ser. A Stat. Soc. 185, S86–S95 (2022).
Flaxman, S. et al. Estimating the effects of non-pharmaceutical interventions on COVID-19 in Europe. Nature 584, 257–261 (2020).
Stan Development Team. Stan modeling language users guide and reference manual, version 2.18.0. http://mc-stan.org/ (2018).
Nei, M. & Li, W.-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl Acad. Sci. USA 76, 5269–5273 (1979).
Zhao, L. & Illingworth, C. J. Measurements of intrahost viral diversity require an unbiased diversity metric. Virus Evol. 5, vey041 (2019).
Hamming, R. W. Error detecting and error correcting codes. Bell Syst. Tech. J. 29, 147–160 (1950).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989).
Yang, Z. Molecular Evolution: a Statistical Approach (Oxford University Press, 2014).
Hoffman, M. D. & Gelman, A. -The No-U-Turn sampler: adaptively setting path lengths in Hamiltonian Monte Carlo. J. Mach. Learn. Res. 15, 1593–1623 (2014).
Phan, D., Pradhan, N. & Jankowiak, M. Composable effects for flexible and accelerated probabilistic programming in NumPyro. arXiv preprint arXiv:1912.11554 (2019).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinforma. 20, 1160–1166 (2019).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Bürkner, P.-C. brms: An R package for Bayesian multilevel models using Stan. J. Stat. Softw. 80, 1–28 (2017).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Sanderson, T. Taxonium, a web-based tool for exploring large phylogenetic trees. eLife 11, e82392 (2022).
Louca, S. & Pennell, M. W. Extant timetrees are consistent with a myriad of diversification histories. Nature 580, 502–505 (2020).
Pagel, M. Detecting correlated evolution on phylogenies: a general method for the comparative analysis of discrete characters. Proc. R. Soc. Lond. Ser. B Biol. Sci. 255, 37–45 (1994).
Paradis, E. & Schliep, K. ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R.Bioinformatics 35, 526–528 (2019).
Hijmans, R. J. geosphere: Spherical trigonometry. R package version 1.5-18. https://CRAN.R-project.org/package=geosphere (2022).
Luxen, D. & Vetter, C. Real-time routing with OpenStreetMap data in Proceedings of the 19th ACM SIGSPATIAL International Conference on Advances in Geographic Information Systems 513–516 (Association for Computing Machinery, 2011).
Louca, S. & Doebeli, M. Efficient comparative phylogenetics on large trees. Bioinformatics 34, 1053–1055 (2017).
Khurana, M. P., Scheidwasser, N. & Curran-Sebastian, J. High-Resolution Epidemiological Landscape from ~290,000 SARS-CoV-2 Genomes from Denmark https://doi.org/10.5281/zenodo.12783498 (2024).
Acknowledgements
The authors express gratitude to Statens Serum Institut and The Danish Health Data Authority for their efforts in gathering and granting access to the data. Additionally, appreciation is extended to the Danish Covid-19 Genome Consortium for conducting the sequencing of SARS-CoV-2 samples; the full list of members and their affiliations is listed in the Supplementary Information. SB and CM acknowledge funding from the MRC Centre for Global Infectious Disease Analysis (reference MR/X020258/1), funded by the UK Medical Research Council (MRC). This UK-funded award is carried out in the frame of the Global Health EDCTP3 Joint Undertaking. SB is funded by the National Institute for Health and Care Research (NIHR) Health Protection Research Unit in Modelling and Health Economics, a partnership between the UK Health Security Agency, Imperial College London and LSHTM (grant code NIHR200908). Disclaimer: ‘The views expressed are those of the author(s) and not necessarily those of the NIHR, UK Health Security Agency or the Department of Health and Social Care.’. S.B. acknowledges support from the Novo Nordisk Foundation via The Novo Nordisk Young Investigator Award (NNF20OC0059309) which also supports FPL. S.B. acknowledges the Danish National Research Foundation (DNRF160) through the chair grant which also supports M.P.K., J.C.S. and N.S. S.B. acknowledges support from The Eric and Wendy Schmidt Fund For Strategic Innovation via the Schmidt Polymath Award (G-22-63345) which also supports C.M. J.L.J. is supported by the Carlsberg Foundation, grant CF21-0342.
Author information
Authors and Affiliations
Consortia
Contributions
M.P.K., J.C.S., N.S. and S.B. conceived and designed the study. D.A.D., L.H.M. and S.B. supervised. M.P.K., J.C.S., N.S. and C.M. performed the analyses. M.R., J.F., M.S., M.H.E.T., J.L.J., L.A.E.H., F.T.M., M.A., P.J., S.L., T.G.K., H.U. and L.H.M. were responsible for data curation and management. M.U.G.K. and L.D.P. supervised the statistical analyses. All authors contributed to data collection and editing the original draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was conducted using data from national registers only. According to Danish law, ethics approval is not needed for this type of research under the Scientific Ethical Committee Act. All data management and analyses were carried out on Statistics Denmark’s secure research servers. The study only contains aggregated results and no personal data.
Peer review
Peer review information
Nature Communications thanks Joshua Levy, who co-reviewed with Maryam Ahmadi Jeshvaghane, James Otieno and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khurana, M.P., Curran-Sebastian, J., Scheidwasser, N. et al. High-resolution epidemiological landscape from ~290,000 SARS-CoV-2 genomes from Denmark. Nat Commun 15, 7123 (2024). https://doi.org/10.1038/s41467-024-51371-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51371-0
- Springer Nature Limited