

Abstract
Plasmids are small DNA molecules that enable bacteria to share beneficial traits, influencing microbial communities. However, their role within the human gut microbiome remains largely unknown. In this study, we investigate the gut microbiomes of 34 mother-child cohorts, employing a plasmid analysis workflow to understand the impact of plasmids on the gut microbiome. We create a plasmid phylogenetic tree, devise a method for assigning plasmid hosts, and examine potential plasmid transfer networks. Our research discovers a wide variety of previously unidentified plasmid sequences, indicating that current databases do not fully represent the gut plasmidome. Interestingly, infants display greater plasmid diversity compared to mothers and other healthy adults. We find that Bacteroidota, a major bacterial phylum, serves as the primary host for gut plasmids and plays a dominant role in gut plasmid transfer events. Additionally, plasmids broaden the genetic capabilities of bacteria, with their influence on bacterial function becoming more apparent as children’s gut microbiomes develop. This study sheds light on the role of plasmids in the infant gut microbiome, making a significant contribution to our understanding of plasmid biology.
Similar content being viewed by others


Introduction
Plasmids are self-replicating extrachromosomal genetic elements that are broadly present in bacteria and archaea. These molecules show remarkable diversity in size1, copy number2, GC content3, replication mechanism4, transmission mode5,6, DNA topology (circular or linear)7, genetic cargo, and host range8, among other features. Importantly, plasmids carry both backbone or ‘core’ genes that are instrumental to their vertical and horizontal transmission and self-replication5,9,10,11 as well as genetic cargo that codes for genes involved in virulence, ecological interactions, anti-phage systems, and antibiotic resistance, and many unknown functions7,12,13,14.
Plasmids play a key role in bacterial ecology and evolution, especially via horizontal gene transfer (HGT). Plasmids are key drivers of HGT and can be transferred at high rates through a variety of mechanisms, including conjugation (including plasmid mobilization and conduction), transduction, transformation, and vesiduction5,15,16,17. As a result, beneficial traits are rapidly transferred within and between species of bacteria, eventually contributing to increased host fitness18. In addition, high copy numbers of plasmids cause gene dosage effects that increase gene expression19 as well as gene variability through gene maturation and recombination20,21. However, plasmids also impose a burden on their bacterial host from the demands of the plasmid life cycle (for example, plasmid conjugation22, replication, and gene expression23) and from conflicts between chromosomal-encoded and plasmid-encoded proteins within the host bacterium24.
Studies on plasmids have historically focused on single bacterial isolates or mathematical modeling. For example, researchers have investigated plasmid traits in specific bacteria13,25, studied plasmid-mediated antibiotic resistance genes (ARG)26,27, and performed theoretical studies of plasmid persistence and dynamics12,28. Recently, developments in high-throughput metagenomic sequencing have made it possible to study the plasmid metagenome, known as the plasmidome, and some studies have used this approach to explore plasmid community in the gut environment29,30,31. However, our understanding of plasmids is still limited by methodological challenges, including the reliable identification of plasmids and the detection of their individual bacterial hosts in a metagenomic catalog.
Moreover, we lack knowledge about the biology of the human plasmidome beyond archetypal pathogen-associated plasmids. In particular, there has been no comprehensive assessment of the dynamics of the gut plasmidome in early life, even though numerous studies in the last decade have emphasized the importance of the gut microbiome for healthy infant development32,33,34 and plasmids are known to improve the fitness and environmental adaptability of their bacterial hosts13,35. It is plausible that plasmid assemblages in the human gut could shape the colonization and development of bacterial communities during microbiome establishment and maturation. As a starting point, what is needed to test this hypothesis is an overview of the gut plasmidome and plasmid hosts in the human gut that characterizes how they interact, coexist, and develop.
To obtain an overview of the plasmidome and plasmid ecology in the human gut in early life, we analyzed microbiome data obtained from 9520 fecal samples worldwide, mainly from infants, using an in-house ensemble workflow (https://github.com/Wanli-HE/Plaspline) that integrates benchmarked tools for plasmid identification36. With this, we successfully constructed an early life gut plasmid catalog of 75,239 complete plasmid genomes, of which only 24% are found in the current databases. We then reconstructed phylogenetic relationships in the gut plasmid community based on the identification of 633 distinct replicase groups. Using sequence similarity networks, we linked plasmids to their most likely bacterial hosts, which highlighted their prevalence in species in genus Bacteroides that are currently not well represented in reference databases. Based on these putative host annotations, we constructed a potential horizontal transfer network of plasmids in the human gut, which was dominated by plasmids associated with phylum Bacteroidota. Finally, we uncovered evidence of the substantial contribution of gut plasmids to bacterial functions and found that plasmids expand bacterial gene repertoires, especially as children grow older. Overall, this study provides the first overview of the gut plasmidome in early life, describing the ecology of plasmids in the human gut and shedding light on their potential roles in the gut microbiota of infants.
Results
The gut plasmidome in early life
Based on 9520 fecal samples from children (n = 7749), mothers (n = 673), and other healthy adults (n = 1098), we used Plaspline36 to create a comprehensive gut plasmid catalog containing 75,239 circular plasmids and 190,237 plasmid contigs. The most notable feature of this catalog was that only 24% of the 75,239 complete circular plasmids were found in the currents databases (Supplementary Fig. 2c), even with a low BLAST similarity threshold (coverage 0.5 and identity 0.55). This indicates that plasmid diversity to date has been greatly underestimated, and the plasmids recovered in the present study considerably expand our current knowledge of these genetic elements. To obtain a systematic measure of gut plasmid diversity and improve the accuracy of the downstream analysis, subsequent analysis focused only on contigs that contained plasmid backbone genes, which were classified into 633 groups (rep-groups) based on the replicase gene.
Within each rep-group, plasmids were further clustered based on the type of replicase gene present and their mobility, respectively. We found that the clusters “inc” (incompatibility group) and “rep_cluster” (uncharacterized types) were the dominant groups in the gut plasmidome (Fig. 1a) and were significantly phylogenetically different from each other (phylogenetic signal, Pagel’s λ: 0.93; p-value (based on LR test) <0.001). We then attempted to determine if certain rep-groups were associated with certain pregnancy or feeding conditions. Overall, though, we found that 88.7% (547 of 617), 80.6% (489 of 607), and 73.3% (434 of 592) of rep-groups were present in samples associated with more than one feeding pattern, delivery mode, or pregnancy term, respectively (Fig. 1a; Supplementary Fig. 2a). This suggests that the majority of these plasmids are common in the human gut under a variety of conditions.

a Phylogenetic tree of gut plasmids based on the plasmid replicase gene, from which 633 rep-groups were identified. The innermost layers of the heatmap surrounding the tree indicate the plasmids that were unique to the gut of children characterized by a certain delivery mode (vaginal or section), term (full-term or premature), or feeding pattern (breastmilk, mix or formula); the next layer depicts the abundance of each plasmid in samples of different ages, from newborns to mothers and adults (inner to outer); the outer layer represents the richness of plasmids from each mobility group (conjugative, mobilizable, or non-transmissible). b Venn diagram of the shared and unique rep-groups in children, mothers, and adults. c The number of rep-groups whose abundance was highest at a given timepoint. d The number of rep-groups (n = 9520) that were found in children at all timepoints (long-term) versus rep-groups that were not, and their richness of transmissible plasmids, Stars indicate statistical significance (Wilcoxon rank-sum test, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001), the black line denotes the median value (50th percentile), while the lower and upper bounds of the box represent the 25th to 75th percentiles. The lower and upper whiskers represent the 5th and 95th percentiles, respectively. e The richness of rep-groups in samples from all ages of children (nt0d = 418, nt4d = 440, nt1w = 672, nt2w = 429, nt3w = 756, nt1m = 529, nt2m = 627, nt4m = 681, nt6m = 353, nt9m = 503, nt12m = 1173, nt18m = 437, nt24m = 333, nt36m = 155), mothers (n = 673), and adults (n = 1098) (‘sample size’ means read number per sample, same to later text), the black line denotes the median value (50th percentile), while the lower and upper bounds of the box represent the 25th to 75th percentiles. The lower and upper whiskers represent the 5th and 95th percentiles, respectively.
Gut plasmids are highly diverse in early life
Of the 633 rep-groups we identified, 621 were present in children. Notably, 234 rep-groups were exclusive to children, which presented a marked contrast with the samples from adults and mothers (1 and 10 exclusive rep-groups, respectively) (Fig. 1b). Compared to adults and mothers, rep-group diversity was significantly higher in children, particularly in the first nine months of life (Fig. 1b), with the highest diversity found in the plasmid communities of 1-week-old to 2-month-old children (Fig. 1e; Wilcoxon rank-sum test, FDR-corrected; Supplementary data 1). Additionally, the majority of rep-groups were most abundant in the first four months of life (Fig. 1c). These findings indicate that, compared to adults, children harbor a much more diverse plasmid community in their gut, with particular enrichment in the early stages of life.
Plasmid mobility plays an important role in persistence
A notable finding was the consistent presence of 74 rep-groups throughout the first three years of life, and 179 rep-groups between 1 and 3 years of age (Fig. 1a, d). Persistent rep-groups, i.e., those found at all sampled time points in children, contained a higher number of transmissible plasmids (i.e. plasmids containing genes essential for mobilization or conjugation) compared to more-transient rep-groups (Fig. 1d; Wilcoxon rank-sum test; p(only <=1m to only 2m~1y)=0.3e−4; p(only <=1m to =3y)=1.4e−9; p(=3y to only 2m~1y)=0.4e−4), suggesting that plasmid mobility may contribute to persistence. Additionally, these higher-mobility rep-groups were more closely clustered on the phylogenetic tree than expected by chance, indicating a significant phylogenetic influence (phylogenetic signal, Pagel’s λ: 0.75; p-value (based on LR test): 3.5e−32).
Bacteroidota harbor most plasmids in the human gut
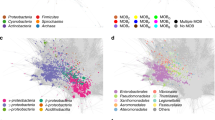
To investigate the relationship between plasmids and their potential bacterial hosts, we compared the draft and circular plasmids obtained here with published plasmids whose bacterial hosts are known. To overcome database bias—since PLSDB contains thousands of plasmids from Escherichia and Klebsiella, but only 85 from Bacteroides and 41 from Bifidobacterium (Supplementary Fig. 3a)—we also recruited whole-genome sequences of 6310 gut isolates to calculate their genomic similarity to the plasmids in this study (Supplementary Fig. 3b). Using this approach, we were able to identify bacterial host (potential host) of 36.03% of the assembled gut plasmids and, based on this, to construct networks depicting plasmid-bacteria associations (Fig. 2a). Most of these connections were between plasmids and members of the Bacteroidota or Proteobacteria (Fig. 2b), indicating that these two phyla may be the dominant plasmid hosts in the human gut. When we calculated the number of plasmids linked to each bacterial host, we found that numerous plasmids in our samples demonstrated similarity to published complete plasmids and draft genomes from Bacteroides, Phocaeicola, Parabacteroides, Escherichia, and Klebsiella. Moreover, this pattern held even when we restricted the analysis to only plasmids with backbone genes (Fig. 2c). These bacteria, especially Bacteroides, thus seem to be the main hosts for the infant gut plasmidome in this study.

a Associations between gut plasmids detected in this study, plasmids in PLSDB, and genomes of gut isolates. Nodes representing PLSDB plasmids and isolate genomes are colored by host phylum; nodes representing the gut plasmids from this study are colored in black (only the 50 bacteria (at genus level) with the highest number of plasmids with backbone are shown). b The number of edges connecting plasmids in this study with different bacterial hosts, grouped by phylum. c) From the left, the first heatmap depicts the richness of plasmids found in different bacterial host genera. ‘Total’ indicates richness of all plasmids; ‘Backbone’,’Con’, ‘Mob’, and ‘Non-trans’ indicate plasmids with backbone genes and those that are conjugative, mobilizable, and non-transmissible, respectively. The second and third heatmaps represent the mean richness and abundance, respectively, of plasmids in children, mothers, and adults (from left to right), normalized by bacterial host copy number and read number (per cell); to the right of each heatmap are the results of statistical comparisons, with “C to M” representing children compared to mothers and “C to A” children compared to adults. Stars indicate statistical significance (Wilcoxon rank-sum test, two-sided, FDR-corrected, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
Bacteria in the phyla Actinobacteriota, Firmicutes, Bacteroidota, and Proteobacteria were found to harbor more diverse and abundant plasmids in children compared to in mothers and adults (Fig. 2c), indicating that these bacteria contain more plasmids in early life. However, Firmicutes A showed the opposite tendency, with the majority of bacteria in this phylum harboring fewer plasmids in children than in mothers and adults (Fig. 2c). Interestingly, for hosts in the genera Bacteroides, Colinsella, Holdemanella, Agathobacter, and Fusicatenibacter, both plasmid diversity and abundance were higher in mothers than in children, but lowest in adults. In adults, instead, the highest diversity of plasmids was found in Escherichia (Fig. 2c). Taken together, these data suggest that relationships between plasmids and their bacterial hosts fluctuate across different human life stages.
Plasmid dynamics within different bacteria at different stages of life
To gain a deeper understanding of how plasmid assemblages change within gut bacteria at various life stages, we turned our focus to evaluating both the abundance and diversity of plasmids within individual bacterial genera across various time points. Bacteroides consistently hosted the most diverse and abundant plasmids, with diversity increasing with age in children but decreasing in adults (Fig. 3a). Similar trends were observed in other bacteria such as Phocaeicola, Parabacteroides, and Streptococcus. However, certain bacteria, such as Citrobacter, Enterobacter, and Klebsiella, exhibited a different pattern, with plasmid diversity peaking in early childhood and decreasing over time. Instead, bacteria like Turicibacter and Dorea harbored plasmids only in later childhood stages (Fig. 3a). These observations imply that relationships between gut plasmids are dynamic, with Bacteroidota and Proteobacteria playing prominent roles in this process.

a Gut plasmid dynamics in early life. b Variation in plasmid communities as explained by the indicated factors, estimated using the PERMANOVA method. The terms ‘vfm’ (n = 476), ‘vff’ (n = 465), and ‘vfb’ (n = 597) mean children born vaginally and at full-term who were fed with mix, formula, or breastfeeding, respectively; ‘vm’ (n = 815), ‘vf’ (n = 801), and ‘vb’ (n = 795) represent vaginally born children who were fed with mix, formula, or breastfeeding, respectively; ‘fm’ (n = 914), ‘ff’ (n = 755), and ‘fb’ (n = 876) represent children born at full-term who were fed with mix, formula, or breastfeeding, respectively; ‘vf’ represent children born vaginally at full-term. c Venn diagram indicating the number of plasmid rep-groups whose variation was significantly explained by age, delivery mode, term, feeding pattern, and microbiome, as estimated using the linear regression method (FDRF-test < 0.05). d Spearman correlations between the abundance of a bacterial host and its associated plasmids, the abundance of the bacterial host and the richness of its plasmids, and the richness of the bacterial host and its associated plasmids at phylum level, respectively. ‘cor’ is the Spearman coefficient; p values are FDR-corrected (two-sided). The line indicates the regression line, and the marked area is the 95% confidence interval, which surrounds the regression line on both sides. e Variation in each bacterium and its associated plasmids as explained by the indicated factors, as estimated using a PERMANOVA analysis of Bray-Curtis distances, the frequency represents the total number of plasmids that are impacted by that factor. f The relationships between the abundance of a given plasmid and the abundance of its own bacterial host or other bacteria (n = 9492). The figure depicts all correlations for which the Spearman coefficient (absolute value) was larger than 0.3 and p-value (two-sided, FDR-corrected) was less than 0.05. ‘Total plasmid’ represents all plasmids of that host, ‘plasmid with single host’ is the plasmids that were annotated with one host, and ‘plasmid with broad host’ is the plasmids that have at least two bacterial hosts. The y-axis is the value of the Spearman coefficient (≧ |0.3| with p values (FDR-corrected) ≦ 0.05), while the x-axis shows the groups of correlations. Blue represents negative correlations, while red represents positive correlations. the black line denotes the median value (50th percentile), while the lower and upper bounds of the box represent the 25th to 75th percentiles. The lower and upper whiskers represent the 5th and 95th percentiles, respectively.
Microbiome and feeding pattern play key roles in shaping the gut plasmidome
To identify influences that might be shaping gut plasmid communities during early life, we scrutinized the relative contributions of key factors such as microbiome composition, feeding patterns, pregnancy term, and delivery mode. Our findings revealed that, compared to the other factors examined, the effect of the microbiome was the most pronounced (Fig. 3b); As expected, microbiome composition was found to exert an impact on variations in the relative abundance of plasmid rep-groups (Fig. 3c; linear regression; FDRF-test <0.05). This underscores the pivotal role of the microbiome as the paramount influence shaping the dynamics of the gut plasmidome. Beyond the microbiome, infant’s feeding pattern also emerged as an influential determinant of gut plasmid assemblages, with formula feeding having the highest explanatory power, followed by mix feeding and breastfeeding (Fig. 3b). Similarly, after microbiome and age, feeding pattern had the strongest influence on variations in plasmid rep-groups (Fig. 3c). Collectively, these findings highlight the multifaceted impact of the microbiome and infant feeding patterns on the nuanced dynamics of the gut plasmidome during the early stages of life.
Plasmid richness and abundance are correlated with host abundance
To further investigate the impact of microbiome composition on the gut plasmidome, we explored the relationship between plasmids and their potential bacterial hosts. We found a significant positive correlation between potential bacterial host abundance and plasmid abundance and richness, especially for hosts affiliated with Actinobacteriota, Bacteroidota, Proteobacteria, and Firmicutes_A, for which correlation coefficients exceeded 0.4 (Fig. 3d; p values in Supplementary data 1). On the other hand, for certain groups of bacteria—Firmicutes C, Firmicutes, Verrucomicrobiota, and Synergistota—we detected a significant, strong correlation between potential bacterial host abundance and plasmid abundance, but the correlation between potential bacterial host abundance and plasmid richness was much weaker or even negligible (Fig. 3d; p values in Supplementary data 1). Notably, the correlation between potential bacterial host richness and plasmid richness was robust only for members of Firmicutes_A and Synergistota (Fig. 3d; p values in Supplementary data 1). This implies that potential bacterial host abundance, and not its richness, is the primary factor influencing plasmid diversity.
At the species level, a plasmid’s abundance was largely positively correlated with that of its bacterial hosts (mean coefficient: 0.46), with only two species exhibiting a negative correlation (Fig. 3f). When we extended the analysis to higher taxonomic levels (e.g., genus level, relating plasmid abundance to that of bacteria in the same genus but not necessarily the same species as their bacterial host), suggesting plasmids are influenced not only by the abundance of their bacterial hosts, but also their hosts‘ relatives, this pattern was observed again both on plasmids with either narrow or broad host ranges (Fig. 3f). In addition, bacterial host abundance had a more significant influence on single-host plasmid groups compared to the multiple-host plasmid group (Fig. 3f), which is expected and indicates that our plasmid host annotation method is trustworthy.
Plasmids can better reflect environmental change than their bacterial hosts do
Given the degree to which inter-individual variation in the gut plasmidome was associated with factors other than microbiome composition (Fig. 3b, c), we hypothesized that environmental factors may exert a direct influence on gut plasmids, rather than (or in addition to) indirect effects that are mediated by the bacterial hosts. Intriguingly, our analyses revealed many instances in which environmental factors significantly influenced plasmid assemblages without any corresponding impact on the bacterial hosts. Moreover, this scenario was three times more prevalent than one in which only the bacterial hosts exhibited significant differences while the corresponding plasmid community did not (Fig. 3e; Supplementary Fig. 4). This finding suggests that plasmids may serve as more sensitive indicators of environmental changes than their bacterial hosts.
Gut plasmids have broad host ranges
Next, we looked more specifically at the host range of the different plasmids in the infant gut. Most plasmids were relatively host-specific, with many being restricted to a single bacterial host. When grouped at the contig level, most plasmids were found in no more than 10 different bacteria; when grouped into rep-groups, most had a maximum of 20 hosts. However, our investigation also revealed plasmids with an exceptionally expansive host range, with documented interactions involving over 70 bacterial species at the contig level and exceeding 100 at the rep-group level, which showed a similar trend to the plasmids in PLSDB (Fig. 4a). This discovery challenges existing assumptions and underscores the potential for a considerably broader spectrum of plasmid host ranges within the human gut ecosystem.

a The host range of gut plasmids, when grouped at the contig and rep-group level, respectively. The inter-density plot showed the host range of plasmids in PLSDB at rep-group level. b Networks of potential plasmid transfer (pPT) between species were constructed for all children, mothers, and other healthy adults. The outermost strip represents phylum-level host assignments. c Differences in pPT between different age stages, calculated as the later stage minus the earlier stage. Only significant differences are shown, p-value was calculated after FDR-corrected and Wilcoxon rank-sum test (two-sided). d The richness and abundance per cell of transmissible plasmids within each bacterial host (normalized by reads number and log-10 transformed) (nActinobacteriota=5662; nFirmicutes = 6698; nFirmicutes_A = 5503; nFirmicutes_C = 2270; nFusobacteriota = 18; nProteobacteria = 6560; nSynergistota = 0; nVerrucomicrobiota = 1894), the black line denotes the median value (50th percentile), while the lower and upper bounds of the box represent the 25th to 75th percentiles. The lower and upper whiskers represent the 5th and 95th percentiles, respectively. e The OLS (ordinary least squares) was calculated based on frequency of pPT between two bacterial hosts and their corresponding Spearman coefficient, ‘***’ indicates p values (two-sided, FDR-corrected) lower than 0.001.
Plasmid transfer network in the human gut
To investigate plasmid-mediated connections among bacterial hosts in the human gut, we focused on plasmids associated with the bacterial hosts identified in Fig. 3. We defined potential plasmid transfer (pPT) events as cases in which the same plasmid was found in different bacterial hosts. The majority of pPT events occurred in the phyla Bacteroidota, Firmicutes, and Proteobacteria (Fig. 4b). A significant proportion of pPT events took place within a single phylum (Fig. 4b), suggesting that plasmid transfers predominantly occurred between related bacteria.
Notably, we detected the highest prevalence of pPT events in phylum Bacteroidota, particularly in the genera Bacteroides and Phocaeicola (Fig. 4b; Supplementary Fig. 5). Interestingly, these two genera were implicated in the majority of pPT events that extended beyond a single phylum, unlike nearly all the other bacteria we examined.
To determine the factors that might contribute to the broad pPT networks within phylum Bacteroidota, we conducted a comprehensive examination of all transmissible plasmids detected within each bacterium. Of the different phyla considered, we found that members of the Bacteroidota possessed the highest diversity and abundance of transmissible plasmids (Fig. 4d; Wilcoxon rank-sum test; p values in Supplementary data 1). These findings reveal the unique role played by bacteria in phylum Bacteroidota in orchestrating and dominating pPT events in the human gut.
The colonization and development of pPT in the human gut
To understand how bacterial colonization in the early gut might influence the developmental trajectory of pPT, we analyzed pPT networks across developmental stages, including children (born vaginally and at full-term), mothers, and adults. As children progressed through the first years of life, we observed an increasing frequency of pPT events orchestrated by members of phylum Bacteroidota. Significant increases in these pPT events were evident during transitions from 4 days old to 1 week old, 1 week old to 2 weeks old, 9 months old to 12 months old, and 12 months old to 18 months old (Fig. 4c and Supplementary Fig. 5). However, only minimal variations in pPT events involving Bacteroidota were identified between 36-month-old individuals and either mothers or adults, hinting that this developmental stage may be relatively mature with respect to pPT dynamics.
The majority of cross-phylum plasmid transfers occurred between bacteria in the phyla Bacteroidota and Firmicutes (Fig. 4b). This type of interphylum transfer was notably more prevalent during the first 6 months of life and increased from 9 months of age to 24 months of age, but exhibited comparatively less variation in 36-month-old children (Fig. 4c; Supplementary Fig. 5). Conversely, only slight alterations were observed in the frequency of pPT events involving Proteobacteria, primarily in the first month of life, with marginal increases from 1 month to 6 months of age and from 9 months to 18 months of age (Fig. 4c). Notably, the frequency of these events in newborns was generally lower than that observed in mothers (Fig. 4c).
pPT promotes bacterial connections in the human gut
We hypothesized that pPT events might actively contribute to the enhancement of bacterial connections. To evaluate this idea, we conducted a detailed analysis of the correlation between bacteria-bacteria interactions (Spearman |r| ≥0.3 and p ≤ 0.05) and their corresponding pPT events (Fig. 3). This revealed a statistically significant correlation between pPT events and bacteria-bacteria interactions across all life stages, which was particularly prominent during the early developmental phases (Fig. 4e; OLS test, p < 0.001, Supplementary data 1). This result provides evidence of a robust and quantifiable association between pPT dynamics and the intricate network of bacterial interactions within the human gut milieu, and suggests that pPT may play a potentially pivotal role in actively promoting and modulating bacterial connectivity, particularly during the nascent stages of life.
Plasmids expand bacterial gene repertoires
We next evaluated the functional capacity of plasmids compared to chromosomes. The total number of chromosomal genes in the bacterial metagenome was nearly 26 times larger than the number of genes carried on plasmids. While most plasmid open reading frames (ORFs) were also found on chromosomes, around 10% (over 89,030) were exclusively found on plasmids (Fig. 5a). Notably, 10.5% of plasmid genes of unknown function were only found on plasmid contigs. Additionally, of the ORFs that were successfully annotated with an identifiable function (using KEGG, CARD, VF, or BacMet2), between 6.6% and 8.3% were located exclusively on plasmids (Fig. 5c), indicating that plasmids made a unique contribution to bacterial gene repertoires by providing specific genes that would not otherwise have been present. Overall, though, only 33.7% of plasmid ORFs received functional annotation, while 43.3% annotation rate for chromosomal ORFs (Fig. 5b). This suggests that plasmids tend to carry more unknown genes than chromosomes, likely representing genes with novel and uncharacterized functions.

a Open reading frames (ORFs) located on plasmids (pink) were compared to those located on chromosomes (blue) (nucleotide sequence BLAST: coverage >0.8; identity >0.85). b Overall annotation rate (ring diagrams) and database-specific annotation rate (bar diagram) for plasmid and chromosome ORFs. c A stacked bar plot of the functional groups identified in plasmid genes. ‘C’ represents plasmid genes also found on chromosomes; ‘P’ represents plasmid-specific genes. d The richness of different functional groups, displayed according to whether they were located on plasmids, chromosomes, or the non-redundant combination of the two (n = 4758), the black line denotes the median value (50th percentile), while the lower and upper bounds of the box represent the 25th to 75th percentiles. The lower and upper whiskers represent the 5th and 95th percentiles, respectively. e The increasing rate of gene richness was calculated as the richness of genes only encoded by plasmids divided by chromosomal gene richness (nt0d = 200, nt4d = 253, nt1w = 331, nt2w = 55, nt3w = 190, nt1m = 65, nt2m = 104, nt4m = 247, nt6m = 140, nt9m = 199, nt12m = 620, nt18m = 261, nt24m = 239, nt36m = 88). OLS (ordinary least squares) was used to test the linear correlation between the increasing rate of gene richness (median value) and a child’s age. The black line denotes the median value (50th percentile), while the lower and upper bounds of the box represent the 25th to 75th percentiles. The lower and upper whiskers represent the 5th and 95th percentiles, respectively. The black line indicates the regression line, and the gray area is the 95% confidence interval, which surrounds the regression line on both sides. f Total genes of different functional groups at different stages. The colors in the heatmap represent plasmid gene abundance divided by chromosomal gene abundance, minus 1; pink represents higher plasmid abundance while blue depicts higher chromosomal abundance. Stars indicate significant differences (Wilcoxon signed-rank test, two-sided, FDR-corrected, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). g Correlation between the abundance of each type of functional gene encoded on plasmids and on chromosomes, assessed using OLS (ordinary least squares, two-sided); ‘***’ represent p values (FDR-corrected) less than 0.001. h Bar diagrams showing the number of genes that were differentially abundant (Wilcoxon rank-sum test, two-sided, FDR < = 0.05; only |-log2FC| larger than 1 and −log10P larger than −log10(0.05) are shown) in children with different feeding patterns. Pink represents genes found only on plasmids, blue represents genes found only on chromosomes, and orange represents genes found on both (non-redundant ORFs); ‘bre’ represents breastfeeding, ‘mix’ represents mix feeding, and ‘for’ represents formula feeding.
When we removed functionally redundant genes from the analysis, we observed a significant reduction in gene richness within plasmids compared to chromosomes within each individual (Wilcoxon signed-rank test; pkegg < 2.2e−16; pVF < 2.2e−16; pCard < 2.2e−16; pBacmet2 < 2.2e−16; Fig. 5d). However, when we combined these two gene pools together and removed redundancy, there was a significant increase in gene richness (Wilcoxon signed-rank test; pkegg = 5.2e−15; pVF = 5.5e−3; pCard = 5.6e−10; pBacmet2 = 4.9e−09; Fig. 5d; Supplementary Fig. 7). This suggests that, although the initial plasmid gene pool is smaller than that of chromosomes, these plasmid genes act to expand bacterial gene repertoires. This same pattern was evident when we examined the functional annotations of the different groups of genes: the addition of plasmid-only genes to chromosomal genes increased the numbers of functional genes present, with mean increasing rates of gene richness of 10.1% for KEGG-annotated genes, 7.4% for VF-annotated genes, 47.6% for CARD-annotated genes, and 22.0% for BacMet2-annotated genes (Fig. 5e). Taken together, these results emphasize the role of plasmids in significantly enhancing bacterial gene diversity, particularly in the context of genes that confer resistance to antibiotics, antibacterial biocides, and metals.
Plasmids shape bacterial gene repertoires as children grow
The proportion of plasmid gene richness that related to chromosomal gene richness (IRGR) across all children was increased with children’s age, in particular for resistance genes at 36 months of age (Fig. 5e). Moreover, chromosomal genes, as expected, predominated in most life stages, while total plasmid genes abundance across all children is higher than chromosomal genes at 36 months of age, especially for resistance genes and many metabolism-related genes (Wilcoxon signed-rank test; p values in Supplementary data 1; Fig. 5f; Supplementary Fig. 8). This suggests a progressive divergence between plasmid and chromosome encoded genes as children mature. To assess this divergence, we analyzed the correlation between plasmid and chromosomal genes abundance. As anticipated, most plasmid gene assemblages showed a significant correlation with chromosomal gene assemblages in the majority of life stages, with the notable exception of 36 months of age (Fig. 5g; OLS test). With respect to genes with KEGG or CARD annotations, we detected no significant relationship between the composition of plasmid genes and that of chromosomal genes at either 24 or 36 months of age. All of these data suggest that plasmid genes become increasingly dissimilar to chromosomal genes as children grow, and highlight the importance of plasmids in expanding bacterial gene repertoires as the gut microbiota develops.
Plasmids exhibit enrichment in specific traits
In general, functional genes were more abundant on chromosomes than on plasmids. However, these patterns shifted when we compared samples obtained under different feeding conditions. Here, plasmids were characterized by enrichment in specific traits, particularly in comparisons of mixed feeding and breastfeeding, formula feeding and breastfeeding, and mixed feeding alone. These differences were less pronounced, however, for genes that were encoded on both plasmids and chromosomes (Wilcoxon signed-rank test FDR; Fig. 5h). This enrichment serves as further evidence that plasmids play a role in shaping bacterial gene repertoires in response to environmental changes.
Discussion
In this study, we investigated the early acquisition and development of the plasmid community in the infant gut—with particular emphasis on the role of plasmids in the larger bacterial community—using a gut metagenomic dataset obtained from more than 9500 samples from 34 studies and 22 countries, including children (7749), mothers (673), and adults (1098). For this, we developed a new bioinformatics pipeline for the isolation and annotation of plasmids from metagenomic data. The vast majority of circular plasmid sequences recovered were not previously identified and have no close relatives in current reference databases, and thus considerably expand our knowledge of the genetic diversity of plasmids. We constructed a phylogenetic tree of plasmids based on their conserved backbone replicon genes and developed a strategy for the identification of potential bacterial hosts based on genomic similarity to known plasmids. Our findings highlight the importance of plasmids for the gut microbiota in early childhood.
One of the key findings of this study is that plasmids are disproportionately abundant in the infant gut in early life (Fig. 1a,b,c,e). Importantly, it showed a different trend comparing to that of gut microbiota (Supplementary 2b). This makes sense considering previous reports that plasmids play an important role in bacterial evolution and adaptability by transferring beneficial traits within and between species of bacteria, positively contributing to host fitness8. The establishment of the early gut microbiome is dominated by stochastic processes that favor quickly proliferating generalist bacteria37. The strength of the competitive pressure in such an environment should favor rapid adaptation, likely increasing the importance of mobile genetic elements carrying adaptive traits. It is plausible that some of the bacteria that are randomly seeded at birth may lack specific colonization factors and may therefore depend on transmissible vectors for these important functions, which would explain the increased abundance and diversity of plasmids observed at 1 week to 2 months of age. Then, as the bacterial community matures, it tends to evolve towards a more diverse, specialized, and constrained microbiota37, causing the necessity for an extensive pan-genome of mobile elements diminishes. This would explain the reduced diversity of the plasmid community that we observed after 12 months of age, as bacterial assemblages begin to resemble those of adults (Fig. 1e). One possible factor in this decline could be the fitness cost of plasmids, which is the main limit to their persistence in bacterial populations19,23,38. As children grow and the functionality of the gut ecosystem gradually stabilizes, the community composition becomes more fixed and a network of mutualisms is developed37,39,40, meaning that the benefits of plasmids for bacteria—i.e., rapid adaptation to stochastic changes—may start to be outweighed by their fitness costs, promoting plasmid loss. Another indisputable factor is breastfeeding, as an earlier study showed that the maturation of the infant gut microbiome was directly associated with the cessation of breastfeeding40. Indeed, we observed that breastfeeding and mix-feeding children harbored more unique plasmids than formula-feeding children (Supplementary Fig. 2). This finding is consistent with previous research on the importance of maternal breast milk for the infant gut microbiome and mobile genetic elements41,42,43, but, in general, this effect is poorly understood. Specifically, it is unknown if breast milk is the main source or driver of the infant gut microbiome, and how this might compare to other types of maternal-infant transfer.
Mobility genes are critically important for plasmids because they facilitate horizontal transfer among bacterial cells5. Here, we also found that plasmid mobility can affect prevalence (Fig. 2a), corroborating previous suggestions that a fast transfer rate can compensate for fitness costs and plasmid loss12,44. Moreover, we found that mobility genes are not distributed randomly in the plasmid population: there was a clear phylogenetic signal (relatedness) in the replicon groups in which these genes were found (Fig. 2a).
One interesting possibility raised by this study is the potential transfer of plasmids from children to their mothers. Previous studies have demonstrated how infants might inherit their microbiota from the mother39,45,46; consistent with this idea, we found that mothers and children shared a more diverse assemblage of plasmids than either group shared with unrelated adults (Fig. 1c). This implies a plausible mechanism for plasmid transfer between mother and child. However, there is no direct evidence arguing that this transfer is unidirectional (i.e., only from mother to child). Here, although plasmid diversity was significantly higher in infants than in mothers, it was also significantly higher in mothers than in other healthy adults (Fig. 1e). This temporal alignment suggests the possibility that children’s plasmids contribute to the heightened diversity observed in mothers. While it is obvious that mothers and children have numerous intimate contacts that might enable the exchange of microbiota, it is crucial to acknowledge the absence of direct evidence substantiating this hypothesis. Further investigations are needed to elucidate the ways in which the microbiota, and therefore plasmids, might be transferred between mothers and children and to ascertain whether this process is indeed bidirectional.
In the literature, plasmids are well known from the intensively studied bacteria in family Enterobacteriaceae (phylum Proteobacteria), especially Enterococcus, Escherichia, and Klebsiella47,48,49,50, which has led to the overrepresentation of these examples in databases such as PLSDB (Supplementary Fig. 3a)51. However, a key finding of the present study is that the main hosts of plasmids in the human gut are members of phylum Bacteroidota, such as Bacteroides, Phocaeicola, and Parabacteroides (Fig. 3d). Moreover, construction of a pPT network revealed that—in children of all ages, mothers, and adults—pPT was most frequent in Bacteroidota (Fig. 4b, Supplementary Fig. 5), possibly because this phylum contained the highest diversity and abundance of transmissible plasmids (Fig. 3d). This result supports previous findings that many interspecies and intergenus pPT events are facilitated by Bacteroides 30 and, indeed, that almost half of the pPT events in the human gut are mediated by members of phylum Bacteroidota30. Likewise, our finding that Bacteroidota have the most widespread pPT networks (Figure 4ab, Supplementary Fig. 5) makes sense given previous research revealing that plasmids are very prevalent in members of this phylum8. Importantly, we found that the frequency of putative pPT events decreased with increasing phylogenetic distance between bacteria, which is consistent with the fact that plasmid transfers are more frequent between closely related individuals and species and occurs more rarely between distant relatives52. Overall, coherence between our findings and previous experimental work support reliability of our in-silico plasmid-host linkage method, which in the future may serve as an important tool for identifying plasmid hosts in metagenomes of the gut environment. More importantly, though, these results expand our knowledge on the ecology of gut plasmids, highlighting that the members of Bacteroidota are the main plasmid hosts and the dominant players involved in pPT events in the human gut and correcting previous biases towards Escherichia and Klebsiella due to limited representation of Bacteroidota plasmid in current plasmid sequence database.
Another important finding was that plasmid community assemblages may be more sensitive than their bacterial host communities to environmental variation (Fig. 3e; Supplementary Fig. 4). Although unexpected, this is consistent with previous studies reporting that, due to their higher diversity and copy number2, plasmid-encoded genes evolve faster than chromosomal genes20,21. Plasmids may therefore be able to adapt faster to changing environmental conditions. This could mean that, compared to bacteria, plasmid colonization of the infant gut is less influenced by inheritance from the mother, because the newborn gut environment differs from that of the mother in significant ways (e.g., bacterial density, oxygen concentration, host modulation); this could also potentially explain why plasmid diversity in newborns was significantly higher than in mothers (Fig. 1c, e). In this way, plasmids might serve as mechanisms for rapid adaptation to environmental variation. This phenomenon could potentially be responsible for the heightened frequency of pPT events in the early stages of life, along with the discernible surges in pPT occurrences we observed throughout infancy (Fig. 4c and Supplementary Fig. 5). Most importantly, our observations underscore the crucial role of pPT events in fostering inter-bacterial connections, with the significance of this function diminishing as children age (Fig. 4e). However, there is an alternative explanation for our results that cannot be excluded: rather than plasmid diversity being higher than bacterial diversity, it is possible that the better resolution we detected in plasmid community composition reflects individual strain-level specificity which cannot be accounted for with precision using short-read metagenomics. When viewed as a whole, though, our results suggest that plasmids serve as mechanisms for rapid adaptation to environmental variation for their bacterial host. These explanations are also not exclusive of the fact that assembling plasmids from metagenomes allows to discretize samples with a finer taxonomic resolution where plasmids are proxy of strain-level resolution, while short-reads metagenomes only a broader genus and species-level abundances.
The results presented here highlight the role of plasmids in expanding the gene repertoires of their bacterial hosts. We detected a larger number of previously unidentified genes with unknown function on plasmids than on chromosomes (Fig. 5c), and certain groups of functional genes were only found on plasmids (Fig. 5c). These findings could possibly be due to the typically high copy number of plasmids, which promotes a higher mutation rate5,35. Moreover, of the genes that were found on both plasmids and chromosomes, some were found in higher abundance on plasmids, especially antibiotic resistance genes (Fig. 5f, h; Supplementary Fig. 8), which supports previous studies reporting that plasmid genes usually have higher gene dosage effects than chromosomal genes19. It could also be that certain genes have a stronger fitness effect for the host bacterium when present on a plasmid compared to a chromosome. This might be one reason why, compared to the numbers of genes found only on chromosomes, the percentage of genes encoded only on plasmids increased as children grew (Fig. 5e), resulting in a progressive divergence between the gene communities found on plasmids and those found on chromosomes (Fig. 5g). This finding is notable because it marks the first instance of a known distinction in the developmental functionality between plasmids and chromosomes during early life. Future work in this area will improve our understanding of plasmid function in the early stages of life, which has almost certainly been considerably underestimated thus far.
The methodological approach in this work mainly relies on our previously developed plasmid analysis pipeline, with some improvements in performance and reliability. Still, though, only a limited number of plasmids were identified, which we believe is likely to be a function of sequencing bias. In the future, efforts should be made to improve plasmid sequencing technology in order to isolate more plasmid genomes and enable more comprehensive analyses of plasmids in the gut. Notably, although our method was able in many cases to identify the hosts of plasmids based on their genetic similarity to examples in the PLSDB database and gut isolates, we were inherently constrained by the limitations of the database: only 36% of the plasmids in our sample were successfully linked to potential bacterial hosts. Furthermore, because it relies on previously published knowledge, this method cannot be used to detect novel plasmid-carrying bacteria. Future work to improve detection algorithms and expand the plasmid database will help in this respect. Although our current understanding of gut plasmid biology is still limited, this study provides strong evidence that plasmids play an indispensable role in the colonization and development of the gut microbiota in early life.
Methods
Subject details
This research complies with all relevant ethical regulations. No new datasets were generated in the study, all datasets are publicly available.
Study population and sampling
Metagenomic data were obtained from NCBI. The final dataset represented a total of 9520 fecal samples (children: 7749, mothers: 673, adults: 1098) obtained from 34 studies and 22 countries (Supplementary Fig. 1a). All metagenomic sequencing data were quality controlled using KneadData (version: 0.7.6) with default parameters.
Reconstruction of plasmid catalog
The gut plasmid catalog was constructed using Plaspline (version: 1.4) (https://github.com/Wanli-HE/Plaspline.git)36. First, de novo metagenome assembly was performed using metaSPAdes53, with sequence reads (filtered) as the input and default parameters applied (k-mer 21,33,55) for each sample. Only contigs longer than 2 kb were used in downstream analysis. Next, circular contigs were reconstructed by metaSPAdes (version: 3.14.1) using the “-plasmid” option54 and SCAPP (version: 0.1.4)55 with a max k-mer of 55 and “--use_scores” set as false; following processing by SCAPP, circular contigs were filtered by Plasforest (version: 1.3)56. To remove non-plasmid circular elements from the set of circular contigs, we used viralVerify (https://github.com/ablab/viralVerify; version: 1.1). We then generated a non-redundant catalog of circular plasmid genomes by considering sequence identity as well as bidirectional sequence coverage, in order to avoid grouping plasmids with very similar backbone genes but different accessory regions; for this, we employed mmseqs (version: 12.113e3)57 with sequence identity >0.9 and coverage >0.9 in bidirectional mode “--cov-mode 0” (“easy-cluster”). To obtain a more comprehensive catalog of plasmids, particularly at the gene level, the next step of the Plaspline workflow utilized Plasforest (version: 1.3)56 and Platon (version: 1.4.0)58 to detect plasmid fragments from all assembled contigs. After merging the results of these two tools, phage genomes were removed using Phamb (version 1.0.1)59, and a non-redundant draft catalog of linear plasmid contigs was generated using mmseqs (version: 12.113e3) with sequence identity >0.9 and coverage >0.9 (“--cov-mode” 2). Finally, the linear draft catalog was compared to the circular catalog using BLAST (version: 2.9.0 + ), and any contigs in the former that matched those in the latter were removed (identity >0.85, coverage >0.8). The circular and the draft catalogs were then merged to create the final plasmid catalog.
Plasmid genome and gene abundance calculations
To calculate the abundance of plasmid genomes and plasmid genes, Plaspline uses Bwa (version: 0.7.17)60, Samtools (version: 1.9)61, and Msamtools (https://github.com/arumugamlab/msamtools; version: 0.9). First, reads were filtered based on mapped length (>60 bp), identity (90%), and read coverage (80%). Then, contigs and complete plasmid sequences with less than 55% mapped coverage were removed. Abundance was calculated and normalized based on the number of fragments per kilobase of sequence mapped per million reads (FPKM). A gene’s abundance was calculated as the sum of the abundance of all plasmid sequences that contained that gene in that sample.
Annotation of plasmid catalog
To classify plasmids, Plaspline makes use of MOB-typer modules from the software MOB-suite (version: 2.0.1)62. These modules classify putative plasmids from metagenome contigs by searching for replicase genes (rep), mobilization proteins (relaxase), genes encoding the mate-pair formation system (MPF), and origin of transfer (oriT) sites. Gut plasmids are then annotated based on the presence or absence of these plasmid markers.
The novel plasmids were detected by comparing to plasmids in the current databases, which are PLSDB (version: 2023-11-03_v2), a plasmid catalog predicted by Plasx63 and JGI64 (version: IMG_VR_2023-08-08_1) though BLAST (version: 2.9.0+) with lower thresholds (identity is less than 55.0 and coverage is less than 0.5).
Plasmid phylogenetic tree
A plasmid phylogenetic tree was constructed using the plasmid replicase marker gene. MAFFT (version: 7.475)65 was used to perform multiple alignment of plasmid replicase protein sequences. The multiple alignment results were then fed into IQ-TREE (version: 2.0.3)66 to reconstruct a phylogeny of plasmids, with the parameter “-MFP” to determine the best-fit model of the SH-like approximate likelihood ratio test using 1000 bootstrap replicates. The final phylogenetic tree was visualized and annotated using iTOL (version: 4)67.
Plasmid host linking
For each plasmid reconstructed from the gut metagenomes, we inferred the most probable host based on sequence similarity with a collection of gut isolate genomes51 and known plasmids from PLSDB (version: 2021-06-23), which in most cases includes information on the host from which the plasmid was isolated. The gut bacterial isolate genomes were obtained from three studies (Forster et al.68; Poyet et al.69; Zou et al.70) and were downloaded from NCBI. We estimated genomic similarity by calculating the Jaccard Index (JI) based on shared 21-bp k-mers, with a minimum JI threshold of 0.3, as described in Acman et al.71. JI values were calculated using Bindash (version 1.0)72. If a plasmid matched with more than one host, all matching hosts were recorded. Finally, each bacterial host was annotated with information on its linked plasmids in PLSDB (NCBI taxa), which was transferred to their corresponding GTDB-TK taxa by using the python package “taxopy” (version: 0.10.2) with database of “ncbi-taxdump” and “gtdbtk-taxdump-r202”. Gut isolate genomes were directly annotated with GTDB-TK (version: 1.6.0).
Taxonomic annotation and calculation of abundance
Abundance calculation and taxonomic profiling of reads was carried out using MetaPhlAn (version: 3.0)73 at the species level.
Gene annotation. Gene functions were annotated using eggNOG-mapper (version: 2.1.7)74. Virulence factors were annotated based on information in the VFDB database (version: before JUN 2022)75. Antibiotic resistance genes were annotated using the CARD Resistance Gene Identifier (RGI, version: 5.2.1)76. BacMet2 was employed for the annotation of antibacterial biocide and metal resistance genes (version 2.0)77.
Construction of potential plasmid transfer network
Based on the number and identity of plasmids associated with different hosts, we constructed a network of putative horizontal plasmid transmission events. Specifically, a plasmid transfer event was presumed to have occurred if plasmids with the same contig were found in two or more bacterial hosts. We then refined the HGT networks to examine children of different ages, mothers, and adults. In these analyses, the number of HGT events, and the number of non-transmissible and transmissible plasmids that were involved in HGT events, was calculated in each individual and then compared between different groups.
Statistical analysis
To comprehensively assess plasmid dynamics within the human gut, we divided the age continuum of children into 14 groups: 0 days (newborn), 4 days (1–4 days), 1 week (4–7 days), 2 weeks (7–14 days), 3 weeks (14–21 days), 1 month (21–30 days), 2 months (30–60 days), 4 months (60–120 days), 6 months (120–180 days), 9 months (180–270 days), 12 months (270–360 days), 18 months (360–540 days), 24 months (540–720 days), and 36 months (over 720 days) (Supplementary Fig. 1b). Samples obtained from mothers were separated into 4 groups: 0 days (sampled at infant’s birth), 1 week (1 to 7 days after delivery), 1 month (8 to 30 days after delivery), and over 1 month (over 30 days after delivery).
All statistical analyses were conducted using Python (version: 3.7.6) with the packages “scipy.stats” (version: 1.7.3) and “skbio” (version: 0.5.6). Phylogenetic signal (Pagels’ λ) was computed using the R package “phytools” (version: 2.0-3). Alpha and beta diversity of plasmid and bacterial communities were determined using the functions “skbio.diversity.alpha_diversity” and “skbio.diversity.beta_diversity”. Wilcoxon rank-sum tests were executed with “skbio.stats.ranksums”, and Wilcoxon signed-rank tests were carried out using “skbio.stats.wilcoxon”. Spearman correlations were assessed using “scipy.stats.spearmanr”. Ordinary least squares (OLS) regression was performed with “statsmodels.api.OLS” (version: 0.11.0). Permutational multivariate analysis of variance (PERMANOVA) and testing were conducted using “skbio.stats.ordination.pcoa” and “skbio.stats.distance.permanova” (permutations = 999). Finally, the resulting p-values were adjusted using “statsmodels.stats.multitest.fdrcorrection” as indicated in the text.
When calculating the impact of microbiome structure on gut plasmids, we clustered the microbiome community into 92 groups based on their abundance in all samples, using “sklearn.cluster.AgglomerativeClustering” (version: 0.0) with “distance threshold = 0.7”.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The metagenome datasets used in the study are downloaded from NCBI SRA based on the accession numbers published in each included study, PRJNA716780, PRJNA557731, PRJNA524703, PRJNA698986, PRJNA549787, PRJNA916952, PRJEB6456, PRJNA322188, PRJNA345144, PRJNA396794, PRJNA475246, PRJNA352475, PRJNA379120, PRJNA489090, PRJNA473126, PRJNA376566, PRJNA327106, PRJNA273761, PRJNA294605, PRJEB32631, PRJNA301903, PRJNA542703, PRJNA290380, PRJNA436562, PRJEB15257, PRJNA384716, PRJNA390646, PRJNA543520, PRJNA272371, PRJNA339914, PRJNA287207, PRJNA296814, PRJNA231909, PRJNA715601, PRJEB39223(adults). The early life plasmid catalog and its gene catalog generated in this study have been deposited in the zenodo database under accession code [https://doi.org/10.5281/zenodo.10653816]. All processed data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Code availability
The original code is an in-house plasmid analysis pipeline was Plaspline (https://github.com/Wanli-HE/Plaspline.git). Any additional information required to reanalyze the data reported in this paper is available from sjs@bio.ku.dk; joseph.nesme@bio.ku.dk.
References
Hall, J. P. J., Botelho, J., Cazares, A. & Baltrus, D. A. What makes a megaplasmid? Philos. Trans. Roy. Soc. B: Biol. Sci. 377, (2022).
Del Solar, G. & Espinosa, M. Plasmid copy number control: an ever-growing story. Mol. Microbiol. 37, 492–500 (2002).
Nishida, H. Comparative analyses of base compositions, DNA sizes, and dinucleotide frequency profiles in archaeal and bacterial chromosomes and plasmids. Int. J. Evol. Biol. 2012, 1–5 (2012).
Carattoli, A. et al. Identification of plasmids by PCR-based replicon typing. J. Microbiol. Methods 63, 219–228 (2005).
Smillie, C., Garcillan-Barcia, M. P., Francia, M. V., Rocha, E. P. C. & de la Cruz, F. Mobility of plasmids. Microbiol. Mol. Biol. Rev. 74, 434–452 (2010).
Ramsay, J. P. et al. An updated view of plasmid conjugation and mobilization in Staphylococcus. Mob. Genet. Elem. 6, e1208317 (2016).
Pilla, G. & Tang, C. M. Going around in circles: virulence plasmids in enteric pathogens. Nat. Rev. Microbiol. 16, 484–495 (2018).
Rodríguez-Beltrán, J., DelaFuente, J., León-Sampedro, R., MacLean, R. C. & San Millán, Á. Beyond horizontal gene transfer: the role of plasmids in bacterial evolution. Nat. Rev. Microbiol. 19, 347–359 (2021).
Nordström, K. Plasmid R1-replication and its control. Plasmid 55, 1–26 (2006).
Wein, T. & Dagan, T. Plasmid evolution. Curr. Biol. 30, R1158–R1163 (2020).
Norman, A., Hansen, L. H. & Sorensen, S. J. Conjugative plasmids: vessels of the communal gene pool. Philos. Trans. R. Soc. B: Biol. Sci. 364, 2275–2289 (2009).
Lopatkin, A. J. et al. Persistence and reversal of plasmid-mediated antibiotic resistance. Nat. Commun. 8, 1689 (2017).
Davray, D., Deo, D. & Kulkarni, R. Plasmids encode niche-specific traits in Lactobacillaceae. Micro. Genom. 7, mgen000472 (2021).
Pinilla-Redondo, R. et al. Type IV CRISPR-Cas systems are highly diverse and involved in competition between plasmids. Nucleic Acids Res. 48, 2000–2012 (2020).
Clark, A. J. & Warren, G. J. Conjugal transmission of plasmids. Annu. Rev. Genet. 13, 99–125 (1979).
Chen, J. et al. Genome hypermobility by lateral transduction. Science 362, 207–212 (2018).
Erdmann, S., Tschitschko, B., Zhong, L., Raftery, M. J. & Cavicchioli, R. A plasmid from an Antarctic haloarchaeon uses specialized membrane vesicles to disseminate and infect plasmid-free cells. Nat. Microbiol. 2, 1446–1455 (2017).
Li, L. et al. Plasmids persist in a microbial community by providing fitness benefit to multiple phylotypes. ISME J. 14, 1170–1181 (2020) https://doi.org/10.1038/s41396-020-0596-4.
San Millan, A. et al. Integrative analysis of fitness and metabolic effects of plasmids in Pseudomonas aeruginosa PAO1. ISME J. 12, 3014–3024 (2018).
Rodríguez-Beltrán, J. et al. High recombinant frequency in extraintestinal pathogenic escherichia coli strains. Mol. Biol. Evol. 32, 1708–1716 (2015).
Couce, A., Rodríguez-Rojas, A. & Blázquez, J. Bypass of genetic constraints during mutator evolution to antibiotic resistance. Proc. R. Soc. B: Biol. Sci. 282, 1–9 (2015).
Baltrus, D. A. Exploring the costs of horizontal gene transfer. Trends Ecol. Evol. 28, 489–495 (2013).
San Millan, A. & Maclean, R. C. in Microbial Transmission (eds. Baquero, F., Bouza, E., Gutiérrez-Fuentes, J. A. & Coque, T. M.) 65–79 (Wiley, 2019).
Hall, J. P. J. et al. Plasmid fitness costs are caused by specific genetic conflicts enabling resolution by compensatory mutation. PLOS Biol. 19 (2021).
Sun, J. et al. Plasmid-encoded tet(X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat. Microbiol. 4, 1457–1464 (2019).
Villa, L. & Carattoli, A. Plasmid-mediated quinolone resistance and β-lactamases in Escherichia coli from healthy animals from Nigeria. J. Antimicrob. Chemother. 66, 1269–1272 (2011).
Millan, A. S. et al. Positive selection and compensatory adaptation interact to stabilize non-transmissible plasmids. Nat. Commun. 5, 1–11 (2014).
Hernández-Beltrán, J. C. R., Millán, San, Fuentes-Hernández, A. & Peña-Miller, A. R. mathematical models of plasmid population dynamics. Front. Microbiol. 12, 1–18 (2021).
Brown Kav, A. et al. Unravelling plasmidome distribution and interaction with its hosting microbiome. Environ. Microbiol. 22, 32–44 (2020).
Stockdale, S. R., Harrington, R. S., Shkoporov, A. N., Khokhlova, E. V. & Daly, K. M. Metagenomic assembled plasmids of the human microbiome vary across disease cohorts. Sci. Rep. 12, 9212 (2022).
Yu, A. M. K., Fogarty, E. C. & Eren, A. M. The genetic and ecological landscape of plasmids in the human gut. bioRxiv https://doi.org/10.1101/2020.11.01.361691 (2022).
Houghteling, P. D. & Walker, W. A. Why is initial bacterial colonization of the intestine important to infants’ and children’s health? J. Pediatr. Gastroenterol. Nutr. 60, 294–307 (2015).
Parekh, P. J., Balart, L. A. & Johnson, D. A. The influence of the gut microbiome on obesity, metabolic syndrome and gastrointestinal disease. Clin. Transl. Gastroenterol. 6, e91–12 (2015).
Roswall, J. et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 29, 765–776.e3 (2021).
Billane, K., Harrison, E., Cameron, D. & Brockhurst, M. A. Why do plasmids manipulate the expression of bacterial phenotypes? Philos. Trans. R. Soc. B: Biol. Sci. 377, 20200461 (2022).
Yu, Z. et al. Insights into the circular: the cryptic plasmidome and its derived antibiotic resistome in the urban water systems. Environ. Int 183, 108351 (2024).
Guittar, J., Shade, A. & Litchman, E. Trait-based community assembly and succession of. Nat. Commun. 10, 512 (2019).
Vogwill, T. & Maclean, R. C. The genetic basis of the fitness costs of antimicrobial resistance: a meta-analysis approach. Evol. Appl 8, 284–295 (2015).
Ferretti, P. et al. Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 24, 133–145.e5 (2018).
Bäckhed, F. et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17, 690–703 (2015).
Pärnänen, K. et al. Maternal gut and breast milk microbiota affect infant gut antibiotic resistome and mobile genetic elements. Nat. Commun. 9, 1–11 (2018).
Jost, T., Lacroix, C., Braegger, C. P., Rochat, F. & Chassard, C. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ. Microbiol. 16, 2891–2904 (2014).
Moossavi, S. et al. Composition and variation of the human milk microbiota are influenced by maternal and early-life factors. Cell Host Microbe 25, 324–335.e4 (2019).
Brockhurst, M. A. & Harrison, E. Ecological and evolutionary solutions to the plasmid paradox. Trends Microbiol. 30, 534–543 (2022).
Tochitani, S. Vertical transmission of gut microbiota: Points of action of environmental factors influencing brain development. Neurosci. Res. 168, 83–94 (2021).
Patangia, D. V., Ryan, C. A., Dempsey, E., Stanton, C. & Ross, R. P. Vertical transfer of antibiotics and antibiotic resistant strains across the mother/baby axis. Trends Microbiol. 30, 47–56 (2021).
Partridge, S. R., Kwong, S. M., Firth, N. & Jensen, S. O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31, 1–61 (2018).
Dobiasova, H. & Dolejska, M. Prevalence and diversity of IncX plasmids carrying fluoroquinolone and β-lactam resistance genes in Escherichia coli originating from diverse sources and geographical areas. J. Antimicrobial Chemother. 71, 2118–2124 (2016).
Ramirez, M. S., Traglia, G. M., Lin, D. L., Tran, T. & Tolmasky, M. E. Plasmid-mediated antibiotic resistance and virulence in gram-negatives: the klebsiella pneumoniae paradigm. Microbiol. Spectr. 2, 1–15 (2014).
León-Sampedro, R., Novais, C., Peixe, L., Baquero, F. & Coque, T. M. Diversity and evolution of the Tn5801-tet(M)-like integrative and conjugative elements among Enterococcus, streptococcus, and Staphylococcus. Antimicrob. Agents Chemother. 60, 1736–1746 (2016).
Galata, V., Fehlmann, T., Backes, C. & Keller, A. PLSDB: a resource of complete bacterial plasmids. Nucleic Acids Res. 47, D195–D202 (2019).
Andam, C. P. & Gogarten, J. P. Biased gene transfer in microbial evolution. Nat. Rev. Microbiol. 9, 543–555 (2011).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Antipov, D., Raiko, M., Lapidus, A. & Pevzner, P. A. Plasmid detection and assembly in genomic and metagenomic data sets. Genome Res. 29, 961–968 (2019).
Pellow, D. et al. SCAPP: An algorithm for improved plasmid assembly in metagenomes. bioRxiv https://doi.org/10.1101/2020.01.12.903252 (2020).
Pradier, L., Tissot, T., Fiston-Lavier, A. S. & Bedhomme, S. PlasForest: a homology-based random forest classifier for plasmid detection in genomic datasets. BMC Bioinform. 22, 349 (2021).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Schwengers, O. et al. Platon: identification and characterization of bacterial plasmid contigs in short-read draft assemblies exploiting protein sequence-based replicon distribution scores. Microb. Genom. 6, mgen000398 (2020).
Johansen, J. et al. Genome binning of viral entities from bulk metagenomics data. Nat. Commun. 13, 965 (2022).
Li, H. & Durbin R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–60 (2009).
Li, H. et al. The sequence slignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Robertson, J. & Nash, J. H. E. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 4, e000206 (2018).
Yu, M. K., Fogarty, E. C. & Eren, A. M. Diverse plasmid systems and their ecology across human gut metagenomes revealed by PlasX and MobMess. Nat. Microbiol. 9, 830–847 (2024).
Camargo, A. P. et al. IMG/PR: a database of plasmids from genomes and metagenomes with rich annotations and metadata. Nucleic Acids Res. 52, D164–D173 (2024).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Flouri, T. et al. The phylogenetic likelihood library. Syst. Biol. 64, 356–362 (2015).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Forster, S. C. et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 37, 186–192 (2019).
Poyet, M. et al. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med. 25, 1442–1452 (2019).
Zou, Y. et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 37, 179–185 (2019).
Acman, M., van Dorp, L., Santini, J. M. & Balloux, F. Large-scale network analysis captures biological features of bacterial plasmids. Nat. Commun. 11, 1–11 (2020).
Zhao, X. BinDash, software for fast genome distance estimation on a typical personal laptop. Bioinformatics 35, 671–673 (2019).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with biobakery 3. Elife 10, 1–42 (2021).
Cantalapiedra, C. P., Hern̗andez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Liu, B., Zheng, D., Zhou, S., Chen, L. & Yang, J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917 (2022).
Alcock, B. P. et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525 (2020).
Pal, C., Bengtsson-Palme, J., Rensing, C., Kristiansson, E. & Larsson, D. G. J. BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 42, 737–743 (2014).
Acknowledgements
This research was partly funded by the European Union’s Horizon 2020 Research and Innovation Program under grant agreement No.818431 (SIMBA, Sustainable Innovation of Microbiome Applications in the Food System) and the NNF Data Science Initiative through the grant pTracker (NNF200C0062223 awarded to SJS). This work was performed using the Danish National Life Science Supercomputing Center, Computerome.
Author information
Authors and Affiliations
Contributions
W.L.H. processed the bioinformatic and statistical analysis, produced the figures, and wrote the manuscript; J.R. gave recommendations for bioinformatic and statistical analyses; F.K. gave recommendations for data collection; J.N. and S.J.S. reviewed the results and provided suggestions for bioinformatic analyses. All authors commented on and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Itzhak Mizrahi, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, W., Russel, J., Klincke, F. et al. Insights into the ecology of the infant gut plasmidome. Nat Commun 15, 6924 (2024). https://doi.org/10.1038/s41467-024-51398-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51398-3
- Springer Nature Limited