

Abstract
Industrial CO2 electrolysis via electrochemical CO2 reduction has achieved progress in alkaline solutions, while the same reaction in acidic solution remains challenging because of severe hydrogen evolution side reactions, acid corrosion, and low target product selectivity. Herein, an industrial acidic CO2 electrolysis to pure HCOOH system is realized in a proton-exchange-membrane electrolyzer using an acid-tolerant Bi-based metal-organic framework guided by a Pourbaix diagram. Significantly, the Faradaic efficiency of HCOOH synthesis reaches 95.10% at a large current density of 400 mA/cm2 with a high CO2 single-pass conversion efficiency of 64.91%. Moreover, the proton-exchange-membrane device also achieves an industrial-level current density of 250 mA/cm2 under a relatively low voltage of 3.5 V for up to 100 h with a Faradaic efficiency of 93.5% for HCOOH production, which corresponds to an energy consumption of 200.65 kWh/kmol, production rate of 12.1 mmol/m2/s, and an energy conversion efficiency of 38.2%. These results will greatly aid the contemporary research moving toward commercial implementation and success of CO2 electrolysis technology.
Similar content being viewed by others

Introduction
CO2 electrolysis technology (also known as the electrochemical carbon dioxide reduction reaction, eCO2RR) is expected to aid the global carbon-neutral target by eliminating excessive CO2, especially when it is driven by renewable energy such as solar, wind and geothermal1,2,3,4. Progress has been made for alkaline eCO2RR in terms of current density, selectivity, and energy conversion efficiency through catalyst design, microenvironmental regulation and reactor modification5,6,7, since alkaline media can suppress the competitive hydrogen evolution reaction (HER) because of a decreased proton concentration and large water dissociation barrier8,9,10. However, the alkaline/neutral eCO2RR has not become commercially successful because CO2 gases react with hydroxide ions and alkaline cations producing severe carbonate/bicarbonate salts and there is inherent bicarbonate in neutral media11,12. These side reactions decrease the CO2 single-pass conversion efficiency (SPCE) via the eCO2RR, cover the electrode surface, and jam the pores of the gas diffusion layer (GDL), thus harming the durability of the CO2 electrolysis system.
Therefore, acidic electrolytes are used to avoid side reactions that generate carbonate/bicarbonate salts, and thus address the carbon loss and stability issues13. In addition, acidic media enable the eCO2RR to be adaptable to commercial proton-exchange-membrane (PEM)-type electrolyzers and greatly promote its industrialization14. Nevertheless, in weak acids (pH ≥ 3), carbonate/bicarbonate salts are still present locally since rapid consumption of protons by industrial-level eCO2RR leads to high-concentration OH− and thus salt precipitation15,16. Strong-acid (pH ≤ 1) electrolytes could alleviate such issues; however, they also introduce challenges related to acid corrosion of electrocatalysts and electrolyzers. More importantly, severe HER occurs because of the extra-high concentration of protons. In particular, for eCO2RR to formic acid (HCOOH), acidic media (pH ≤ 3.75) can improve the CO2 SPCE and decrease the high separation cost associated with different product forms (formic acid/formate) in media with different pH values17. Therefore, electrolytes with proper pH values must be carefully selected to obtain the best trade-off between CO2 SPCE and product selectivity, which is highly related to the feasibility of electrocatalysts, electrolyzers, and product cost18. Bismuth-based electrocatalysts show great potential in eCO2RR, because of their high selectivity, long-lasting stability, and wide operating potential/current density windows19. For industrial applications, relevant parameters such as formic acid concentration, energy consumption, and energy efficiency values should also be mentioned20.
Herein, a PEM-type CO2 electrolyzer was constructed using acidic electrolytes and acid-tolerant bismuth metal-organic frameworks (Bi-MOFs) as electrocatalysts for the eCO2RR to produce HCOOH. A Pourbaix diagram was proposed to theoretically guide the preparation of acid-stable Bi-MOFs, which can also benefit the future design of acidic electrocatalysts. At an industrial-level current density of 400 mA cm−2, a pH 2 electrolyte was used to suppress both HER and local carbonates. A high Faradaic efficiency of formic acid (FEHCOOH) of 95.10% and a large SPCE for HCOOH production of 64.91% were simultaneously obtained. The PEM device operated at an industrial current density of 250 mA cm−2 and a relatively low voltage of 3.5 V, achieved an excellent FEHCOOH of 93.5%, an energy consumption of 200.65 kWh kmol−1 and a product rate of 12.11 mmol m−2 s−1. Furthermore, a solar-cell-driven PEM-electrolyzer prototype was assembled to obtain a superior energy conversion efficiency from solar to electricity and subsequently chemical energy (STC, 15.94%). This work would greatly promote the commercialization of CO2-consuming and energy-efficient eCO2RR technology.
Results
Electrolyte choice for industrial eCO2RR
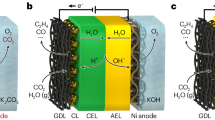
As shown in Fig. 1a, b, the reaction microenvironment of eCO2RR differs in alkaline and acidic media. Typically, CO2 gases pass through the GDL to reach catalysts; electrons also travel to the catalysts via a current collector; protons are generated by water dissociation (H2O → H+ + OH−) or bicarbonate dissociation (CO2 + OH− → HCO3−, HCO3− → CO32− + H+) in alkaline and directly available as free protons (H+) or hydrated protons (H3O+, H5O2+, etc.) in acids9,21. Then CO2 and protons are electrochemically reduced on the catalyst surface (known as proton-coupled electron transfer, PCET) and generate products such as HCOO−. Otherwise, CO2 transforms carbonate/bicarbonate, which greatly reduces CO2 utilization via the eCO2RR. This phenomenon is much more severe in alkaline environments than in acidic environments with alkaline cations used to suppress the HER.

Schematics of the eCO2RR microenvironment in a alkaline and b acidic electrolytes; GDL gas diffusion layer. c, d Simulated CO32− and CO2 concentrations in a pH-2 electrolyte. Source data are provided as a Source Data file.
Therefore, the eCO2RR local environment in acidic media was simulated using a one-dimensional (1D) model in a broad current density range (0–500 mA cm−2, Fig. 1c, d, Supplementary Note 1 and Figs. 1–4). Initially, in acidic electrolytes with a pH range of 4–2, the local CO2 concentration is much lower than that of the bulk electrolyte under industrial-level current densities above 250 mA cm−2 due to the rapid CO2 consumption during the eCO2RR. However, in a strongly acidic electrolyte at pH 1, the CO2 concentrations in the local and bulk electrolytes are similar because carbonates are quickly converted back to CO2 by high-concentration protons. The CO32− concentration shows a different trend, which is much greater locally than in bulk electrolytes with pH values of 4–2 above 250 mA cm−2, and the difference becomes increasingly significant with decreasing distance from the bulk to the electrode surface. However, the CO32− concentration is notably low in the pH-1 electrolyte, and no obvious difference is observed in the entire current-density range (Supplementary Fig. 3). In detail, the local CO32− concentration in the pH-2 electrolyte is significantly lower than that in the pH-3 and -4 electrolytes at a current density of 250 mA cm−2 (Supplementary Fig. 4), which increases with increasing current density because of faster proton consumption. Thus, the corresponding carbon loss in the pH-2 electrolyte is less than that in the pH-3 and -4 electrolytes, and there should be a threshold value of electrolyte pH (pH 2) and current density (250 mA cm−2) that can simultaneously achieve a high CO2 SPCE, product selectivity and industrial-level eCO2RR rate.

a XRD patterns and b FTIR spectra of Bi-TDC MOFs and the 2,5-H2TDC ligand. c High-angle annual dark-field TEM and EDS mapping images of Bi, C, S and O in the Bi-TDC@DMF. Scale bars, 500 nm. The colors cyan, green, yellow and red represent the elements bismuth, carbon, sulfur and oxygen, respectively. d XANES spectrum of the Bi L3-edge, e R-space of the EXAFS spectrum and f wavelet transform of the Bi L3-edge of the Bi-TDC MOFs and Bi foil reference. g Schematic configurations of the Bi-TDC@DMF MOF. Source data are provided as a Source Data file.

a Surface Pourbaix diagram derived from DFT calculations of thermodynamically stable hydrogenations on Bi-TDC@DMF. b Polarization curves plotted using steady-state current densities. Potentials are not iR corrected. c FEHCOOH in electrolytes with pH values of 2 and 3. Error bars represent the standard deviation of three independent experiments. d FEHCOOH and corresponding SPCE of the Bi-TDC@DMF-promoted eCO2RR at 400 mA cm−2 in an electrolyte with pH 1, 2, and 3 at different CO2 flow rates. e Comparison of the CO2RR partial current density, FEHCOO− product, electrolyte pH, potential and SPCE. SHE standard hydrogen electrode, RHE reversible hydrogen electrode, FE Faraday efficiency. Source data are provided as a Source Data file.

a Polarization curves plotted using steady-state current densities. b FEHCOOH and corresponding SPCE against CO2 flow rates of the eCO2RR at 250 mA cm−2. c Chronoamperometry testing at 250 mA cm−2. d Plot of the current against time under chopped solar illumination, the right panel shows the FEHCOOH and e the i-V curve of Si-solar cells and polarization curve of the eCO2RR. The electrocatalyst was Bi-TDC@DMF, and the electrolyte was a mixed solution of H2SO4 with K2SO4 at pH 2. f Techno-economic analysis. The voltage is not iR corrected. FE Faraday efficiency. Source data are provided as a Source Data file.
Synthesis and characterization of Bi-TDC electrocatalysts
To adapt PEM-type electrolyzers, acid-tolerant eCO2RR electrocatalysts were designed and prepared. Two types of Bi-based MOFs were synthesized by using 2,5-thiophenedicarboxylic acid (2,5-H2TDC) through a solvothermal process using N, N-dimethylformamide (DMF) and ethanol (denoted Bi-TDC@DMF and Bi-TDC@EtOH, respectively). In Fig. 2a and Supplementary Fig. 5, these MOFs exhibit similar X-ray diffraction (XRD) patterns with a (−111) peak decreasing from the EtOH to DMF sample, possibly because the steric hindrance of DMF is greater than that of ethanol, which increases the interplanar spacing. The Fourier transform infrared spectra (FTIR, Fig. 2b) of the Bi-TDC MOFs, exhibit revealed peaks at approximately 1641, 1401, and 745 cm−1, assigned to the stretching vibrations of C = O, C-O from -COOH, and C-S-C, respectively22. Due to coordination between O from carboxylic groups of the ligand and Bi atoms, MOFs exhibit a weaker ν(C = O) peak than 2,5-H2TDC, with that of Bi-TDC@DMF weaker than the EtOH sample. Thus, Bi atoms in the Bi-TDC@DMF MOF may coordinate with more O atoms, which is consistent with the Raman results (Supplementary Fig. 6).
According to transmission electron microscopy (TEM, Fig. 2c), field emission scanning electron microscopy (FESEM), energy-dispersive spectroscopy (EDS) mapping images (Supplementary Figs. 7, 8), and atomic force microscopy (AFM, Supplementary Fig. 9), the assembled morphology of the Bi-TDC@DMF MOF shows a nanosheet shape (a thickness of 1.8 nm) with a homogeneous dispersion of Bi, C, S and O.
The element valence and coordination of Bi-TDC MOFs were also characterized using X-ray photoelectron spectroscopy (XPS, Supplementary Fig. 10). In both samples, peaks ascribed to Bi, C, S, and O were observed in the survey scan. In detail, deconvolution of the high-resolution Bi 4f spectrum of Bi-TDC@DMF revealed 4f5/2 and 4f7/2 peaks at approximately 164.5 eV and 159.2 eV, respectively, with ΔE of 5.3 eV, which can be assigned to Bi3+ species23,24,25. The Bi 4f5/2 peak of Bi-TDC@EtOH shifts by 0.08 eV to a lower binding energy, which suggests that the coordination state of Bi-O in the two MOFs might vary, which is consistent with the FTIR results. The high-resolution S 2s, O 1s and C 1s spectra of the Bi-TDC MOFs are deconvoluted, which confirms the presence of Bi-O coordination and corresponding ligands.
The synchrotron-irradiated X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) of Bi MOFs were also analyzed (Fig. 2d–f and Supplementary Figs. 11, 12). As shown in Bi L3-edge XANES spectra (Fig. 2d), Bi-TDC@DMF shows a slightly greater Bi valence than the EtOH sample according to the slightly greater photon energy, which suggests that Bi in the DMF MOF might coordinate with more oxygen atoms than that in the EtOH MOF. Fourier transform of EXAFS (Fig. 2e and Supplementary Fig. 12) was also performed to evaluate the coordination environment of Bi atoms, and a peak at ∼1.60 Å can be attributed to the Bi-O coordination of Bi-TDC@EtOH, slightly smaller than that of the DMF MOF26. As displayed in the wavelet transformed (WT) contour plots of Bi L3-edge EXAFS (Fig. 2f), Bi-TDC MOFs display only one intensity maximum at ∼1.55 Å, also assigned to the Bi-O coordination. To clearly illustrate the coordination state, the intense peak is finely fitted that the Bi atom of Bi-TDC@DMF coordinates with O atom with a coordination number of 8.23 ± 0.36, larger than that of the Bi-TDC@EtOH sample (7.07 ± 0.71, Supplementary Table 1). Based on the above results, it can be concluded that the Bi atom in Bi-TDC@DMF is highly coordinated with eight O atoms, which results in greater stability than the less-coordinated Bi-TDC@EtOH. Specifically, the Bi3+ ion in Bi-TDC@DMF can be considered a 4-connected node, and the TDC2− ligand as a 2-connected node. Therefore, the Bi-TDC@DMF MOF can be described as a 2-fold interpenetrated topology (Fig. 2f).
Electrochemical measurements of the acidic eCO2RR
Before the eCO2RR performance is quantified in electrolytes with different pH values, the stability of Bi-TDC MOFs in such media is simulated by establishing a Pourbaix diagram using density functional theory (DFT) calculations (Fig. 3a), which is proposed based on a hydrogen-adsorbed catalyst surface as a function of pH and the potential27,28,29. Specifically, integration of the surface formation energy (SFE) formalism in an ensemble Pourbaix diagram provides a measure of confidence in predicted stability by quantifying consistency between functionals30,31. The Pourbaix diagram shows that two terminations are the most favorable in the potential range of −1.00 to 2.00 V at variable pH. The SFE of Bi-TDC@DMF is thermally stable at potentials below 1.67 V even in media with extra-low pH values, and that of the hydrogen-adsorbed material (*H+Bi-TDC@DMF) decreases to 1.49 V, which implies that Bi-TDC@DMF can remain stable in acids. In great contrast, the corresponding calculation model of the Bi-TDC@EtOH MOF collapses during computation despite various tested approaches, which suggests that it is unstable in acids. In addition to the above theoretical studies, experimental stability verification was conducted (Supplementary Figs. 13–22). The chemical states of Bi-TDC MOFs were tested using XPS (Bi 4f and S 2s peaks). Considering the XRD and FTIR results, the Bi-TDC@DMF MOF can be stable in strong acids but transform to Bi2CO5 and Bi2O3 in alkaline conditions, whereas the Bi-TDC@EtOH MOF sample cannot withstand strong acids. Thus, the highly saturated coordination between Bi and O atoms of Bi-TDC@DMF MOF results in a robust skeleton structure, whereas the undercoordinated EtOH MOF is not durable. In short, the thermodynamic acidic stability of the Bi-TDC@DMF MOF material was confirmed by the theoretical Pourbaix diagram and experiment.
According to the aforementioned results, eCO2RR measurements were conducted in a three-chamber flow-type electrolytic cell using electrolytes with different pH values, and as-prepared catalyst constructed gas diffusion electrodes (GDEs) as working electrodes (Supplementary Fig. 23). Gas and liquid products were detected by gas chromatography (GC) and nuclear magnetic resonance (NMR) spectroscopy (Supplementary Fig. 24). The eCO2RR performance was tested using various electrochemical techniques (Supplementary Fig. 25). Chronopotentiometry (v-t) curves were collected at current densities of 50–700 mA cm−2 (Supplementary Fig. 26; no iR corrections if not specified) in different electrolytes, and polarization curves were plotted using the above steady-state current density and potential (Fig. 3b). The corresponding FEs of eCO2RR products were analyzed three times at applied current densities in electrolytes with various pH values (Fig. 3c, d, Supplementary Figs. 27–29 and Supplementary Table 2).
As a result, the Bi-TDC@DMF constructed electrode shows superior eCO2RR performance, where HCOO− is the dominant product over the entire pH range (1–14). Specifically, the FEHCOOH concentration in a pH-2 electrolyte is above 90% over a wide current density/potential range (50–500 mA cm−2, and the corresponding potential is −1.32 to −3.20 V vs RHE). The total FE of eCO2RR products (HCOO− and CO) exceeds 97% in the range of 100–400 mA cm−2, which suggests that the competitive HER has been greatly suppressed even in acidic electrolytes. To compare the eCO2RR activity of two Bi-TDC MOFs, pH-2 and pH-3 electrolytes are used, ensuring both materials are stable in the test media so that Bi-TDC@DMF exhibits a greater amount of FEHCOO− over the entire range of applied current densities (Fig. 3c and Supplementary Figs. 30–32). This wide working range of current density or potential will greatly benefit the direct connection of the CO2 electrolyzer with intermittent solar cells and enable an efficient and stable STC conversion system.
Theoretically, acidic electrolytes used in the eCO2RR can prevent carbon loss by inhibiting carbonate/bicarbonate salts and promote the global carbon-neutral target. To experimentally confirm this assumption, the CO2 SPCE was examined in different electrolytes. As shown in Fig. 3d and Supplementary Fig. 33, at an industrial-level current density of 400 mA cm−2, the SPCE for HCOOH production reaches a high value of 64.91% in a pH-2 medium by gradually reducing the inlet flow rate of CO2 from 50 to 3 standard cubic centimeter per minute (sccm), which is larger than that of the pH-14 (21.34%), pH-3 (43.47%) and even pH-1 media (51.25%). In particular, isotope-labeling experiments with 13CO2 were performed to test if the carbon source of the eCO2RR product was from the gaseous reactant or organic ligands of Bi MOFs. Liquid products of H13COOH are identified, suggesting they are from 13CO2 gases via the eCO2RR, instead of ligands or other contaminants (Supplementary Fig. 34). According to the XRD, FTIR and XPS results, within the testing period (1200 s), the Bi3+ nodes in Bi-TDC@DMF could maintain their electrocatalytic activity (Supplementary Fig. 35). Supplementary Table 3 summarizes the recent research on the eCO2RR to HCOO− products mainly in acidic systems, where bismuth-based catalysts have shown excellent performance. The as-prepared Bi-TDC@DMF electrocatalyst ranks among the best for the acidic eCO2RR to HCOOH (Fig. 3e, Supplementary Table 3)14,18,25,31,32,33,34,35,36,37,38,39,40,41.
Even decent FEHCOOH and CO2 SPCE have been achieved in the previous three-chamber electrolyzer; to implement practical applications, the eCO2RR performance should be evaluated in a real PEM-type electrolyzer (Supplementary Fig. 36). The polarization curve was plotted using the steady-state current density against the cell voltage (Fig. 4a), showing an industrial current density of 250 mA cm−2 at a relatively low voltage of 3.5 V. In addition, a superior FEHCOOH (93.5%), energy consumption (200.65 kWh kmol−1), product rate (12.11 mmol m−2 s−1) and energy conversion efficiency (38.2%) for HCOOH production are obtained (Supplementary Table 4). At this high current density in the pH 2 medium, the FEHCOOH could be maintained above 82.1% decreasing the gas flow rate from 50 to 3 sccm, achieving the highest SPCE of 51.16% at 3 sccm, as shown in Fig. 4b. Furthermore, FEHCOOH could remain above 90% during chronoamperometry testing at 250 mA cm−2 for up to 100 h with an even slightly lower driving voltage (Fig. 4c). Then, the two-electrode PEM-electrolyzer prototype was directly powered by solar cells, to evaluate the energy conversion efficiency from solar to electricity and subsequently to HCOOH (Supplementary Fig. 37). According to the i-V curve of the GaInP/GaInAs/Ge triple-junction cell and the polarization curve of eCO2RR, the optimal working voltage and current are 2.30 V and 10.29 mA, respectively. Importantly, the current remains at approximately 10.25 mA, with an average FEHCOOH of 92.47% within 1600 s. On average, the STC efficiency is 15.94%, with an electrical-chemical energy conversion efficiency of 57.49% (Fig. 4d, e), possibly due to the extraordinary eCO2RR activity and its flexible adaptation because of the wide working window in acidic media.
Furthermore, techno-economic analysis (TEA) was conducted to compare PEM and anion-exchange membrane (AEM, which uses alkaline electrolytes)-type CO2 electrolysis42. The details are described in Supplementary Information Note 2. The parameters for TEA are as follows: a full-cell voltage of 3.5 V, a current density of 250 mA cm−2, and an SPCE of 51.16%, which are achieved in this work (Supplementary Table 4). As a result, the estimated cost for AEM-type HCOOH production is 0.895 $ kg−1, with a product separation of ∼$8,333,333 million per year, pressure swing adsorption (PSA) for CO2 recovery of ∼$721,810 per year, electricity of ∼$11,921,983 per year, and equipment maintenance of ∼$217,436 per year. However, the PEM-type HCOOH production shows a much lower cost (0.470 $ kg−1), even less than the market price in China of 0.683 $/kg. The cost breakdown includes a separation of ∼$5,221,433 million per year, a PSA of ∼$190,350 per year, electricity of ∼$6,623,584 million per year, and maintenance at ∼$45,855 per year. Obviously, the downstream separation costs for the eCO2RR for HCOOH are significant, and those for alkaline (electrodialysis followed by distillation) are much greater than those for acid (pressure swing distillation, 0.250 vs 0.157 $ kg−1). Therefore, the direct industrial PEM-type CO2 electrolysis to HCOOH has high techno-economic feasibility attributed to the large CO2 SPCE and feasible product form, which is highly competitive with the commercial BASF method.
Reaction mechanism of the acidic eCO2RR to HCOOH
The reaction mechanism was investigated using both DFT calculations (the atomic coordination of optimized computational models is provided in Supplementary Data 1, and DFT details are described in Supplementary Information Note 3) and in situ techniques. During a cathodic process, the eCO2RR and competitive HER occur. As shown in Supplementary Fig. 38a–d, the HER mechanism was investigated first, including an initial Volmer step (*H production) followed by a Tafel (a combination of two *H to *H2) or Heyrovsky step (*H reacts with free hydrogen to produce *H2). In detail, the Gibbs free energy of HER is firstly calculated by assuming a pH-0 electrolyte; then, the calculation was corrected to pH 2, which is the actual value of the working media. In both mechanisms, the Volmer step of HER catalyzed by Bi-TDC@DMF shows a more negative Gibbs energy than that of the Bi-TDC@EtOH sample (−0.55 vs −0.18 eV), which suggests a stronger adsorption energy for hydrogen. The Heyrovsky step of DMF sample displays a much higher energy barrier than that of the EtOH sample (3.29 vs 1.87 eV), and the Tafel step exhibits the same trend (1.78 vs 0.64 eV), which indicates that the potential determining step (PDS) for HER promoted by the DMF and EtOH samples is the *H2 generation and the former shows lower HER activity. The kinetic Tafel slopes of the HER promoted by the Bi-TDC@DMF and EtOH catalysts are 334 and 184 mV dec−1, which indicates that the HER proceeds via the Heyrovsky step possibly because surface adsorbed K+ ions reduce the *H concentration (Supplementary Fig. 39)43. In other words, as compared to the Bi-TDC@EtOH MOF, the DMF sample shows lower HER activity but stronger adsorption of hydrogen and benefits the later PCET steps of the eCO2RR process, which is also supported by the calculated charge density difference (Supplementary Figs. 40–42).
The free-energy diagrams of the eCO2RR to HCOOH and CO were computed (Fig. 5a, b and Supplementary Figs. 43, 44) using free H+ as the proton source, which is identical to the HER simulation. In detail, for the eCO2RR to HCOOH process catalyzed by Bi-TDC@DMF, after CO2 been adsorbed (* + CO2 → *CO2, the adsorption energy ∆Eads is −0.67 eV, Fig. 5b and Supplementary Fig. 41), the first PCET step from *CO2 to *OCHO (C is hydrogenated) is exothermic with a Gibbs energy change (∆G) of −0.86 eV. Then the second PCET from *OCHO to *HCOOH (*OCHO + H+ + e → *HCOOH) is uphill (∆G, +0.95 eV), followed by the desorption of *HCOOH to produce HCOOH (∆G, +0.46 eV). However, for the competing eCO2RR to CO pathway, ∆G of the first PCET step from *CO2 to *COOH (O is hydrogenated) is −0.01 eV, and the uphill second PCET from *COOH to *CO (*COOH + H+ + e → *CO + H2O, ∆G of +1.94 eV), followed by desorption of *CO to the CO product with ∆G of +1.3 eV. Both PDS for the eCO2RR to HCOOH and the CO pathway constitute the second PCET step, where the largest ∆G values are +0.95 eV and 1.94 eV, respectively. Consequently, for the eCO2RR to HCOOH, PDS might also be the second PCET step but with a much smaller energy barrier than the CO pathway (0.95 eV vs 2.84 eV), which suggests that the eCO2RR promoted by Bi-TDC@DMF energetically prefers HCOOH production. Supplementary Fig. 45 shows a schematic diagram of eCO2RR to CO.

a Calculated reaction pathways for the eCO2RR in a pH-0 electrolyte. b Reaction pathways for the eCO2RR to HCOOH in a pH 2 electrolyte. c Charge density difference of *OCHO and *HCOOH adsorbed on the Bi-TDC@DMF and Bi-TDC@EtOH configurations. The purple and yellow regions represent charge accumulation and depletion, respectively. d Operando Raman spectra of the acidic eCO2RR (pH = 1, 2, 3) promoted by Bi-TDC@DMF. e, f In situ FTIR spectra in a pH-2 electrolyte. V is the potential relative to RHE in (d–f). Source data are provided as a Source Data file.
Notably, the effect of pH on the reaction diagram was simulated over the entire pH range (Supplementary Figs. 43, 44). ∆G of PDS (∆GPDS) for the eCO2RR highly depends on the pH, and a smaller pH leads to a smaller ∆G and a decreased energy barrier. ∆GPDS of the Bi-TDC@DMF-promoted eCO2RR in the pH-14 electrolyte increases to +1.78 eV from +0.95 eV in the pH-1 electrolyte, both of which are lower than those of Bi-TDC@EtOH catalyst (+2.72 eV and +1.39 eV). In particular, in the pH-2 medium, ∆GPDS of Bi-TDC@DMF is +1.07 eV, which is much smaller than the value of +2.01 eV of the EtOH sample (Fig. 5b). To elucidate the underlying reasons, the charge density difference was calculated based on the amount of absorbed intermediates during the crucial step (*OCHO + H+ + e− → *HCOOH, Fig. 5c). The charge transfer between *OCHO and Bi sites in Bi-TDC@DMF is more pronounced than that in the EtOH sample, which favors the second PCET step. Therefore, it could be concluded that the pH-corrected ∆G does not alter the reaction pathway of the eCO2RR to HCOOH44, but regulates the energy barrier of its PDS (Supplementary Fig. 46).
In addition to theoretical investigations, operando Raman spectroscopy has been employed to inspect key intermediates and reaction microenvironment of the eCO2RR to HCOOH by sequential application of potentials from −0.2 to −0.6 V vs RHE (Fig. 5d and Supplementary Figs. 47a, 48). In detail, a peak attributable to SO42− is observed at approximately 1000 cm−1 in all electrolytes (pH = 1–3); the peaks of CO32− (1065 cm−1) and/or HCO3− (1016 cm−1) are observed in the pH 3 medium with increasing overpotential while absent in the pH-1 and −2 electrolytes. Thus, carbonate and bicarbonate ions may not form in strong-acid electrolytes (pH 1 and 2) during eCO2RR, which benefits the CO2 SPCE. The situation in the pH-2 medium is different from the simulation result (Fig. 1c), possibly due to the lower current density during testing, which confirms that a pH-2 medium is a good candidate for balancing the by reaction HER, CO2 SPCE, and industrial current density45,46,47. In addition, the O–C–O antisymmetric mode from *HCOO− appears at 1558 cm−1, and the broad peak at 2820–2950 cm−1 can be ascribed to the C–H stretching vibration of *HCOO−, where the peak intensity gradually increases with increasing overpotential12,32, which confirms the HCOOH formation from eCO2RR.
Next, in situ attenuated total reflectance (ATR)-FTIR measurements were conducted to capture possible intermediates during the eCO2RR (Fig. 5e, f and Supplementary Fig. 47). With the applied potential ranging from −0.1 to −1.4 V vs RHE, the O–H stretching vibration frequency of interfacial H2O in the eCO2RR catalyzed by Bi-TDC@DMF shows a more pronounced redshift (3695 → 3486 cm−1) than the EtOH catalyst. This indicates the competitive adsorption of water molecules on the catalyst surface is weakened, which favors CO2 adsorption. Additionally, the dissolved CO2 exhibits a noticeable blueshift (2341 → 2364 cm−1) and effectively suppresses the HER. Signals from *CO intermediates are not observed possibly due to the low FECO43,48. Two characteristic IR peaks at 1400 and 2912 cm−1 appear at −0.1 V vs RHE and progressively increase with increasing overpotential, which can be ascribed to the C–H bending and C–H stretching of *OCHO, respectively, and confirm the increased concentration of key intermediates. The peak at 1349 cm−1 is assigned to the adsorbed HCOO−, which verifies the formation of HCOOH species and corresponds well with operando Raman49. Based on the above results, the reaction pathway of the eCO2RR to HCOOH catalyzed by Bi-TDC@DMF is proposed and shown in Fig. 5g and Supplementary Figs. 47, 48, where CO2 is first absorbed on the catalyst surface and forms *CO2; then, the first PCET generates *OCHO, followed by the second PCET to *HCOOH and finally to HCOOH products.
According to the above results, the pH-1 electrolyte is expected to obtain the largest CO2 SPCE, since no carbonates are theoretically or experimentally observed. Meanwhile, the highest value of 64.91% is achieved in the pH-2 medium, possibly because the strong HER in the pH-1 medium (51.25%) compromises the eCO2RR and decreases the CO2 consumption37. In addition, even when the acid tolerance of electrocatalysts is good, pH-2 media have better compatibility with electrocatalysts and devices than pH-1 media, which makes them the best choice to obtain decent CO2 SPCE and simultaneously suppress HER at industrial-level current densities.
Based on the proposed Pourbaix diagram, an acid-stable highly coordinated Bi-MOF is developed displaying unexpectedly superior eCO2RR to HCOOH performance, which is even better than that of the low-coordinated Bi-MOF. MOF electrocatalysts with saturated coordination are commonly stable but inactive since they cannot efficiently activate reacting gases such as CO2, while those with undersaturated coordination behave oppositely, which leads to a trade-off between stability and activity50,51,52. This study appears to mitigate this limitation by developing a highly stable and active Bi-TDC@DMF electrocatalyst, which has a coordination number of 8.23 ± 0.36 and shows improved eCO2RR to HCOOH activity, as compared to Bi-TDC@EtOH with a coordination number of 7.07 ± 0.71. Therefore, this work proposes a “Semi-Activation-Semi-Transformation (SAST)” mechanism, where coordination determines the reactant activation capability of electrocatalysts, and the intermediate transformation efficiency impacts the overall reaction. This concept divides the reaction into a reactant activation segment and a product formation segment, suggesting a nuanced understanding of the relationship between electrocatalyst stability and activity. In other words, the highly stable electrocatalyst (high-coordinate Bi-TDC@DMF) unobtrusively activates CO2 (the activation energy calculated by the transition state of the CO2 molecule (TS1) is 0.72 eV, as shown in Supplementary Fig. 46a, and the bond lengths of C-O1 and C-O2 in CO2 are 1.357 Å and 1.360 Å, respectively). Then, the electrocatalyst absorbs CO2 with a consistent ∆Eads of −0.67 eV. These high energies couple with a low transformation barrier from *CO2 to *OCHO (0.71 eV for TS2), followed by the free energy of *OCHO intermediates of −1.53 eV (Fig. 5a and Supplementary Fig. 46a). The overall PDS is *HCOOH desorption (0.95 eV). In contrast, the low-stability sample (low-coordinate Bi-TDC@EtOH) strongly activates CO2 (the activation energy of TS1 is 0.44 eV, and the bond lengths of C-O1 and C-O2 in CO2 are 1.361 Å and 1.362 Å, respectively), and ∆Eads is −0.83 eV. The transformation energy barrier of TS2 from *CO2 to *OCHO is 1.08 eV and the free energy of *OCHO is −2.22 eV, which are against those of Bi-TDC@DMF, which proposes that efficient activation of the CO2 reactant does not essentially match the targeted transformation of intermediates to form final products. Additionally, the high conversion barrier for *HCOOH formation is 1.88 eV, as shown in Fig. 5a and Supplementary Fig. 46a. Based on the Sabatier principle, this SAST concept builds a possible relationship between the moderate bonding of adsorbates and energy barriers of intermediate transformation, which can circumvent the trade-off between stability and activity and may be applicable in other electrocatalytic reactions (Fig. 6).

Schematic diagram of the eCO2RR to HCOOH process promoted by Bi-TDC@DMF and the activity-stability trade-off.
Discussion
In this study, a highly techno-economically feasible and carbon-consuming acidic eCO2RR to HCOOH system was fabricated in a PEM-type electrolyzer. An acid-tolerant Bi-TDC@DMF MOF was designed and prepared according to a Pourbaix diagram, which can guide the future design of acidic electrocatalysts. The eCO2RR promoted by Bi-TDC@DMF MOF in acidic media (pH 2) displayed an optimal FEHCOOH of 95.10%, a high SPCE for HCOOH production of 64.91% at 400 mA cm−2, and a broad working window of 50–500 mA cm−2 in a three-chamber flow cell. The constructed PEM-type CO2 electrolyzer achieved an industrial-level current density of 250 mA cm−2 at a cell voltage of only 3.5 V with a FEHCOOH of 93.5%, a product rate of 12.11 mmol m−2 s−1, an energy consumption of 200.65 kWh kmol−1 and a superior STC of 15.94% directly driven by solar cells. According to the theoretical DFT calculations and experimental operando Raman and in situ ATR-FTIR results, the second PCET is the PDS for the eCO2RR to HCOOH catalyzed by Bi-TDC MOFs. Interestingly, a “SAST” concept was proposed to explain the circumvented trade-off relationship between stability and activity of the saturated-coordinated Bi-TDC@DMF MOF for the eCO2RR. In practice, the performance of the industrial-level acidic eCO2RR to HCOOH and high techno-economic feasibility will greatly aid the commercial success of eCO2RR technology and promote the global carbon-neutral target.
Methods
Chemicals
2,5-thiophenedicarboxylic acid (C6H4O4S, ≥ 98%), absolute ethanol (CH3CH2OH, ≥ 99.5%), N,N-dimethylformamide (C3H7NO, ≥ 99.9%), methanol (CH3OH, ≥ 99.9%) isopropyl alcohol (C3H8O, ≥ 99.7%) and dimethyl sulfoxide (C2H6SO, ≥ 99.8%) were purchased from Aladdin (China). Bismuth nitrate pentahydrate (Bi(NO3)3·5H2O, ≥ 99%), potassium sulfate (K2SO4, ≥ 99%) and sulfuric acid (H2SO4, 95.0‒98.0%) were purchased from Sinopharm Chemical Reagent Co. Ltd (China). Deuterium oxide (D2O, 99.9 atom%D) was purchased from Acros Organics. Potassium hydroxide (KOH, 95%) and methanol (CH4O, 99.5%) were purchased from Macklin. The CO2 gas (99.999%) was supplied by Nanjing Wenda Special Gas Co., Ltd. All materials were used as received without further purification. Deionized (DI) water was purified with a UPT-II-20T ultrapure water system.
Synthesis of Bi-TDC@DMF
Bi(NO3)3·5H2O (120 mg, 0.25 mmol), C6H4O4S (170 mg, 1 mmol), DMF (6 ml), methanol (6 ml) and water (6 ml) were sealed in a 50 ml vial and heated at 115 °C for 16 h. After cooling down, the solution was collected, centrifuged, washed with EtOH and deionized water, and freeze-dried for 24 h to obtain the white powder product (90 mg, percentage yield is 56%).
Synthesis of Bi-TDC@EtOH
Bi(NO3)3·5H2O (120 mg, 0.25 mmol), C6H4O4S (170 mg, 1 mmol), EtOH (6 ml), methanol (6 ml) and water (6 ml) were sealed in a 50 ml vial and heated at 115 °C for 16 h. After cooling down, the solution was collected, centrifuged, washed with EtOH and deionized water, and freeze-dried for 24 h to obtain the white powder product (80 mg, percentage yield is 48%).
Characterizations
XRD was conducted on a Bruker D8 ADVANCE X-ray diffractometer with a Cu Kα resource. XPS spectra were carried out on a Thermo Scientific K-Alpha X-ray Photoelectron Spectrometer. The morphology of the as-synthesized materials was measured by TEM (FEI TalosF200x). The morphology and thicknesses of on-chip films were measured by SEM (GeminiSEM300) and AFM (Bruker Multimode 8). The pH of electrolytes was measured by a pH meter (FiveEasy Plus FE28). The Raman spectrum was measured by the confocal Raman microscope (LabRAM HR-Evolution), with a focused 532 nm laser for light excitation. Attenuated total reflection-infrared spectroscopy (ATR-FTIR) was performed using a Bruker INVENIO spectrometer equipped with a diamond ATR element. The X-ray absorption fine structure (XAFS) spectra at Bi L3-edge were collected from the Shanghai synchrotron radiation facility (SSRF).
Synchrotron-based X-ray absorption measurements
The X-ray absorption fine structure (XAFS) spectra at Bi L3-edge were collected from the Shanghai synchrotron radiation facility (SSRF). The acquired XAFS data were processed according to the standard procedures using the Athena module of Demeter software packages. To obtain the quantitative structural parameters around central atoms, least-squares curve parameter fitting was performed using the Artemis module of Demeter software packages.
Local species profile modeling
One-D model was set up to analyze the local Carbonate distribution by using COMSOL Multiphysics software. The following local species, such as CO2, HCO32−, CO32−, and H+, are considered in the process. The model includes a catalyst layer (about 300 nm) and a surrounding diffusion layer (100 µm). More simulation details are discussed in the Supplementary Information (Supplementary Note 1).
Electrocatalytic measurements in the three-compartment flow cell
The flow cell is composed of three chambers, including the gas chamber, catholyte chamber and anolyte chamber. The Ag/AgCl electrode (filled with saturated KCl) was used as the reference electrode, commercial Ni foams (for use under alkaline conditions) or Iridium oxide-loaded titanium meshes (Ti-IrO2, for use under acidic conditions) were used as the counter electrode, and carbon papers (YLS-30T, size: 2.5 × 2.5 cm2, thickness: 0.23 nm) loaded with cathodic catalysts were used as the working electrode. In detail, the Bi-TDC catalyst dispersion was prepared by stirring and sonicating a 2 ml mixed solution of isopropyl alcohol and deionized water (volume ratio: 3:1) with 20 mg of Bi-TDC and 80 µl of Nafion solution. Subsequently, 2 ml of the dispersion was spray-coated on a hydrophobic carbon paper with a microporous layer using a semi-automatic airbrush. The catalyst layer area of the working electrode was 1 × 1 cm2 after spray-coating through the mold with a catalyst loading of 2.5 mg cm−2. Iridium oxide-loaded titanium meshes (Ti-IrO2) were prepared by etching a Ti mesh, dipping it in an isopropanol solution of IrCl3, and calcining at 500 °C for 10 min53. Anion exchange membrane (Fumasep FAB-PK-130, size: 2.5 × 2.5 cm2, thickness: 130 µm) and proton exchange membrane (Nafion N117, size: 2.5 × 2.5 cm2, thickness: 183 µm) were used as the ion exchange membranes. The proton exchange membrane was immersed in 0.5 M H2SO4 for about 1 h before use. The catholytes with different pH (1–5) were prepared using 0.5 M K2SO4 and H2SO4. For alkaline tests, 1 M KHCO3 and 1 M KOH solution were used. All solutions were saturated with argon to exclude interference from other soluble gases. The pH value was determined by a calibrated pH meter. The primary source of uncertainty is the inherent error of the pH meter, with an accuracy of ±0.01 pH units. The flow rates of catholyte and anolyte were both set to be at 5 ml·min−1 via a peristaltic pump (EC200-01). CO2 gas was delivered into the cathode chamber at a flow rate of 50 ml·min−1 using a mass flow controller (FMA-2617A-VOL). The current density reported was based on the geometric surface area. The SPCE measurements were only performed once. If not otherwise stated, all potentials are converted to the RHE scale via the equation (without IR compensation):
Gas-phase products were quantified using a gas chromatograph (Agilent 8890). Liquid products were quantified by an AVANCE III 500 MHz NMR spectrometer with water suppression using the excitation sculpting method. The NMR sample was prepared by mixing 300 µl of the electrolyte with 75 µl of D2O and 18.7 mM (or 10.7 mM) dimethyl sulfoxide as an internal standard.
In HER measurements, linear sweep voltammetry (LSV) tests were performed with iR compensation (The resistance of 6 Ω × 85% was used to calculate the iR-correction), and the presented potential values were calibrated to the reversible hydrogen electrode (RHE). The cell resistance was measured by the electrochemical workstation. All electrochemical tests were performed at room temperature and pressure.
Electrocatalytic measurements in the PEM-type electrolyzer
For the eCO2RR test in a PEM-type electrolyzer, a custom-made PEM-type electrolyzer was used. The Bi-TDC@DMF-loaded layer (catalyst loading area: 4 × 5 mm for stability test, catalyst loading mass: 2.5 mg cm−2), a cation exchange membrane (Nafion N117, size: 2 × 2 cm2) and Ti-IrO2 were compressed to form the PEM electrode. A mixed solution of 0.5 M K2SO4 and H2SO4 (pH = 2) served as the anolyte. The flow rate of humidified CO2 gas was 50 to 3 sccm during the eCO2RR, and the liquid products in the outlet gas were collected with deionized water (2 ml per collection). In consideration of requirements for long-term operation of the device, 10 mg of PTFE particles were dispersed in 1 ml of isopropanol solution by sonication for 30 min, and 100 µl of the dispersion was spray-coated onto the surface of carbon paper loaded with Bi-TDC@DMF. The periodic testing scheme of running at 1 mA cm−² for 20 min every 20 h is implemented to address the water management issue during the operation of PEM-type electrolyzer. All electrochemical tests were performed at room temperature and pressure.
Calculation of the eCO2RR performance
The product FE in eCO2RR were all obtained with bare catalysts without surface coating. Once we identified the best-performing catalyst, we coated the catalyst with a PTFE layer for the long-term stability test in the PEM-type electrolyzer. It showed the same performance as that without the carbon layer coating. The coating of the PTFE layer only made the electrical field more homogeneous during long-term operation.
HCOO− Faradaic efficiency was determined as below:
Where n is the moles of liquid product in the cathodic compartment, N is 2, F = 96,485 C mol−1, t is the reaction time, j is the current density.
For the single-pass conversion efficiency of HCOOH or formate, we only measured the Faradaic efficiency of the formic acid product. The single-pass carbon efficiency (SPCE) of CO2 toward HCOOH was calculated as follows at 25 °C, 1 atm:
Where I represents the partial current of HCOOH or formate, N stands for electron transfer and is 2 for HCOOH.
The energy consumption per kmol of the target product is defined as the amount of energy needed to produce the desirable product. The calculation formula is as follows:
where j, A and M are also the current density supplied to the PEM cell, the geometric electrode area and the number of moles of the target product generated, respectively, and V is the cell potential of the PEM cell in each experiment.
The electricity to formic acid energy conversion efficiency is defined as:
Solar-driven electrochemical CO2 reduction system (pH 2)
The GaInP/GaInAs/Ge triple-junction solar cell, model HGSC-A100B, is available for purchase from Hasunopto and has a geometric area of 0.85 cm². An Oriel Instruments 75 W Solar Simulator provided the illumination for laboratory testing. Details of the testing setup can be found in Supplementary Fig. S37. Real-time monitoring and recording of the solar cell’s voltage and current outputs were carried out using a data acquisition system. Measurements for the solar-driven eCO2RR were conducted as a single set of experiments.
The S = solar-to-fuel efficiency is described as:
In this context, JGDE represents the current density (mA cm−2) associated with the gas diffusion electrode (GDE), ΔUrxn denotes the thermodynamic potential (V) required for the complete reaction, fFE,HCOOH is the Faradaic efficiency, C indicates the light concentration factor, and Plight refers to the incident light irradiance (mW cm−2) prior to concentration. Additionally, AGDE and APV are the respective areas of the GDE and photovoltaic cell (PV) measured in cm². For the cathodic reaction converting CO2 to formic acid and the anodic reaction involving the oxygen evolution from water, ΔUrxn is set to 1.43 V.
For our system with AGDE = 0.1 cm2 and APV = 0.85 cm2, C = 1, fFE,HCOOH = 92.47%, we determined:
The electricity to HCOOH energy conversion efficiency is defined as:
Techno-economic analysis (TEA)
Assume that the project life cycle of the CO2 electrolytic reduction plant is 20 years, with a daily output of 100 tons of formic acid and an annual operation of 8000 h. The voltage of the membrane electrode assembly cell is 3.5 V, the current density is 250 mA cm−2, FE of HCOOH is 82.1%, and the single-pass conversion rate is 51.16%. For the alkaline system, since the product is formate rather than formic acid, according to the work of Mahinder Ramdin17, the method of electrodialysis and then distillation is used for the purification of formate, and the cost of liquid phase separation is obtained by referring to the calculation cost. For the rest, please refer to the calculation formula of the acidic system. More computational details are discussed in the Supplementary Information (Supplementary Note 2).
Operando Raman spectroscopy characterization of the eCO2RR on catalyst surface
In situ Raman was conducted on a HORIBA LabRAM HR Evolution Raman spectrometer with a 532 nm solid laser as an excitation source. The measurements were carried out in a homemade flow cell with a quartz window to detect the signal from cathode GDE. For each scan, the Raman spectrum was accumulated by two acquisitions (20 s per acquisition). A peristaltic pump was used to pass 0.5 M K2SO4 (pH = 2) into the liquid chamber at a flow rate of 5 ml per minute. CO2 gas was introduced to the back of the GDE at 50 sccm by a mass flow controller (FMA-2617A-VOL). Working electrodes were fabricated by airbrushing catalysts onto GDE. A platinum sheet was used as the counter electrode.
In situ attenuated total reflection-infrared spectroscopy (ATR-IR)
For electrochemical studies, in situ ATR-SEIRAS was carried out with a Nicolet iS50 infrared spectrometer equipped with an MCT-A detector that is cooled using liquid nitrogen. Unpolarized infrared radiation was employed to capture spectra, with a resolution of 4 cm−1. Spectral data were displayed in absorbance, calculated as −log(R/R0), with R being the spectrum of the sample and R0 the reference spectrum. The electrochemical workstation used for controlling potential and measuring current was a CHI 660 model from CH Instruments, Inc. A gold-coated semi-cylindrical prism, 20 mm in diameter, was used as the conductive substrate for the catalysts and as the infrared reflection component. Catalyst suspensions were applied onto the Au/Si surface to prepare the working electrode. ATR-IR spectra were obtained in situ during changes in the working electrode potential54.
DFT calculations
Density functional theory (DFT) computations were performed using the Perdew-Burke-Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) in the VASP package code55,56,57. The ionic cores were described using the projector-augmented wave (PAW) method. A plane wave expansion with a cut-off energy of 400 eV was utilized, optimized from a range of cut-off energies. The electronic self-consistent-loop criterion was set at 10−5 eV. During geometry optimization, structures were relaxed until forces on all atoms were smaller than 0.05 eV/Å. Gaussian smearing with a 0.50 eV width was applied, and K-points for structural optimization in all models were set to 1 × 1 × 1. More computational details are discussed in the Supplementary Information (Supplementary Note 3).
The detailed calculation of Gibbs free energy for the eCO2RR has been conducted as follows:
where G, E and CP represent the chemical potential (partial molar Gibbs free energy), electronic energy and heat capacity, respectively. The entropy term can be defined as the combination of translational, rotational, vibrational, and electronic contributions, expressed as:
In addition, intrinsic zero-point energy (ZPE) and extrinsic dispersion (D) corrections can be finally obtained as follows:
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and Supplementary Information files. Source data are provided with this paper.
References
Yang, Y. et al. Operando studies reveal active Cu nanograins for CO2 electroreduction. Nature 614, 262–269 (2023).
Monteiro, M. C. O. et al. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 4, 654–662 (2021).
Wang, Z. et al. Advanced catalyst design and reactor configuration upgrade in electrochemical carbon dioxide conversion. Adv. Mater. 35, 2303052 (2023).
Chi, L. P. et al. Stabilizing indium sulfide for CO2 electroreduction to formate at high rate by zinc incorporation. Nat. Commun. 12, 5835 (2021).
Kim, C. et al. Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy 6, 1026–1034 (2021).
Seger, B., Robert, M. & Jiao, F. Best practices for electrochemical reduction of carbon dioxide. Nat. Sustain. 6, 236–238 (2023).
Niu, Z.-Z. et al. CO2-assisted formation of grain boundaries for efficient CO–CO coupling on a derived Cu catalyst. Natl. Sci. Open 2, 20220044 (2023).
Huang, J. E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Chen, X. et al. Promoting water dissociation for efficient solar driven CO2 electroreduction via improving hydroxyl adsorption. Nat. Commun. 14, 751 (2023).
Li, Z. et al. Electron‐rich Bi nanosheets promote CO2− formation for high‐performance and pH‐universal electrocatalytic CO2 reduction. Angew. Chem. Int. Ed. Engl. 62, e202217569 (2023).
Zheng, M. et al. Electrocatalytic CO2-to-C2+ with ampere-level current on heteroatom-engineered copper via tuning *CO intermediate coverage. J. Am. Chem. Soc. 144, 14936–14944 (2022).
Wu, Z. et al. Engineering bismuth-tin interface in bimetallic aerogel with a 3D porous structure for highly selective electrocatalytic CO2 reduction to HCOOH. Angew. Chem. Int. Ed. Engl. 60, 12554–12559 (2021).
Zhang, L. et al. Atomically dispersed Ni-Cu catalysts for pH-universal CO2 electroreduction. Adv. Mater. 35, e2209590 (2023).
Fang, W. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).
Endrődi, B. et al. Operando cathode activation with alkali metal cations for high current density operation of water-fed zero-gap carbon dioxide electrolysers. Nat. Energy 6, 439–448 (2021).
Bondue, C. J., Graf, M., Goyal, A. & Koper, M. T. M. Suppression of hydrogen evolution in acidic electrolytes by electrochemical CO2 reduction. J. Am. Chem. Soc. 143, 279–285 (2021).
Ramdin, M. et al. High-pressure electrochemical reduction of CO2 to formic acid/formate: effect of pH on the downstream separation process and economics. Ind. Eng. Chem. Res. 58, 22718–22740 (2019).
Chi, L. P. et al. Efficient and stable acidic CO2 electrolysis to formic acid by a reservoir structure design. Proc. Natl Acad. Sci. USA 120, e2312876120 (2023).
Fernández-Caso, K., Díaz-Sainz, G., Alvarez-Guerra, M. & Irabien, A. Electroreduction of CO2: advances in the continuous production of formic acid and formate. ACS Energy Lett. 8, 1992–2024 (2023).
Díaz-Sainz, G. et al. Improving trade-offs in the figures of merit of gas-phase single-pass continuous CO2 electrocatalytic reduction to formate. Chem. Eng. J. 405, 126965 (2021).
Zhao, Y. et al. Conversion of CO2 to multicarbon products in strong acid by controlling the catalyst microenvironment. Nat. Synth. 2, 403–412 (2023).
Wang, C.-P. et al. Self-optimized metal-organic framework electrocatalysts with structural stability and high current tolerance for water oxidation. ACS Catal. 11, 7132–7143 (2021).
Yue, X., Cheng, L., Li, F., Fan, J. & Xiang, Q. Highly strained Bi‐MOF on bismuth oxyhalide support with tailored intermediate adsorption/desorption capability for robust CO2 photoreduction. Angew. Chem. Int. Ed. Engl. 61, e202208414 (2022).
Wang, Y. et al. BiPO4‐derived 2D nanosheets for efficient electrocatalytic reduction of CO2 to liquid fuel. Angew. Chem. Int. Ed. Engl. 60, 7681–7685 (2021).
Yang, Q. et al. Novel Bi-doped amorphous SnOx nanoshells for efficient electrochemical CO2 reduction into formate at low overpotentials. Adv. Mater. 32, e2002822 (2020).
Liang, X., Fu, N., Yao, S., Li, Z. & Li, Y. The progress and outlook of metal single-atom-site catalysis. J. Am. Chem. Soc. 144, 18155–18174 (2022).
Rodriguez-Olguin, M. A. et al. Chlorine in NiO promotes electroreduction of CO2 to formate. Appl. Mater. Today 28, 101528 (2022).
Kramer, D., Wang, Y. & Wharton, J. Mechano-electrochemistry effects due to deformation of copper oxide films. Faraday Discuss. 180, 137–149 (2015).
Hansen, H. A., Rossmeisl, J. & Nørskov, J. K. Surface Pourbaix diagrams and oxygen reduction activity of Pt, Ag and Ni(111) surfaces studied by DFT. Phys. Chem. Chem. Phys. 10, 3722–3730 (2008).
Zhang, J. et al. Accelerating electrochemical CO2 reduction to multi-carbon products via asymmetric intermediate binding at confined nanointerfaces. Nat. Commun. 14, 1298 (2023).
Shen, H. et al. Acidic CO2-to-HCOOH electrolysis with industrial-level current on phase engineered tin sulfide. Nat. Commun. 14, 2843 (2023).
Yao, D. et al. The controllable reconstruction of Bi-MOFs for electrochemical CO2 reduction through electrolyte and potential mediation. Angew. Chem. Int. Ed. Engl. 60, 18178–18184 (2021).
Ma, L. et al. In situ-activated indium nanoelectrocatalysts for highly active and selective CO2 electroreduction around the thermodynamic potential. ACS Catal. 12, 8601–8609 (2022).
Chen, X. et al. Boron dopant induced electron‐rich bismuth for electrochemical CO2 reduction with high solar energy conversion efficiency. Small 17, 2101128 (2021).
Ye, K. et al. In situ reconstruction of a hierarchical Sn-Cu/SnOx core/shell catalyst for high-performance CO2 electroreduction. Angew. Chem. Int. Ed. Engl. 59, 4814–4821 (2020).
Qiao, Y. et al. Engineering the local microenvironment over Bi nanosheets for highly selective electrocatalytic conversion of CO2 to HCOOH in strong acid. ACS Catal. 12, 2357–2364 (2022).
Gu, J. et al. Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium. Nat. Catal. 5, 268–276 (2022).
Sun, B. et al. Unveiling pH‐dependent adsorption strength of *CO2− intermediate over high‐density Sn single atom catalyst for acidic CO2‐to‐HCOOH electroreduction. Angew. Chem. Int. Ed. Engl. 63, e202318874 (2024).
Yu, X. et al. Coverage enhancement accelerates acidic CO2 electrolysis at ampere-level current with high energy and carbon efficiencies. Nat. Commun. 15, 1711 (2024).
Yan, T., Pan, H., Liu, Z. & Kang, P. Phase-inversion induced 3D electrode for direct acidic electroreduction CO2 to formic acid. Small 19, e2207650 (2023).
Xue, H., Zhao, Z.-H., Liao, P.-Q. & Chen, X.-M. “Ship-in-a-bottle” integration of ditin(IV) sites into a metal–organic framework for boosting electroreduction of CO2 in acidic electrolyte. J. Am. Chem. Soc. 145, 16978–16982 (2023).
Zhao, B.-H. et al. Economically viable electrocatalytic ethylene production with high yield and selectivity. Nat. Sustain. 6, 827–837 (2023).
Ma, Z. et al. CO2 electroreduction to multicarbon products in strongly acidic electrolyte via synergistically modulating the local microenvironment. Nat. Commun. 13, 7596 (2022).
Yang, G. et al. Regulating Fe-spin state by atomically dispersed Mn-N in Fe-N-C catalysts with high oxygen reduction activity. Nat. Commun. 12, 1734 (2021).
Dubessy, J., Geisler, D., Kosztolanyi, C. & Vernet, M. The determination of sulphate in fluid inclusions using the MOLE Raman microprobe. Application to a Keuper halite and geochemical consequences. Geochim. Cosmochim. Acta 47, 1–10 (1983).
Ichinohe, Y., Wadayama, T. & Hatta, A. Electrochemical reduction of CO2 on silver as probed by surface‐enhanced Raman scattering. J. Raman Spectrosc. 26, 335–340 (1995).
Castro, J. L., Otero, J. C. & Marcos, J. I. Anomalous SERS of monocarboxylic acids on silver sols. J. Raman Spectrosc. 28, 765–769 (1997).
Stalder, C. J., Chao, S. & Wrighton, M. S. Electrochemical reduction of aqueous bicarbonate to formate with high current efficiency near the thermodynamic potential at chemically derivatized electrodes. J. Am. Chem. Soc. 106, 3673–3675 (1984).
Cao, C. et al. Metal-organic layers leading to atomically thin bismuthene for efficient carbon dioxide electroreduction to liquid fuel. Angew. Chem. Int. Ed. Engl. 59, 15014–15020 (2020).
Huang, J. et al. Experimental Sabatier plot for predictive design of active and stable Pt-alloy oxygen reduction reaction catalysts. Nat. Catal. 5, 513–523 (2022).
Zeng, Y. et al. Tuning the thermal activation atmosphere breaks the activity–stability trade-off of Fe–N–C oxygen reduction fuel cell catalysts. Nat. Catal. 6, 1215–1227 (2023).
Li, X. et al. Functional CeOx nanoglues for robust atomically dispersed catalysts. Nature 611, 284–288 (2022).
Luc, W., Rosen, J. & Jiao, F. An Ir-based anode for a practical CO2 electrolyzer. Catal. Today 288, 79–84 (2017).
Ye, J.-Y., Jiang, Y.-X., Sheng, T. & Sun, S.-G. In-situ FTIR spectroscopic studies of electrocatalytic reactions and processes. Nano Energy 29, 414–427 (2016).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC 52488201, 52376193 and 92163124) and the Jiangsu Innovative/Entrepreneurial Talent Program. We thank the BL14W1 and BL02U2 beamline of the Shanghai Synchrotron Radiation Facility (SSRF) for providing beamtime. We also thank Shanghai Yuanfang Tech for providing infrared testing services.
Author information
Authors and Affiliations
Contributions
K.Y. was responsible for material characterization, electrochemical testing, and manuscript writing. M.L. handled DFT calculations and done mechanism analysis. T.G. conducted the techno-economic analysis. G.X. performed COMSOL simulations. D.L. took care of the material synthesis. Y.Z. provided discussion. Q.L. and J.D. designed and supervised this project. All the authors discussed the results and edited this manuscript.
Corresponding author
Ethics declarations
Competing interests
J.D. has filed Chinese provisional patent applications (No. 202410704581) based on this work. The other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Min-Rui Gao, Guillermo Díaz-Sainz, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, K., Li, M., Gao, T. et al. An acid-tolerant metal-organic framework for industrial CO2 electrolysis using a proton exchange membrane. Nat Commun 15, 7060 (2024). https://doi.org/10.1038/s41467-024-51475-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51475-7
- Springer Nature Limited