

Abstract
Type 2 alveolar epithelial (AT2) cells of the lung are fundamental in regulating alveolar inflammation in response to injury. Impaired mitochondrial long-chain fatty acid β-oxidation (mtLCFAO) in AT2 cells is assumed to aggravate alveolar inflammation in acute lung injury (ALI), yet the importance of mtLCFAO to AT2 cell function needs to be defined. Here we show that expression of carnitine palmitoyltransferase 1a (CPT1a), a mtLCFAO rate limiting enzyme, in AT2 cells is significantly decreased in acute respiratory distress syndrome (ARDS). In mice, Cpt1a deletion in AT2 cells impairs mtLCFAO without reducing ATP production and alters surfactant phospholipid abundance in the alveoli. Impairing mtLCFAO in AT2 cells via deleting either Cpt1a or Acadl (acyl-CoA dehydrogenase long chain) restricts alveolar inflammation in ALI by hindering the production of the neutrophilic chemokine CXCL2 from AT2 cells. This study thus highlights mtLCFAO as immunometabolism to injury in AT2 cells and suggests impaired mtLCFAO in AT2 cells as an anti-inflammatory response in ARDS.
Similar content being viewed by others


Introduction
Type 2 alveolar epithelial (AT2) cells are sentinel progenitor cells of the alveoli that are essential to alveolar recovery from injury1 and are crucial in regulating immunity to environmental stimuli and micro-organisms2. Importantly, AT2 cells are metabolically active, are enriched for fatty acid synthase3,4, and produce lipoprotein-containing alveolar surfactant to sustain alveolar surface tension5. Reduced lipid synthesis in AT2 cells is associated with various pathological changes in murine lungs and promotes the development of respiratory distress syndrome6,7, lung fibrosis8,9,10, chronic obstructive pulmonary disease4, and acute lung injury (ALI)11.
Mitochondrial long-chain fatty acid β-oxidation (mtLCFAO) is the major pathway for degrading fatty acids, fueling cellular energy production, and mtLCFAO disorders are life-threatening12. In experimental models, impaired mtLCFAO leads to dysfunction of various organ systems, including the liver and heart13. In murine lungs, loss of mtLCFAO via deleting acyl-coenzyme A dehydrogenase long-chain (Acadl) gene, encoding the key enzyme long-chain acyl-CoA dehydrogenase (LCAD), leads to surfactant impairment and reduced lung function14,15, and polymorphisms in Acadl have been linked to respiratory distress syndrome and sudden infant death14. Additionally, mtLCFAO impairment is associated with decreased alveolar inflammation in murine models of bacterial and viral acute lung injury (ALI)16,17. The above findings suggest that mtLCFAO plays a pivotal role in the development and function of the lungs at homeostasis and in response to infection. Yet the role of mtLCFAO in AT2 cells in regulating responses of lungs to injury remains to be determined.
Transport of long-chain acyl-coenzyme A (CoA) from the cytosol into mitochondria is the rate-limiting step in mtLCFAO and is carried out by the enzyme carnitine palmitoyltransferase 1 (CPT1)12. While CPT1 has a demonstrated specialized role in lymphogenesis by lymphatic endothelial cells18,19, glucagon release from pancreatic islet cells20, and the function of various stem cells21,22, little is known about its role in AT2 cell biology and function. Downregulation of CPT1a in AT2 cells has been implicated in a murine model of acute lung injury23, and the ratio of carnitine to palmitoylcarnitine plus oleylcarnitine [C0/(C16 + C18)], an indicator of CPT1 deficiency, is increased in the plasma of individuals with acute respiratory distress syndrome (ARDS)24. However, the functional significance of mtLCFAO in AT2 cells at baseline and in ALI is unclear and requires extensive evaluation.
In this study, we first assessed if mtLCFAO is altered in the alveolar epithelium in the lungs of individuals with ARDS as well as in AT2 cells isolated from murine ALI models. Using transgenic mice with a deletion of Cpt1a selectively in AT2 cells, we show that Cpt1a-deficient AT2 cells display a unique adaptive metabolic response involving homeostatic energy production and phospholipid synthesis upregulation. Through a series of in vivo models, metabolomic, lipidomic, and transcriptomic approaches, this study demonstrates that downregulated mtLCFAO in AT2 cells supports the resolution of alveolar inflammation in ALI by restricting the production of CXCL2 from AT2 cells to limit neutrophilic inflammation. Collectively, our data highlights the pivotal role of AT2 cell-specific mitochondrial metabolism in regulating alveolar immune responses to lung injury.
Results
mtLCFAO is decreased in AT2 cells in human and murine ALI
We first assessed if mtLCFAO is altered in individuals with ARDS with pathological hallmarks of diffuse alveolar damage (DAD)25. According to The Human Protein Atlas26, CPT1a is the main CPT1 isoform expressed in human lungs, while CPT1b and CPT1c are not expressed. Immunohistochemical (IHC) staining was performed to evaluate CPT1a and LCAD expression in surgical sections from healthy controls and those with ARDS and evidence of DAD (Supplementary Fig. 1; Supplementary Table). CPT1a expression, quantified by a histological score (H score), was significantly decreased in the alveolar epithelium in DAD lungs, whereas the H score of LCAD was unaltered (Fig. 1a–c). Immunofluorescent staining of CPT1a and HTII-28027, an apical surface marker of human AT2 cells, was further performed using DAD lung samples with low IHC CPT1a H scores and control lung samples. The results confirmed that CPT1a expression was remarkably decreased in AT2 cells in DAD lungs (Supplementary Fig. 2). We also analyzed publicly available single-cell RNA sequencing (scRNAseq) datasets generated from individuals with coronavirus disease 2019 (COVID-19)-associated ARDS (Supplementary Fig. 3a)28,29. AT2 cell clusters were identified through the positive expression of SFTPA1, SFTPB, and SFTPC, and the negative expression of AGER and PDPN (Supplementary Fig. 3b, c). Gene set enrichment analyses (GSEAs) revealed downregulated expression scores for the gene ontology (GO) pathway fatty acid β-oxidation (GO:0006635) in AT2 cells from individuals with COVID-19-associated ARDS (Fig. 1d). These results suggest that mtLCFAO is downregulated in AT2 cells in ARDS lungs.

a Representative results of immunohistochemistry staining for CPT1a, long-chain acyl-CoA dehydrogenase (LCAD), and cytokeratin (CK) using surgical sections from control and diffuse alveolar damage (DAD) donors (scale bar = 50 μm). b, c The alveolar epithelial levels of CPT1a (b) and LCAD (c) are semi-quantified using the H scores (control n = 18, DAD n = 27; the line indicates median; two-sided p values are calculated by Mann–Whitney U tests). d Expression of fatty acid β-oxidation pathway (GO: 0006635) in AT2 cells in ARDS is analyzed using human lung single-cell RNA sequencing datasets from people with fatal COVID-19 and controls (GSE171524, GSE171668). Expression score for each cell is calculated by averaging the expression of the gene involved in the fatty acid β-oxidation pathway, and the comparison between groups is exhibited using violin plots. Two-sided p values are calculated using Mann–Whitney U tests. e AT2 cells from SftpcCreERt2+/− mice instilled with LPS or ddH2O are isolated for extracellular flux analyses using palmitate alone or after etomoxir pretreatment (Oligo oligomycin, Rot rotenone, AA antimycin A). f Oxygen consumption rates (OCRs) for ATP production are then calculated (palmitate alone, n = 8 replicates per group; palmitate with etomoxir pretreatment, n = 10 replicates per group; both are representative data from two independent experiments). e, f Data are represented as mean ± SEM, and f two-sided p values are calculated by unpaired Student’s t-tests.
Next, we assessed if mtLCFAO is altered in AT2 cells of a murine ALI model. ALI was induced through the intratracheal instillation of lipopolysaccharide (LPS) in control mice. After 24 h, AT2 cells were isolated and mtLCFAO activity was assessed. Extracellular flux analyses showed that glucose-linked ATP production was significantly decreased in AT2 cells from LPS-exposed mice when compared to controls (Supplementary Fig. 4a, b). Palmitate-related oxygen consumption rates (OCRs) of ATP production were also significantly decreased in AT2 cells isolated from LPS-exposed mice when compared to controls. Pretreatment of isolated AT2 cells with etomoxir, an irreversible CPT1a inhibitor, abrogated the differences of palmitate-related OCRs in the control and AT2 cells upon LPS treatment (Fig. 1e, f), suggesting that defects in mtLCFAO are related to decreased expression or activity of CPT1a. Quantitative polymerase chain reaction (qPCR) analyses demonstrated significantly decreased Cpt1a expression, but increased Acadl expression in AT2 cells after LPS instillation (Supplementary Fig. 4c, d). However, immunoblotting for CPT1a and LCAD in isolated AT2 cells revealed no robust changes of CPT1a or LCAD protein expression upon LPS treatment (Supplementary Fig. 4e–g). The above findings suggest that decreased mtLCFAO activity in AT2 cells after LPS instillation may be related to altered in vivo regulation of CPT1a activity30,31.
Targeted deletion of Cpt1a in AT2 cells impairs mtLCFAO
To further explore the role of CPT1a and mtLCFAO in AT2 cells, we first confirmed that murine AT2 cells express CPT1a and LCAD. Single-cell suspensions from murine lungs were generated and fractionated using anti-CD45 and anti-EpCAM antibodies. CPT1a and LCAD were expressed in all the cellular fractions, including CD45−EpCAM+SFTPC+ AT2 cells (Fig. 2a). We next generated transgenic mice with a tamoxifen-inducible AT2 cell-specific deletion of Cpt1a (Cpt1aloxP/loxPSftpcCreERt2+/−, referred to as Cpt1aiΔAT2; Fig. 2b)8. Six weeks after tamoxifen injection, immunoblotting of isolated AT2 cell lysates confirmed CPT1a depletion in Cpt1a−/− AT2 cells when compared to SftpcCreERt2+/− control mice (Fig. 2c). Bulk RNA sequencing (RNASeq) confirmed that Cpt1a is the principal isoform expressed in murine AT2 cells, and loss of Cpt1a does not lead to remarkable upregulation of Cpt1b or Cpt1c expression (Supplementary Fig. 5a–d).

a The immunoblot shows CPT1a and LCAD expression in different cellular populations selected from single-cell suspensions using anti-CD45 and anti-EpCAM (representative data of two independent experiments). b A scheme showing the generation of transgenic mice with tamoxifen-inducible AT2 cell-specific Cpt1a deletion. c Immunoblots of control and Cpt1a−/− AT2 cells isolated 6 weeks after tamoxifen injection (n = 3 mice per group; representative data of two independent experiments). d Intracellular levels of carnitine (C0), palmitoylcarnitine (C16), and stearoylcarnitine (C18) are measured for calculating C0/(C16 + C18) ratio in AT2 cells (n = 5 mice per group; representative data of two independent experiments). e–h Extracellular flux analyses (e) with palmitate alone (Oligo oligomycin, Rot rotenone, AA antimycin A) or g after etomoxir pretreatment in AT2 cells, and oxygen consumption rates (OCRs) f, h are calculated (palmitate, n = 9 replicates per group; palmitate/etomoxir, control n = 7 replicates, Cpt1a−/− n = 8 replicates; representative data of two independent experiments). d–h Data are represented as mean ± SEM, and d, f, h two-sided p values are calculated by unpaired Student’s t-tests.
To confirm that Cpt1a-deficient AT2 cells had impaired mtLCFAO, intracellular C0/(C16 + C18) ratios were measured by lipidomic mass spectroscopy analyses. Consistently, the C0/(C16 + C18) ratio was significantly higher in Cpt1a-deficient AT2 cells when compared to control AT2 cells (Fig. 2d). Palmitate-dependent OCRs were significantly decreased in Cpt1a−/− AT2 cells (Fig. 2e, f), and etomoxir pretreatment eliminated palmitate-dependent OCR differences between Cpt1a−/− and control AT2 cells (Fig. 2g, h). Although previous studies demonstrated that mtLCFAO by CPT1a could regulate the production of ATP and mitochondrial reactive oxygen species (ROS)20,32, neither intracellular ATP levels (Fig. 3a) nor the generation of mitochondrial ROS (Supplementary Fig. 6) were altered in Cpt1a-deficient AT2 cells. The above results confirm that mtLCFAO is impaired in Cpt1a-deficient AT2 cells and that alternative metabolic pathways in Cpt1a−/− AT2 cells may compensate for a loss in mtLCFAO-linked ATP production.

a Intracellular ATP levels in the control and Cpt1a−/− AT2 cells (n = 5 mice per group). b, c Extracellular flux analyses (b) with glucose (Glu), pyruvate (Pyr), and glutamine (Gln) in AT2 cells, and OCRs (c) are calculated (n = 9 per group; representative data of two independent experiments). d, e Glycolysis stress tests (d) in AT2 cells (2DG, 2-deoxyglucose), and e extracellular acidification rates (ECARs) are calculated (n = 10 replicates per group; representative data of two independent experiments). f–k Extracellular flux analyses with (f) glutamine alone, h pyruvate alone, or j both pyruvate and glutamine in AT2 cells, and OCRs for ATP production using (g) glutamine alone (control n = 10 replicates, Cpt1a−/− n = 9 replicates), i pyruvate alone (n = 10 replicates per group), or k both pyruvate and glutamine (control, n = 9 replicates, Cpt1a−/−, n = 7 replicates) are calculated (f–k), all are representative data of two independent experiments. l A cartoon illustrating the metabolic adaptations in Cpt1a−/− AT2 cells (TCA tricarboxylic acid, OXPHOS oxidative phosphorylation). a–k Data are represented as mean ± SEM, and a, c, e, g, I, k two-sided p values are calculated by unpaired Student’s t-tests.
AT2 cells deficient in CPT1a maintain ATP production by utilizing substrates other than glucose
We next assessed if CPT1a-deficient cells had alterations in energy-generating metabolic pathways to sustain ATP production. Extracellular flux analyses with glucose, pyruvate, and glutamine showed increased OCRs for ATP production in Cpt1a−/− AT2 cells when compared to controls (Fig. 3b, c). However, extracellular acidification rates (ECARs) with glucose were significantly downregulated in Cpt1a−/− AT2 cells (Fig. 3d, e). To explore the effects of a loss of CPT1a on glycolysis further, we deleted Cpt1a in the murine AT2 cell line MLE12 by CRISPR/Cas9 (clustered regularly interspaced palindromic repeats and CRISPR-associated protein 9) gene editing (Supplementary Fig. 7a). CPT1a-deficient MLE12 cells not only showed decreased mtLCFAO, but also showed metabolic changes similar to those observed in murine AT2 cells, including increased OCRs with glucose, pyruvate and glutamine, and decreased ECARs with glucose (Supplementary Fig. 7b–g). Stable isotope resolved metabolomic analyses using uniformly 13C-labeled glucose ([U−13C] glucose) as the tracer was then performed to evaluate glycolytic activity in CPT1a-deficient MLE12 cells (Supplementary Fig. 8a). After metabolic labeling for 30 min, control and CPT1a-deficient MLE12 cells were collected for analyzing intracellular 13C-labeled metabolic derivatives of glycolysis and the tricarboxylic acid (TCA) cycle. Metabolites of glycolysis, including fructose-6-phosphate, 3-phosphoglycerate, and phosphoenolpyruvate, downstream amino acids (alanine, glutamine), and metabolites in the TCA cycle (citrate/isocitrate, malate) were all significantly lower in CPT1a-deficient MLE12 cells when compared to controls (Supplementary Fig. 8b, c). The data above thus confirm that glycolysis is downregulated in CPT1a-deficient MLE12 cells and thus may not be the pathway contributing to the maintenance of ATP production in CPT1a-deficient AT2 cells.
Glutamine anaplerosis or exogenous pyruvate supplementation can support energy production under glucose deprivation33,34. However, our data showed that Cpt1a−/− AT2 cells could not increase OCRs for ATP production from glutamine (Fig. 3f, g) nor pyruvate alone (Fig. 3h, i), suggesting that sustained ATP production in Cpt1a−/− AT2 cells may require both glutamine and pyruvate. Consistently, OCRs for ATP production utilizing both pyruvate and glutamine were significantly increased in Cpt1a−/− MLE12 (Supplementary Fig. 7h, i) and AT2 cells (Fig. 3j, k). Collectively, our results indicate that adaptative metabolism involving the utilization of substrates other than glucose is activated to maintain energy production in Cpt1a−/− AT2 cells (Fig. 3l).
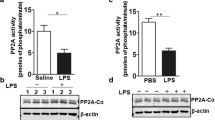
CPT1a deficiency upregulates phospholipid synthesis in AT2 cells
AT2 cells synthesize fatty acids in abundance to generate phospholipid-rich surfactants, in particular phosphatidylcholine (PC) and phosphatidylglycerol (PG)5. Loss of Cpt1a has been shown to result in lipid accumulation in renal tubular cells35, suggesting CPT1a may be important for lipid or phospholipid synthesis in AT2 cells. We first found that intracellular triglyceride levels were higher in the Cpt1a−/− AT2 cells when compared to controls (Supplementary Fig. 9). We next assessed if a loss in CPT1a affects the fundamental ability of AT2 cells to synthesize phospholipids. Using CLICK chemical reactions to label and quantify the amounts of newly synthesized choline-containing phospholipids36, phospholipid synthesis was significantly increased in CPT1a-deficient MLE12 cells (Fig. 4a, b). Conversely, CPT1a overexpression inhibited phospholipid synthesis in MLE12 cells (Fig. 4c–e). As CPT1a-deficient MLE12 cells, AT2 cells isolated from Cpt1aiΔAT2 mice also demonstrated upregulation of phospholipid synthesis (Fig. 4f, g). To assess whether upregulated phospholipid synthesis activity in the absence of CPT1a results in increased intracellular phospholipid levels in primary AT2 cells, lipidomic analyses through ultra-high performance liquid chromatography coupled with tandem mass spectrometry (UHPLC-MS/MS) were performed in Cpt1a−/− and control AT2 cells (Supplementary Data File 1, 2). Surprisingly, Cpt1a−/− AT2 cells showed similar intracellular levels of the top five abundant PC and PG species when compared to control AT2 cells (Fig. 4h, i). AT2 cells secrete mature surfactants into the alveoli, and altered surfactant synthesis in AT2 cells alters extracellular alveolar phospholipid abundance4. To test if a loss of CPT1a in AT2 cells led to changes in the abundance of phospholipids in the bronchoalveolar lavage fluid (BALF), lipidomic analyses were carried out in BALF obtained from Cpt1aiΔAT2 and control mice. Conversely to the AT2 cells, several PG and PC species were significantly increased in BALF in Cpt1aiΔAT2 mice when compared to controls (Fig. 4j, k; Supplementary Data File 3, 4). In summary, the above data shows that CPT1a regulates surfactant phospholipid synthesis in AT2 cells, and deleting Cpt1a in AT2 cells results in increased phospholipid synthesis with increased extracellular abundance of PC and PG species in the alveoli.

a, b Flow cytometric analyses (a) evaluating phospholipid synthesis in Cpt1a−/− and control MLE12 cells. The geometric mean fluorescent intensity (MFI) of phospholipid labeling (b) represents phospholipid synthesis activity (n = 3 replicates per group; representative data of two independent experiments). c–e Immunoblots (c) confirming CPT1a overexpression in Cpt1aOE MLE12 cells, generated through lentiviral transduction (control MLE12 cells generated through lentiviral transduction of empty construct; representative data of two independent experiments). d Flow cytometric analyses evaluating phospholipid synthesis assay of Cpt1aOE and control MLE12 cells, and I phospholipid synthesis activity is compared (n = 3 replicates per group; representative data of two independent experiments). f, g Flow cytometric analyses (f) evaluating phospholipid synthesis in AT2 cells, and the phospholipid synthesis activity (g) is compared (n = 3 replicates per group; representative data of two independent experiments). h, i Intracellular contents of PC and phosphatidylglycerol (PG) species in AT2 cells, and data of the top 5 abundant h PC and i PG species are shown (n = 12 mice per group; data represent results of two independent experiments). j, k The contents of PC and PG species in bronchoalveolar lavage fluid (BALF) samples, and data of the top five abundant j PC and k PG species are demonstrated (control mice n = 10, Cpt1aiΔAT2 mice n = 9; data represent results of two independent experiments). b, e, g, and h–k Data are represented as mean ± SEM, and b, e, g–k two-sided p values are calculated by unpaired Student’s t-tests (h–k, only p values less than 0.05 are shown).
CPT1a deficiency in AT2 cells resolves alveolar inflammation in ALI
We next assessed the physiological ramifications of deleting Cpt1a in AT2 cells in vivo. Six weeks after tamoxifen injection, we did not observe altered alveolar tissue damping and elastance in Cpt1aiΔAT2 mice (Supplementary Fig. 10a). Eight months after tamoxifen injection, we did not find any remarkable histologic changes in inflated lung sections of Cpt1aiΔAT2 mice (Supplementary Fig. 10b). LPS-induced lung injury is one of the most used experimental models for ARDS simulating the neutrophilic inflammatory response observed in individuals with ARDS37. In this model, administration of LPS into the lungs results in robust migration of neutrophils into the lung tissue with resolution 72 h after exposure38. Using this model, we find that the principal immune cells in the BALF at baseline are alveolar macrophages (AMs), whereas, at 24 h, there is a significant dominance of neutrophils which begins to diminish or resolve 72 h post-LPS instillation (Supplementary Fig. 11). We then investigated if CPT1a deficiency in AT2 cells could alter the response of the lung to acute injury induced by intratracheal LPS instillation. Control and Cpt1aiΔAT2 mice were instilled with LPS, and inflammatory and injury markers assessed at 24 or 72 h. Flow cytometric analysis of BALF cells isolated from control and Cpt1aiΔAT2 mice showed there was no significant difference in the percentages of AMs, neutrophils, eosinophils, or T lymphocytes between the control and Cpt1aiΔAT2 mice, either at baseline (Supplementary Fig. 12a, b) or in LPS-induced ALI (24 h; Fig. 5a, b). In the BALF, total cell counts, and protein levels were also similar between the control and Cpt1aiΔAT2 mice after 24 h of LPS (Fig. 5c, d). However, 72 h after LPS instillation, BALF cell numbers were significantly decreased in Cpt1aiΔAT2 mice despite no change in BALF protein levels (Fig. 5e–f). The findings suggest that Cpt1aiΔAT2 mice have a similar initial response to injury but appear to have accelerated resolution of alveolar cellular infiltrates in the LPS-induced ALI model.

a, b At 24 h after intratracheal LPS instillation, flow cytometric analyses (a) are performed to evaluate (b) the percentages of various immune cells, including alveolar macrophages (CD11c+SiglecF+), neutrophils (CD11c−Ly6G+), eosinophils (CD11c−Ly6G−CD3−SiglecF+), and T lymphocytes (CD11c−Ly6G−CD3+SiglecF−), in the CD45.2+ cell population in bronchoalveolar lavage fluid (BALF) (control mice n = 20, Cpt1aiΔAT2 mice n = 16; results of two independent experiments). c–h BALF samples are collected at 24 h (c, d, g) and 72 h (e, f, h). The c, e total cell number and the level of d, f protein and g, h various cytokines/chemokines, including CXCL1, CXCL2, tumor necrosis factor (TNF) α, and interleukin (IL) 6, in BALF are measured (data at 24 h: control mice, c, d n = 8 g n = 4 for ddH2O, c, d, g n = 47 for LPS; Cpt1aiΔAT2 mice, c, d n = 7 g n = 3 for ddH2O, LPS c, d, g n = 35 for LPS; results of four independent experiments; e, f, h data at 72 h: control mice, n = 4 for ddH2O, n = 27 for LPS; Cpt1aiΔAT2 mice, n = 4 for ddH2O, n = 33 for LPS; results of two independent experiments). b, c–h Data are represented as mean ± SEM, and two-sided p values are calculated by (b) unpaired Student’s t-tests or c–h one-way ANOVA with Bonferroni correction.
Tumor necrosis factor (TNF) α, CXCL1, and CXCL2 are essential to neutrophilic inflammation39, and several metabolites, such as lactate40, lysine41, and arginine42, regulate alveolar inflammation in murine ALI models. The improved resolution of alveolar cellular infiltrates in Cpt1aiΔAT2 mice may be associated with altered alveolar inflammatory and metabolomic response in ALI. Cytokine profiling with BALF samples was performed at 24 h after LPS instillation, and Cpt1aiΔAT2 mice showed significantly decreased BALF levels of TNFα, CXCL1, and CXCL2, but similar interleukin (IL) 6, IL1β, CCL2, and CCL5 levels as those in the control mice (Fig. 5g; Supplementary Fig. 13). At 72 h after LPS stimulation, BALF levels of CXCL2, TNFα, and IL6 were significantly decreased in Cpt1aiΔAT2 mice (Fig. 5h). In addition, various amino acids and metabolites of glycolysis and the TCA cycle were measured in BALF via metabolomic analyses by UHPLC-MS/MS. While Cpt1aiΔAT2 and control mice showed a similar alveolar metabolome at baseline, Cpt1aiΔAT2 mice had increased alveolar levels of various amino acids (tryptophan, tyrosine, proline, histidine, arginine, and lysine) and metabolites of glycolysis (glucose, lactate) and the TCA cycle (citrate/isocitrate) in the BALF 24 h after LPS-induced ALI (Supplementary Fig. 14; Supplementary Data File 5). The above data thus indicates that the accelerated resolution of alveolar cellular infiltrates observed in the Cpt1aiΔAT2 mice may be associated with altered mechanisms regulating alveolar inflammatory responses in LPS-induced ALI.
To further confirm if a loss of Cpt1a in AT2 cells resulted in better resolution in neutrophil dominant inflammation in lung injury, ALI was induced in control and Cpt1aiΔAT2 mice by activating toll-like receptor (TLR) 2 through the instillation of lipoteichoic acid and peptidoglycan, which induce neutrophil dominant ALI43. Neutrophils were the major immune cells in BALF at 24 h after instillation (Supplementary Fig. 15a, b). At 24 h, total BALF cell numbers, protein levels, and the percentage of AMs, neutrophils, eosinophils, or T lymphocytes were not significantly different between control and Cpt1aiΔAT2 mice (Supplementary Fig. 15a–d). Cpt1aiΔAT2 mice had significantly decreased levels of BALF CXCL2, but not TNFα, CXCL1, IL1β, IL6, CCL2 or CCL5, at 24 h after TLR2 stimulation (Supplementary Fig. 15e–k). Similar to the findings in the LPS-indued ALI model, at 48 h, Cpt1aiΔAT2 mice had significantly lower total BALF cell numbers (Supplementary Fig. 14l, m). These data confirm our findings in the murine LPS-induced ALI model and indicate that a loss of CPT1a in AT2 cells improves the resolution of alveolar inflammation and cellular infiltrates in ALI induced by either TLR2 or TLR4 stimulation.
Deleting Cpt1a in AT2 cells is associated with increased expression of chemokines in alveolar macrophages
AMs consume surfactant phospholipids5 and are the first-line innate immune cells recruiting neutrophils to alveoli in ALI44. In addition, the contents of several PC and PG species are increased in alveoli in Cpt1aiΔAT2 mice (Fig. 4k, l), and PG has been shown to suppress inflammatory responses of macrophages45. To determine whether AMs are involved in the mechanism by which the Cpt1aiΔAT2 mice had increased alveolar phospholipid levels at baseline and enhanced resolution following ALI, we isolated AMs from Cpt1aiΔAT2 and control mice, through flow cytometric sorting of BALF cells (Supplementary Fig. 16), for RNAseq. At baseline, Cpt1a expression and transcriptomic profiles in AMs from Cpt1aiΔAT2 and control mice were not remarkably different (Supplementary Fig. 17a, b) with GSEAs demonstrating that a loss of Cpt1a in AT2 cells did not alter AMs GM-CSF signaling (GO: 0097012), fatty acid oxidation (GO: 0019395) and lipid biosynthetic process (GO: 0008610) (Supplementary Fig. 17c–f). The results suggest that AMs do not appear to show any signs of altered lipid metabolism indicative of decreased surfactant phospholipid degradation at baseline (Fig. 4j, k).
Four hours after LPS instillation, RNAseq analysis revealed profoundly altered gene expression in annotations related to innate immunity, in particular, immune response, myeloid differentiation, apoptotic signaling, and cell migration (Supplementary Fig. 18a, b). Functional enrichment analyses, based on the comparison of transcriptomics in AMs from Cpt1aiΔAT2 mice to those from the control, demonstrated that Cpt1a deletion in AT2 cells alters immune responses in AMs, and several genes of pro-inflammatory cytokines, such as Tnf, Cxcl1, Cxcl2, Ccl3, and Ccl4, were upregulated in AMs from Cpt1aiΔAT2 mice in ALI (Supplementary Fig. 18c–h). The above data indicate that AMs are not the key cells leading to less alveolar cytokine levels in ALI in Cpt1aiΔAT2 mice but may upregulate signals for neutrophil recruitment into the alveoli.
Alveolar inflammation regulated by Cpt1a-deficient AT2 cells is not through alveolar PG species
Our findings demonstrate that a loss of CPT1a in AT2 cells leads to higher abundance of PG and PC phospholipids in the BALF of mice (Fig. 4j, k). Interestingly, administration of PG-rich surfactant has been shown to reduce neutrophilic inflammation in murine ALI model45,46. To assess if PG species may play a role in AT2 cell-associated inflammation in ALI, we first explored if PG species were altered in the BALF of individuals with ARDS or in the BALF of our murine ALI model. Total BALF PG levels were decreased in individuals with ARDS when compared to healthy controls (Supplementary Table). BALF PG levels were also decreased in mice at 24 h after LPS instillation (Supplementary Fig. 19), suggesting that PG abundance in the alveoli may be important in regulating alveolar inflammation in ALI. To assess if the increased abundance of PG species in the alveoli, as observed in the Cpt1aiΔAT2 mice at baseline (Fig. 4k) may be a mechanism by which these mice have lower inflammation under ALI (Fig. 6a), we carried out lipidomic analyses through UHPLC-MS/MS on BALF isolated from Cpt1aiΔAT2 and control mice at 24 h after LPS instillation. Total BALF levels of measured PG species in ALI were similarly decreased in controls and Cpt1aiΔAT2 mice (Fig. 6b; Supplementary Data File 6), and PG species PG(34:2)_18:2 and PG(36:4)_20:4 were significantly lower in Cpt1aiΔAT2 mice when compared to controls (Fig. 6c). Loss of alveolar PG abundance in ALI may result from increased phospholipase activity, which leads to increased levels of lysophospholipids and free fatty acids (FFAs)47,48. While the lysophosphatidylglycerol (LPG) to PG ratio and the FFAs in the BALF were similarly increased in the Cpt1aiΔAT2 and the control mice 24 h after LPS administration (Fig. 6d, e), BALF LPG(16:0) levels were significantly increased in Cpt1aiΔAT2 mice when compared to controls upon ALI (Fig. 6f). The data suggest that the significant loss of PG abundance observed in the Cpt1aiΔAT2 mice in ALI may be related to enhanced PG breakdown and may not be related to the protection of Cpt1aiΔAT2 mice to ALI.

a A cartoon illustrating the investigation for the link between altered baseline alveolar PG compositions in Cpt1aiΔAT2 mice and the immuno-modulation in ALI. b–f Measuring levels of free fatty acid (FFA), PG, and lysophosphatidylglycerol (LPG) species in bronchoalveolar lavage fluid (BALF) samples. Lipidomic analyses are performed to measure PG and LPG compositions in BALF, and the total measured PG species (b), the top 5 abundant PG species (c) and the ratios of LPG to PG (d) in BALF are compared (control mice, n = 4 for ddH2O, n = 24 for LPS; Cpt1aiΔAT2 mice, n = 4 for ddH2O, n = 20 for LPS; results of two independent experiments). e Free fatty acid (FFA) levels are measured in BALF samples at 24 h after instillation (control mice, n = 4 for ddH2O, n = 23 for LPS; Cpt1aiΔAT2 mice, n = 3 for ddH2O, n = 15 for LPS; results of two independent experiments). f The contents of the top 5 abundant LPG species in BALF samples at 24 h after LPS instillation are demonstrated and compared (control mice, n = 24, Cpt1aiΔAT2 mice, n = 20; results of two independent experiments). g An experimental schema showing intratracheal instillation of LPS with solvent, PG(32:0)_16:0 (~1.5 mM), or PG(34:1)_18:1 (~1.5 mM) to induce ALI with obliteration of alveolar phospholipidomic differences of specific PG species at baseline. BALF samples are collected 24 h after the induction of ALI. h–l The BALF (h) cell number and levels of i protein, j CXCL1, k CXCL2, and l TNFα are measured and compared (control mice, n = 16 for solvent, n = 10 for PG(32:0)_16:0, and n = 10 for PG(34:1)_18:1; Cpt1aiΔAT2 mice, n = 16 for solvent, n = 10 for PG(32:0)_16:0, and n = 10 for PG(34:1)_18:1; results of three independent experiments). b–f and h–l Data are represented as mean ± SEM, and two-sided p values are calculated by (e) one-way ANOVA with Bonferroni correction or b–d, f, h–l unpaired Student’s t-tests (c and f, only p values less than 0.05 are shown).
To further confirm this hypothesis, LPS was instilled into Cpt1aiΔAT2 and control mice together with high-dose PG(32:0)_16:0 or PG(34:1)_18:1 to evaluate if high-dose PG supplement at baseline results in similar alveolar inflammation between Cpt1aiΔAT2 and control mice (Fig. 6g). After instillation of LPS and the vehicle solvent, Cpt1aiΔAT2 mice showed similar protein levels but significantly decreased cell numbers and levels of CXCL1, CXCL2, and TNFα in BALF at 24 h (Fig. 6h–l). Instillation of PG(32:0)_16:0 obliterated the differences in alveolar CXCL1 and TNFα levels, and instillation of PG(34:1)_18:1 obliterated the differences in alveolar CXCL1 levels (Fig. 6j, l). However, alveolar cell numbers and CXCL2 levels remained significantly higher in control than those in Cpt1aiΔAT2 mice (Fig. 6h, k). The results thus indicate that decreased alveolar cellular infiltrates in Cpt1aiΔAT2 mice is not related to altered alveolar PG contents at baseline.
Cpt1a deletion in AT2 cells decreases neutrophilic inflammation in ALI
The above data shows that the mechanism of protection from ALI in the Cpt1aiΔAT2 mice may not be related to AM function or baseline alveolar PG contents. We therefore next assessed if signals from the AT2 cell have an immunomodulatory role aiding resolution from alveolar inflammation after LPS. Alveolar epithelial death promotes the severity of ALI49, and AT2 cells secret inflammatory mediators including neutrophilic chemokines50. We first examined whether a loss of CPT1a alters the viability of AT2 cells upon ALI. We generated Cpt1aiΔAT2 and control (SftpcCreERt2+/−ROSA26tdTomato+/−, referred as controltdTomato−AT2) reporter mice with tdTomato-expressing AT2 cells (Supplementary Fig. 20a). After 24 h of LPS instillation, flow cytometric analyses showed that the viability of tdTomato-labeled AT2 cells was not altered by Cpt1a deletion (Supplementary Fig. 20b, c). We next investigated if AT2 cells increase neutrophilic chemotactic signals, particularly CXCL1 and CXCL2, in LPS-induced ALI. Immunofluorescent staining using lung cryosections from controltdTomato−AT2 mice at 6 and 24 h after LPS instillation (Fig. 7a; Supplementary Fig. 21 and 22a) showed that CXCL1 and CXCL2 were localized to both tdTomato-labeled AT2 cells and CD45+ immune cells. Transcriptomic profiling by bulk-RNAseq in AT2 cells 24 h after LPS instillation revealed that pro-inflammatory signaling pathways, including STAT1, STAT3, RELA, NFKBIA, NFKB1, and IRF7, were activated in AT2 cells (Supplementary Fig. 22b), and functional enrichment analyses further identified annotations related to innate inflammatory responses, in particular leukocyte migration and cytokine production (Fig. 7b). Cxcl1 and Cxcl2 are among the top 10 cytokine and chemokine genes involved in these two annotations and were significantly upregulated in AT2 cells after LPS-induced ALI (Fig. 7c). The findings suggest that pro-inflammatory signaling in AT2 cells regulates neutrophilic chemotaxis to the airspaces in ALI. While Cpt1a−/− AT2 cells showed similar transcriptional changes to LPS as controls (Fig. 7d and Supplementary Fig. 22c), non-significant trends of decreased Cxcl1 and Cxcl2 expression were observed in Cpt1a−/− AT2 cells (Fig. 7e, f). To clarify the above findings that CPT1a deficiency may alter Cxcl1 and Cxcl2 expression in AT2 cells in LPS-induced ALI, qPCR analyses confirmed that the expression of Cxcl2, but not Cxcl1, was significantly downregulated in Cpt1a−/− AT2 cells when compared to controls (Fig. 7g, h).

a At 24 h after LPS instillation to controltdTomato−AT2 mice, cryosections of lungs are obtained for staining of CD45, CXCL1, and CXCL2 and confocal microscopy (representative data from n = 7 mice; arrow, chemokine in AT2 cells; arrowhead, chemokine in immune cells; scale bar = 20 μm). b, c RNA sequencing (RNAseq) in control AT2 cells after LPS instillation (24 h). A functional enrichment map (b) plotted using genes differently expressed (an adjusted p < 0.01 and a fold change > 2) at baseline and in ALI. c The expression levels of cytokine and chemokine genes included in cytokine production (GO: 0001816) and leukocyte migration (GO: 0050900) annotations are demonstrated in transcript per million (TPM) (n = 4 mice for each group). d Heatmaps comparing the expression of genes involved in cytokine production and leukocyte migration annotations in control and Cpt1a−/− AT2 cells (n = 4 mice for each group). The expression levels of e Cxcl1 and f Cxcl2 in AT2 cells are compared across the groups. g, h Quantitative polymerase chain reactions (qPCRs) measuring g Cxcl1 and h Cxcl2 expression in AT2 cells at 24 h after LPS instillation, using Tbp for normalization (data analyzed by 2−ΔΔCt method; control n = 10 mice, Cpt1a−/− n = 11 mice; results of four independent experiments). i After 6 h of tumor necrosis factor (TNF) α (2 nM) activation along with CXCL2 or not, Cxcl2 expression in bone marrow derived neutrophils is measured by qPCR (n = 4 replicates per group; representative data of three independent experiments). j, k qPCRs measuring Cxcl1 and Cxcl2 expression in alveolar neutrophils in ALI (control mice, n = 17, Cpt1aiΔAT2 mice, n = 14; results of three independent experiments). c, e–k Data are represented as mean ± SEM, and two-sided p values are calculated by (e, f) linear modeling and empirical Bayes moderation, adjusted through the Benjamini–Hochberg procedures, g, h, j, k unpaired Student’s t-tests or i one-way ANOVA with Bonferroni correction. b–f The details of RNAseq analyses are described in “Methods” section.
CXCL2 is essential for neutrophilic chemotaxis and self-recruitment for inflammatory maintenance39,51. To confirm whether CXCL2 regulates neutrophilic inflammation just like CXCL139, we isolated bone marrow-derived neutrophils to investigate if CXCL2 can enhance TNFα-induced neutrophilic activation. The results showed that TNFα-induced Cxcl2 expression in neutrophils could be further increased by CXCL2 (Fig. 7i). The data suggest that decreased CXCL2 production in Cpt1a-deficient AT2 cells may result in decreased neutrophilic production of CXCL1 and CXCL2 for self-recruitment51,52. At 24 h after LPS instillation, alveolar neutrophils were isolated through flow cytometric sorting (Supplementary Fig. 16) and were used for RNA extraction and qPCR analyses. Consistently, Cxcl1 and Cxcl2 expression were lower in alveolar neutrophils from Cpt1aiΔAT2 mice (Fig. 7j, k). The findings above thus disclose transcellular regulation between AT2 cells and alveolar neutrophils and show the improved resolution of alveolar cellular infiltrates in Cpt1aiΔAT2 mice in LPS-induced ALI is associated with downregulated signaling for neutrophilic infiltration.
Impaired mtLCFAO in AT2 cells by deleting Acadl protects against alveolar inflammation in ALI
Lastly, although CPT1a loss alters the metabolic state of AT2 cells (Figs. 3, 4), it is unclear whether such metabolic changes are CPT1a-specific or related to mtLCFAO impairment. Moreover, it is also unclear whether downregulated CXCL2 production in Cpt1a-deficient AT2 cells in ALI is CPT1a-dependent or can be suppressed by impairing mtLCFAO (Fig. 8a). For investigation, we generated AcadlloxP/loxP mice (Fig. 8b, c), which were crossed to SftpcCreERt2+/+ mice to generate AcadlloxP/loxPSftpcCreERt2+/− mice (referred to as AcadliΔAT2 mice) (Fig. 8d). At 6 weeks after tamoxifen injection, immunoblots confirmed depletion of LCAD in Acadl−/− AT2 cells (Fig. 8e). Palmitate-dependent OCRs for basal respiration and maximal respiration were significantly decreased in Acadl−/− AT2 cells (Fig. 8f, g), and the findings confirm that LCAD depletion results in impaired mtLCFAO in AT2 cells. In contrast to metabolic changes in Cpt1a−/− AT2 cells, loss of LCAD does not upregulate OCRs with glucose, pyruvate, and glutamine, or alter glycolysis activity in AT2 cells (Supplementary Fig. 23). In addition, Acadl deletion neither alters intracellular TG contents or phospholipid synthesis in AT2 cells nor leads to remarkable changes in alveolar PC and PG compositions as observed in Cpt1aiΔAT2 mice (Supplementary Fig. 24; Supplementary Data File 7, 8). Twenty-four hours after LPS instillation, qPCR analyses in AT2 cells revealed significantly decreased Acadl and Cxcl2 expression, but unaltered Cxcl1 and Tnf expression, in Acadl−/− AT2 cells (Fig. 8h–k). The cell numbers and protein levels in BALF were significantly decreased in Acadl iΔAT2 mice at 72 h after ALI induction (Fig. 8l, m).

a A scheme showing the hypothesis that mtLCFAO in AT2 cells regulates alveolar neutrophilic inflammation in ALI. b The genomic design of AcadlloxP/loxP mice. c Representative genotyping results confirm the successful insertion of loxP sequences in introns 1, 4, and 5 of the Acadl gene in AcadlloxP/loxP mice. d An experimental scheme to induce Acadl deletion in AT2 cells through tamoxifen injection and to investigate whether AT2 cell-specific deletion of Acadl decreases alveolar inflammation in LPS-induced ALI. e An immunoblot showing successful depletion of LCAD, encoded by Acadl, in AT2 cells at 6 weeks after tamoxifen injection (n = 3 mice per group; representative data of two independent experiments). f, g Extracellular flux analyses (f) with palmitate are performed in AT2 cells (Oligo oligomycin, Rot rotenone, AA antimycin A), and g oxygen consumption rates (OCRs) are calculated (n = 9 replicates for each group; representative data of two independent experiments). h–k Quantitative polymerase chain reactions were used to measure the mRNA expression of h Acadl, i Cxcl1, j Cxcl2, and k Tnf in AT2 cells isolated 24 h after LPS instillation. The results are normalized to Tbp expression and analyzed using the 2−ΔΔCt method (control mice, n = 15, AcadliΔAT2 mice, n = 8; results of four independent experiments). l, m At 72 h after LPS administration, cell number (l) and protein level (m) in BALF are measured (control mice, n = 17, AcadliΔAT2 mice, n = 9; results of two independent experiments). f–m Data are represented as mean ± SEM, and g–m two-sided p values are calculated by unpaired Student’s t-tests.
Collectively, although deleting Acadl in AT2 cells impairs mtLCFAO without inducing metabolic alternations as observed in Cpt1a-deficient AT2 cells, AcadliΔAT2 mice show decreased AT2 cell-specific CXCL2 production and less alveolar cellular infiltrates in ALI. The results thus confirm the immuno-regulatory function of CPT1a and LCAD in AT2 cells and suggest that mtLCFAO activity in AT2 cells regulates alveolar inflammation in response to ALI.
Discussion
Here we decipher the critical function of CPT1a in regulating immune responses and surfactant phospholipid synthesis in AT2 cells. Our results showing that inhibiting mtLCFAO through deletion of Cpt1a or Acadl in AT2 cells mitigates alveolar inflammation in ALI further confirm this novel immuno-regulatory role.
The reliance on mtLCFAO for cellular energy production is required in pancreatic islet cells20 but is not found in epithelial cells18 or AT2 cells. Here we show that a loss of CPT1a triggers metabolic adaptative responses to rewire cellular energy production in AT2 cells. While glutamine alone has been shown to support the TCA cycle under glucose and pyruvate deprivation33, our data demonstrate that CPT1a-deficient AT2 cells cannot solely utilize glutamine to maintain the TCA cycle, but require both pyruvate and glutamine to support ATP production. The shortage of carbon sources to sustain the TCA cycle in CPT1a-deficient AT2 cells, due to simultaneous impairment of both mtLCFAO and glycolysis, may explain that a single metabolite, such as pyruvate or glutamine alone, fails to effectively support mitochondrial bioenergetics. Although Cpt1a-deficient AT2 cells can obtain exogenous substrates, such as pyruvate and glutamine, from the circulation under in vivo conditions, it is unclear whether metabolic pathways in Cpt1a-deficient AT2 cells are reprogrammed for de novo generation of metabolites, such as pyruvate and glutamine, to support cellular energy production. Interestingly, the aforementioned bioenergetic adaptations activated in Cpt1a-deficient AT2 cells are not observed in AT2 cells after Acadl deletion. Our data showed that mtLCFAO is impaired but not totally blocked in Acadl−/− AT2 cells, possibly due to the compensation from very long-chain acyl-CoA dehydrogenase12. The differences in bioenergetic alterations in Cpt1a−/− and Acadl−/− AT2 cells may be related to the severity of mtLCFAO defects or due to different levels of mtLCFAO blockade. Although the detailed metabolic landscape in Cpt1a-deficient AT2 cells requires further exploration to clarify, our findings reveal the complexity of the metabolic regulation responsive to mtLCFAO defects in AT2 cells.
To the best of our knowledge, this study is the first to document the differential regulation of intracellular TG levels and phospholipid synthesis in Cpt1a−/− and Acadl−/− AT2 cells. These findings suggest that CPT1a may regulate the intracellular flux of long-chain acyl-CoA to TG and phospholipid synthesis through the carnitine shuttle53. Although impaired mtLCFAO leads to lipotoxicity in hepatocytes and cardiomyocytes13, loss of CPT1a does not cause excessive phospholipid deposition in AT2 cells. The findings suggest the alveolar surfactant pool may serve as a buffer for AT2 cells in regulating the homeostasis of intracellular phospholipid species. Glycolysis downregulation in Cpt1a−/− AT2 cells may also decrease lipogenesis and hence prevent intracellular phospholipid overload. Overall, our data uncover that CPT1a is a pivotal regulator of lipid catabolism and anabolism in AT2 cells.
Alveolar epithelial CXCL2 production has been shown to regulate neutrophilic inflammation in a murine ALI model induced by viral infection54. This study discloses CXCL2 production in AT2 cells is regulated by mtLCFAO activity and confirms that Cpt1a deletion in AT2 cells result in downregulated CXCL1 and CXCL2 production in alveolar neutrophils. However, the transcellular regulation in Cpt1aiΔAT2 mice is complex and may also involve AMs. Increased inflammatory responses in AMs may drive more neutrophil activation and infiltration and may account for a lack of anti-inflammatory phenotypes in Cpt1aiΔAT2 mice at 24 h after LPS instillation. In addition, the AM pool is depleted at 24 h in LPS-induced ALI and is subsequently replenished and reorganized through AM proliferation and monocyte recruitment after 72 h38. Neutrophilic inflammation is critical to monocytic emigration to the inflammatory sites55. The resolution of alveolar cellular infiltrates in Cpt1aiΔAT2 mice may not be solely due to decreased alveolar influx of neutrophils, as suggested by our data, but may be also related to perturbed AM pool restoration after ALI. A recent study further demonstrated that the AM population reshaped by ALI can be long-lasting56, and it is unknown whether Cpt1a deletion in AT2 cells results in altered remodeling of AM population after ALI. Collectively, our data suggest that Cpt1a deletion in AT2 cells regulates AM inflammation and may also affects AM reprogramming after ALI. However, the intricate mechanisms surrounding this transcellular regulation from Cpt1a−/− AT2 cells to AMs in ALI require further exploration.
Little is known regarding the functional and pathophysiological significance of the metabolic alterations in ARDS57. Our study provides evidence to suggest that mtLCFAO is impaired in the alveolar epithelium in DAD lungs, and in AT2 cells in fatal COVID-19. Hyperinflammatory states are associated with increased mortality in ARDS58, and inhibiting mtLCFAO in AT2 cells may halt alveolar inflammation and continuous tissue damage in ARDS. Notably, most of our data did not demonstrate decreased alveolar protein levels in Cpt1aiΔAT2 mice in ALI. While AT2 cells are responsible for alveolar fluid clearance59, the alveolar permeability changes in murine ALI models are also regulated by type 1 alveolar epithelial cells and endothelial cells60,61, and are not solely determined by alveolar influx of immune cells. Moreover, data from murine models of bacterial pneumonia demonstrate that impaired neutrophilic inflammation may compromise bacterial clearance, leading to decreased survival of murine hosts16. Considering the above and the translational gap from murine ALI models and human ARDS62, the clinical implications from our study should be cautiously interpreted. While downregulation of AT2 cell-specific mtLCFAO may halt persistent alveolar inflammation in ARDS, it may not be beneficial to clinical outcomes.
Intratracheal instillation of high-dose PG preparations fails to eliminate the phenotypic differences in alveolar inflammation between the control and Cpt1aiΔAT2 mice in ALI. In addition, impairing mtLCFAO in AT2 cells through Acadl deletion resolves alveolar cellular infiltrates in ALI, without remarkably perturbing basal alveolar PG contents. The findings together suggest that basal alveolar PG contents in Cpt1aiΔAT2 mice may not be the principal determinant suppressing alveolar inflammation. Furthermore, the basal alveolar phospholipidome in Cpt1aiΔAT2 mice is associated with increased alveolar LPG(16:0) in ALI, and lysophospholipids have been shown to enhance neutrophilic inflammation63. While lavage with PG-rich surfactant preparations is proposed as a potential treatment in ARDS46, our data suggest that exogenous PG supplementations may predispose to a pro-inflammatory phospholipidomic microenvironment through increasing alveolar LPG levels in ALI. Therefore, although it is unknown whether lavage with PG-rich surfactant in ARDS results in favorable clinical outcomes, the use of alveolar PG supplements as a therapy must proceed with caution.
This study has some limitations. Our results reveal that a loss of Cpt1a in AT2 cells results in a notable change in the metabolic milieu of the alveoli in response to LPS when compared to control mice (Supplementary Fig. 14; Supplementary Data File 5). In addition, data from previous studies suggest that metabolomic alterations in the alveoli of Cpt1aiΔAT2 mice may form an immuno-modulating environment to promote the resolution of alveolar inflammation40,41,42. However, the role of such metabolic changes in the Cpt1aiΔAT2 mice and how this in turn regulates the response to ALI requires further extensive mechanistic characterization.
In conclusion, through comprehensive explorations, we discover unique metabolic reprogramming in response to CPT1a depletion in AT2 cells and provide evidence supporting the regulatory axis between mtLCFAO and AT2 cell-specific immuno-modulating activity in the alveoli. Our data extrapolate the concept of immunometabolism in immune cells to epithelial cells and form the basis for future studies to clarify the role of AT2 cell-specific mitochondrial metabolism in regulating immune responses of the alveolar microenvironment.
Methods
Human studies
The protocols for all the human studies were approved by the Institutional Review Board (IRB) at each institution (202012115RIND, 201911058RINA, and 201712075RINA in National Taiwan University Hospital [NTUH] and National Taiwan University Hospital Yunlin Branch [NTUH-YL], and FJUH109018 in Fu Jen Catholic University Hospital [FJUH]), and conformed to the standards set by the Declaration of Helsinki. For IHC staining using surgical samples of human lungs, the IRB at NTUH (protocol 202012115RIND) approved waivers of informed consent. Consecutive adult patients (age ≥20 years old) with pathological diagnosis of DAD from January 2004 through November 2020 were identified using the database of the Pathology Index System in the Department of Pathology, NTUH. Patients were enrolled if they had adequate paraffin blocks of surgical lung samples for IHC staining. DAD is determined based on the microscopical features of interstitial edema, collapsed alveoli with hyaline membranes lining the alveolar walls, along with denudation and necrosis of type 1 alveolar epithelial cells and focal hyperplasia of AT2 cells. For all the enrolled patients with DAD, lung sections with hematoxylin and eosin staining were reviewed by pathologists to confirm the diagnosis of DAD. Patients who received lung segmentectomy or lobectomy for lung adenocarcinoma in NTUH from September through November in 2021 were also identified, and the non-tumor parts within the samples served as the staining control. Demographic data, including age and biological sex, of each enrolled participant were obtained from the medical records associated with the surgical lung samples.
After written informed consent was obtained (protocols 201911058RINA and 201712075RINA at NTUH and NTUH-YL, and protocol FJUH109018 at FJUH), BALF samples were collected from ARDS patients and the participants in the control cohort for evaluating cytokine profiles and PG contents in BALF, and demographic data, including age and biological sex, were recorded. Consecutive adult patients with clinical diagnoses of ARDS were enrolled from May in 2020 through December 2022 in medical intensive care units at NTUH and FJUH. ARDS is defined based on Berlin criteria64,65, including (1) hypoxemic respiratory failure developing within 7 days which cannot be explained by cardiogenic lung edema, fluid overload, or deterioration of pre-existing lung diseases, (2) bilateral pulmonary infiltrates revealed by chest images, and (3) a ratio of arterial partial pressure of oxygen to the fraction of inspired oxygen (PaO2/FiO2) less than 300 mmHg, under positive end-expiratory pressure (PEEP) above 5 cmH2O. The exclusion criteria included acute cardiogenic lung edema as the primary diagnosis, massive gastrointestinal bleeding (more than 4 units of pack red blood cell transfusion required within 24 h), uncontrolled hematological malignancies, advanced malignancy with poor pre-admission performance status and inevitable short-term mortality, human immunodeficiency virus infection without immune reconstitution, and refused consent. A recent study revealed that the phospholipid contents in BALF from chronic obstructive pulmonary disease (COPD) patients with preserved lung function are similar to those from healthy people66. Therefore, from August 2019 through February 2022, adult COPD patients with Global Initiative for Chronic Obstructive Lung Disease (GOLD) grade 0–1 were recruited at the outpatient clinics of NTUH-YL and formed the control cohort.
Mice
The animal experiments and procedures, including euthanasia, were approved by the Institutional Animal Care and Use Committee (IACUC) at the National Taiwan University College of Medicine and College of Public Health (protocols 20180128, 20201154, 20210138, 20210401, and 20230151). All the mouse lines in this study were maintained in the National Laboratory Animal Center (NLAC), NARLabs (Taipei, Taiwan), and in Laboratory Animal Center, National Taiwan University College of Medicine (Taipei, Taiwan). Both animal centers are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. All the experimental mice were bred in individually ventilated cages with autoclaved bedding (12-h light:12-h dark cycle with light ON at 8 AM, ambient temperature as 20–24 °C and humidity as 50 ± 20%), and ad libitum fed with sterilized water and irradiated PicoLab® Rodent Diet 20 (catalog 5053, LabDiet, St Louis, MO, USA) in Laboratory Animal Center, National Taiwan University College of Medicine. For murine sample collections, euthanasia was performed through intraperitoneal injection of 0.5 mg xylazine (16~25 mg/kg for mice weighted 20~30 g) and 1.25 mg Zoletil® (40~60 mg/kg for mice weighted 20~30 g; Virbac, Westlake, TX, USA), followed by exsanguination through transection of inferior vena cava and descending aorta, and the euthanasia protocol complies with Guideline for the Care and Use of Laboratory Animals by Council of Agriculture, Executive Yuan, Taiwan. Both male and female mice were used for animal experiments and the reporting of animal experiments follows the ARRIVE guidelines (https://arriveguidelines.org/arrive-guidelines).
Cpt1aloxP/loxP (Cpt1atm1.1Pec) mice were generated and provided by Peter Carmeliet, Vlaams Instituut voor Biotechnologie19. SftpcCreERT2+/+ (B6.129S-Sftpctm1(cre/ERT2)Blh/J) mice and ROSA26tdTomato+/+ (B6.Cg-Gt(ROSA)26Sortm14(CAG−tdTomato)/Hze/J) mice were provided by Augustine MK Choi, Weill Cornell Medicine. To generate mice with tamoxifen-inducible Cpt1a deletion specifically in AT2 cells, Cpt1aloxP/loxP mice were crossed to SftpcCreERt2+/+ mice to generate Cpt1aloxP/loxPSftpcCreERt2+/− (Cpt1aiΔAT2) mice. To induce recombination and deletion of Cpt1a in AT2 cells, five consecutive intraperitoneal injections of tamoxifen (100 mg/kg/dose; catalog T5648, Sigma–Aldrich, St. Louis, MO, USA) in sunflower seed oil (catalog S5007, Sigma–Aldrich) were given around five weeks of age. To access the potential off-target effect by CreERt2, SftpcCreERT2+/− mice were used as the controls8, and were generated through crossing SftpcCreERt2+/+ mice to C57BL/6JNarl from NLAC, NARLabs (Taipei, Taiwan). AcadlloxP/loxP (B6-Acadlem2CJY/J) mice were generated by the CRISPR/Cas9 technology. The online resources, the CRISPick67 and the Cas-OFFinder68 were applied for selecting the sgRNA sequences. The loxP sequences were inserted in the upstream of exon 2 (loxP1), the upstream of exon 5 (loxP2), and the upstream of exon 6 (loxP3) (Fig. 8b), and the sgRNA oligonucleotides targeting at sequences with protospacer adjacent motif (PAM) for inserting the three loxP DNA fragments were 5′-CCATTACAGAGATCTCACCG-3′, 5′-CGTATGAATGTGTACCACGG-3′, and 5′- ATAATCCATCAGCTGCCCAG-3′, respectively. Electroporation of SgRNA and Cas9 protein, both purchased from Synthego Corporation (Redwood City, CA, USA), was performed on the fertilized eggs. Long-range polymerase chain reactions (PCRs) and Sanger sequencing were performed to confirm the correct nucleotide sequences between the loxP fragments. Genotyping is performed through PCRs with gel electrophoresis (Fig. 8c). The nucleotide sequences of the genotyping primers are listed below:
loxP1 forward: 5′-GCTGATGAGGGTCGAAGGTT-3′,
loxP1 reverse:5′-GAGACTCAGCCTTGGAGCAG-3′,
loxP2 forward:5′-AGATGACATCAACAGTGGTGCA-3′,
loxP2 reverse:5′-CCTTTACTCAGGCTGGTTCAAC-3′,
loxP3 forward:5′-CATAGGGGCGGAAACGTGAT-3′, and
loxP3 reverse:5′-ATAAACTATGCTGCCATCCGCT-3′.
The PCRs were performed using GoTeq® Green Master Mixes (catalog M7122, Promega, Madison, WI, USA), and the cycling parameters include the initial denaturation at 95 °C for 5 min, followed by 40 cycles of 95 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s, and the final extension at 72 °C for 7 min. The gel electrophoresis was performed using a 2% agarose gel, under 90 volts for 90 min. AcadlloxP/loxP mice were crossed to SftpcCreERt2+/+ mice to generate AcadloxP/loxPSftpcCreERt2+/− (AcadliΔAT2) mice and deletion of Acadl in AT2 cells was successfully performed through intraperitoneal injections of tamoxifen. All techniques for generating the AcadlloxP/loxP mice were provided by the Transgenic Mouse Model Core Facility of the National Core Facility for Biopharmaceuticals, National Science and Technology Council, Taiwan, and the Animal Resources Laboratory of National Taiwan University Centers of Genomic and Precision Medicine. SftpcCreERT2+/− mice served as the controls for the experiments. For expressing tamoxifen-inducible tdTomato labeling in Cpt1a−/− AT2 cells, ROSA26tdTomato+/+ mice were first crossed to Cpt1aloxP/loxP mice to generate Cpt1aloxP/loxPROSA26tdTomato+/+ mice, which were subsequently crossed to Cpt1aloxP/loxPSftpcCreERt2+/+ mice to generate Cpt1aloxP/loxPSftpcCreERt2+/−ROSA26tdTomato+/− (Cpt1aiΔAT2/tdTomato−AT2) mice8. SftpcCreERT2+/+ mice were crossed to ROSA26tdTomato+/+ mice to generate SftpcCreERt2+/−ROSA26tdTomato+/− (ControltdTomato−AT2) mice, which were used as the controls.
Murine models of ALI
Mice matched for age (10~12 weeks) and sex, both male and female, were used for models of ALI. After induction of anesthesia using 3.5% isoflurane, mice were instilled with 2 mg/kg Escherichia coli O55:B5 LPS (catalog L2637, Sigma–Aldrich) in 50 μL double distilled water (ddH2O)23, or 2 mg/kg TLR2 agonists (peptidoglycan and lipoteichoic acid; catalog 77140 and L2515 respectively, Sigma–Aldrich) in 50 μL ddH2O43 through the intratracheal route8. The instillation of 50 μL ddH2O was administered to mice in the control groups. To investigate the effects of PG species to the severity of LPS-induced ALI, 55.5 μg PG(32:0)_16:0 (catalog 840455 P, Avanti® Polar Lipids, Birmingham, AL, USA) in 3 μL methanol, 57.9 μg PG(34:1)_18:1 (catalog 840457 P, Avanti® Polar Lipids) in 3 μL methanol or 3 μL methanol was given together with LPS in 50 μL ddH2O, through intratracheal instillation. For sample harvest, mice were euthanized at different time points as outlined, after the instillation of the stimulants.
Cell lines
Both MLE12 (CRL-2100) and HEK293T (CRL-3216) cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). MLE12 cell line was maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (catalog 11875093, Gibco) added with 10% fetal bovine serum (FBS) (catalog 1823627, Biological Industries, Cromwell, CT, USA) and 1% penicillin-streptomycin (catalog 15070063, Gibco). Lentiviral transduction using the lentiCRISPRv2 vector (GenScript, Piscataway, NJ, USA) was performed to generate Cpt1a-deficient MLE12 cells, through CRISPR/Cas9 techniques. HEK293T cells were cultured using Dulbecco’s Modified Eagle’s Medium (DMEM) (catalog 11965092, Gibco) containing 10% FBS and 1% penicillin-streptomycin, and were used for lentiviral packaging, through chemical transfection of the lentiviral vector and the psPAX2 (catalog 12260, Addgene) and VSV-G (a gift from David C Chan, Caltech) packaging plasmids. The nucleotide sequences for gRNA targeting at murine Cpt1a was 5′-CACTCACGATGTTCTTCGTC-3′. After lentiviral transduction, the cells are selected using puromycin (2 μg/mL; catalog A11138-03, Gibco) for five days, and monoclonal cell populations were isolated through the limiting dilution method. Sanger sequencing was performed for genotypic screening of selected cell colonies, and immunoblots were performed to confirm the depletion of CPT1a in MLE12 cells. The control MLE12 cell was generated through the transduction of the lentiCRISPRv2 vector. MLE12 cells with CPT1a-Myc-DDK fusion protein overexpression (Cpt1aOE MLE12 cells) were generated through lentiviral transduction using the pLenti-C-Myc-DDK-P2A-Puro vector (catalog MR210568L3, OriGene, Rockville, MD, USA), and the control was generated through transduction of empty vector (catalog PS100092, OriGene). Both the control and Cpt1aOE MLE12 cells were selected using puromycin for two weeks after transduction.
IHC and immunofluorescent staining of human lung surgical samples
Formalin-fixed paraffin-embedded tissue sections of surgical lung samples were used for IHC or immunofluorescent staining. Antigen retrieval was performed at pH9.0 using Epitope Retrieval 2 solution for 20 min at 100 °C. For IHC staining, primary antibodies against human CPT1a (1:10; catalog 12252, Cell Signaling Technology, Danvers, MA, USA), LCAD (1:50; catalog ab152160, Abcam, Cambridge, UK) and CK (PCK26, pre-diluted, Roche VENTANA ULTRA kit, Roche Diagnostics, Basel, Switzerland) were used for IHC staining. The staining of CK was performed using a Roche VENTENA BenchMark autostainer, according to the manufacturer’s instructions. CPT1a and LCAD were stained using a Leica Biosystems BONDMAX autostainer (Leica Microsystems, Wetzlar, Germany). After antigen retrieval, the sections were then subjected to sequential incubation with hydrogen peroxide for blocking endogenous peroxidase, primary antibody for 30 min, post-primary IgG linker reagent localizing mouse antibodies for 8 min, and the polymeric horseradish peroxidase-linker IgG antibody localizing rabbit antibodies for 8 min. The staining was developed with substrate chromogen diaminobenzidine for 10 min, followed by the counterstaining using modified Mayer’s hematoxylin for 5 min. The staining results were interpreted randomly and blindly by a thoracic pathologist in the Department of Pathology, National Taiwan University Hospital, and National Taiwan University Cancer Center Hospital. The intensities of CPT1a and LCAD staining were graded from 0 to 3+, and the H scores were calculated based on the equation below:
For immunofluorescent staining, blocking and permeation of the samples were performed after antigen retrieval, using tris-buffered saline (TBS) containing 5% normal donkey serum (catalog 017-000-121, Jackson ImmunoResearch, West Grove, PA, USA) and 0.3% Triton X-100 (Sigma–Aldrich). The staining was performed using primary antibodies against CPT1a (1:10; catalog 12252, Cell Signaling Technology) and HTII-280 (1:50; catalog TB-27AHT2-280, Terrace Biotech, San Francisco, CA, USA), and secondary antibodies against rabbit IgG (conjugated to Alexa Fluor 647, 1:500; catalog A-31573, Thermo Fisher Scientific), and mouse IgM (conjugated to Alexa Fluor 488, 1:500; catalog A-21042, Thermo Fisher Scientific). Both primary and secondary antibodies were diluted in blocking buffer. The samples were covered with diluted primary antibodies, and were incubated in a humidified chamber overnight at 4 °C. After 24 h, the cryosections were washed with TBS and were then incubated with diluted secondary antibodies for one hour under room temperature. After washing with TBS, Hoechst 33342 (1:1000 dilution in TBS; catalog 62249, Thermo Fisher Scientific) was used for nuclear staining. The slides were mounted using Prolong Gold antifade solution (catalog P36930, Invitrogen), and the images were acquired in the Microscopy Core Facility, Department of Medical Research, National Taiwan University Hospital, using a Zeiss Axio Observer 7 inverted microscope (Oberkochen, Germany). A 40×/0.95 objective was used, and the image was processed using ZEN 3.4 (blue edition) software (version 3.4.91.00000).
Isolation of murine AT2 cells
After euthanasia, the mouse lungs were perfused using phosphate-buffered saline (PBS) through the right ventricle. The lungs were then inflated using 1.5 mL dispase (catalog 354235, BD Biosciences, San Jose, CA, USA) and 0.5 mL 1% low-melting point agarose (catalog 16520-050, Invitrogen, Waltham, MA, USA). After ice-cooling for 2 min, the lungs were excised and transferred to a 50 mL polypropylene tube containing 2 mL dispase. After 45-min digestion at room temperature, the lungs were homogenized manually using the plunger of a 1 mL syringe in a 10-cm petri dish containing 9 mL DMEM and 200 U/mL DNase (catalog D-4527, Sigma–Aldrich). After sequential filtration through 100 μm, 40 μm (catalog 352360 and 352340 respectively, BD Biosciences), and 20 μm (catalog SCNY00020, Merk Millipore, Burlington, MA, USA) strainers, the whole lung cell suspension was obtained after centrifugation at 500 × g for 5 min at 4 °C. The cells were resuspended using MACS buffer, constituted by PBS added with 0.5% bovine serum albumin (catalog 130-091-376, Miltenyi Biotec, Bergisch Gladbach, Germany) and 2 mM EDTA (catalog AM9260G, Invitrogen), and were then negatively selected by anti-CD45 microbeads (catalog 130-052-301, Miltenyi Biotec), followed by positive selection by streptavidin microbeads (catalog 130-048-102, Miltenyi Biotec) and biotin-conjugated anti-EpCAM antibody (catalog 13-5791-82, eBioscience, San Diego, CA, USA), through MACS separation columns (catalog 130-042-401, Miltenyi Biotec). The CD45−EpCAM+ cells are mainly composed of AT2 cells in a purity of ~94%8, and are used for experiments.
Immunoblots
Protein extraction was performed using RIPA buffer (catalog 9806) with protease inhibitor cocktail (catalog 5872, Cell Signaling Technology). The protein concentration was determined through BCA protein assay (catalog 23225, Thermo Fisher Scientific, Waltham, MA, USA), and the proteins were resolved using 8~12% tris-glycine gels. The primary antibodies against CPT1a (1:1000; catalog ab128568, Abcam), LCAD (1:1000; catalog ab129711, Abcam), SFTPC (1:1000; catalog ABC99, Merk Millipore), TOM20 (1:1000; catalog sc11415, Santa Cruz, Dallas, TX, USA), DDK/FLAG (1:1000; catalog TA50011-100, OriGene), and β-actin (1:5000; catalog MAB1501, Merk Millipore), and secondary antibodies, including anti-rabbit IgG (1:5000; catalog 406401, BioLegend, San Diego, CA, USA) and anti-mouse IgG (1:5000; catalog 405306, BioLegend), were used for immunoblotting. Densitometric analyses for semi-quantification of the bands were performed using FIJI running ImageJ software (version 1.51w; https://fiji.sc/).
Extracellular flux analyses
Extracellular flux analyses, including Mito Stress and glycolysis stress tests, were performed using a Seahorse XF24 analyzer (Agilent Technologies, Santa Clara, CA, USA). For assays in MLE12 cells, 4 × 104 cells were plated per well in a Seahorse 24-well microplate (catalog 102342-100, Agilent Technologies), and were maintained in complete RPMI1640 medium. After incubation at 37 °C for 24 h, the medium was replaced with the assay medium required for the assays. To prepare the Seahorse 24-well microplate for extracellular flux analyses in primary AT2 cells, coating solution containing 23 μg/mL Cell-TakTM (catalog 354240, Corning, Corning, NY, USA) was prepared using neutral buffer solution containing 0.1 M NaHCO3 (pH 8.0). In a Seahorse 24-well microplate, 60 μL coating solution was added to each well for coating at room temperature for 20 min. The coating solution was then removed, and the well was washed twice using sterile ddH2O. After isolation, AT2 cells were resuspended in the assay medium, and 2 × 105 cells were added to each Cell-TakTM-coated well in a Seahorse 24-well microplate. The assay medium for Mito Stress tests was constituted by XF Base medium (catalog 103334-100, Agilent Technologies) supplemented with 10 mM glucose, 1 mM pyruvate (catalog 11360070, Gibco) and 2 mM glutamine (catalog A2916801, Gibco) (assay medium for tests with glucose, pyruvate and glutamine), 10 mM pyruvate and 2 mM glutamine (assay medium for tests with pyruvate and glutamine), 10 mM glutamine (assay medium for tests with glutamine alone), or 10 mM pyruvate (assay medium for tests with pyruvate alone). The assay medium for glycolysis stress tests was made using XF Base medium supplemented with 2 mM glutamine. In MLE12 cells, Mito Stress assays were performed using 2 μM oligomycin, 0.5 μM FCCP, and 0.5 μM rotenone and antimycin A (catalog 103015-100, Agilent Technologies), and glycolysis stress tests were performed using 10 mM glucose, 2 μM oligomycin, and 50 mM 2-deoxyglucose (2DG) (catalog 103020-100, Agilent Technologies). In AT2 cells, Mito Stress assays were performed using 2 μM oligomycin, 1 μM FCCP, and 0.5 μM rotenone and antimycin A, and glycolysis stress tests in AT2 cells were performed using 10 mM glucose, 1 μM oligomycin, and 50 mM 2DG.
To evaluate mtLCFAO activity in MLE12 cells, 2 × 105 cells were plated to each well in a Seahorse 24-well microplate and were incubated in substrate-limited medium, constituted by DMEM (Gibco) supplemented with 0.5 mM glucose, 1 mM GlutaMAX (catalog 35050061, Gibco), 0.5 mM carnitine (catalog C0283, Sigma–Aldrich) and 1% FBS. After incubation at 37 °C for 24 h, the medium was changed to mtLCFAO assay medium, containing 111 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 2 mM MgSO4, 1.2 mM NaH2PO4, 0.5 mM carnitine and 0.5 mM HEPES (catalog 15630080, Gibco) in ddH2O (final pH adjusted to 7.4). For evaluating mtLCFAO activity, isolated AT2 cells were first incubated in substrate-limited medium at 37 °C for one hour. The AT2 cells were then resuspended in mtLCFAO assay medium, and 2 × 105 AT2 cells were added to each Cell-TakTM-coated well in a Seahorse 24-well microplate. Extracellular flux analyses with 200 μM bovine serum albumin-conjugated palmitate (catalog 102720-100, Agilent Technologies) in both MLE12 and AT2 cells were performed using 2 μM oligomycin, 1.5 μM FCCP, and 0.5 μM rotenone and antimycin A. Pretreatment with 40 μM etomoxir (catalog E1905, Sigma–Aldrich) was performed to inhibit CPT1a activity in AT2 cells 15 min before the addition of palmitate.
The results of extracellular flux analyses in MLE12 cells were normalized using nucleic acid contents, measured by CyQUANTTM Cell Proliferation Assays (catalog C7026, Invitrogen), and normalized OCR and ECAR per plate were calculated using the equation below:
where NA is the nucleic acid content in each well, n is the total well number in the plate and \(\overline{{NA}}\) is the average nucleic acid content of all the wells in the plate. All the data were analyzed using the Wave software (version 2.6.3.5, Agilent Technologies) (https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897).
Quantitative polymerase chain reactions
Quantitative polymerase chain reactions (qPCR) were performed to evaluate the mRNA expression levels of Cxcl1, Cxcl2, Tnf, Cpt1a and Acadl. TRIzol Reagent (catalog 15596-018, Invitrogen) and TriRNA Pure Kit (catalog TRP100, Geneaid, New Taipei, Taiwan) were used for RNA extraction. Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (catalog 4368814, Applied Biosystems, Waltham, MA, USA). Power SYBR Green PCR Master Mix (catalog 4368577, Applied Biosystems) was used for qPCRs in an Applied Biosystems QuantStudio 7 Real‐Time PCR System (Thermo Fisher Scientific). The nucleotide sequences of the primers are listed as below:
Cxcl1-forward 5′-CCGAAGTCATAGCCACACTCAA-3′,
Cxcl1-reverse 5′-GCAGTCTGTCTTCTTTCTCCGTTA-3′,
Cxcl2-forward 5′-CATCCAGAGCTTGAGTGTGACG-3′,
Cxcl2-reverse 5′-GGCTTCAGGGTCAAGGCAAACT-3′,
Tnf-forward 5′-GGTGCCTATGTCTCAGCCTCTT-3′,
Tnf-reverse 5′-GCCATAGAACTGATGAGAGGGAG-3′,
Cpt1a-forward 5′-GGCATAAACGCAGAGCATTCCTG-3′,
Cpt1a-reverse 5′-CAGTGTCCATCCTCTGAGTAGC-3′,
Acadl-forward 5′-GGCGATTTCTGCCTGTGAGTTC-3′,
Acadl-reverse 5′-GCTGTCCACAAAAGCTCTGGTG-3′,
Tbp-forward 5′-CTACCGTGAATCTTGGCTGTAAAC-3′, and
Tbp-reverse 5′-AATCAACGCAGTTGTCCGTGGC-3′.
The RNA expression of the target gene was calculated relative to that of Tbp, and the 2−ΔΔCt method was applied to analyze the results.
Measurements of intracellular ATP
Luminescent ATP detection assay (catalog ab113849, Abcam) was used to measure the intracellular levels of ATP. For each mouse, 1 × 106 AT2 cells from the lungs were prepared in 500 μL PBS after isolation and were used for ATP quantification. The measurements were performed according to the manufacturer’s instructions, using a SpectraMax iD5 plate reader (Molecular Devices, San Jose, CA, USA).
Phospholipid synthesis assay by CLICK labeling
Phospholipid synthesis assay kit (catalog ab241025, Abcam) was used to quantify newly synthesized choline-related phospholipids in MLE12 cells, through choline analog and fluorescent labeling by CLICK chemistry. For the assay, 1 × 105 cells were added to a well in a 24-well plate and were maintained in complete RPMI1640 medium. After culture at 37 °C overnight, the medium was replaced. Phospholipid label (1:1000 dilution) was added to the medium for the positive control but was not added to that for the negative control. After incubation at 37 °C for 24 h, MLE12 cells were trypsinized, fixed, and permeated, using the fixation solution and permeabilization buffer in the kit. The click reaction mix was prepared based on the manufacturer’s protocol and was used to label newly synthesized choline-related phospholipids in MLE12 cells at room temperature for 30 min.
Phospholipid synthesis assay in AT2 cells was performed based on previously published method36. After isolation, AT2 cells were resuspended in XF Base medium (Agilent Technologies) supplemented with 10 mM glucose, 1 mM pyruvate (Gibco), and 2 mM glutamine (Gibco), and 1.2 × 105 cells were used per test. Propargylcholine (200 μM; catalog CLK-066, Jena Bioscience, Jena, Thuringia, Germany) was added to the medium for the positive control but was not added to that for the negative control. After incubation at 37 °C for 3 h, AT2 cells were fixed using 4% paraformaldehyde (PFA) (catalog 15710, Electron Microscopy Sciences, Hatfield, PA, USA), permeated with 0.5% Triton X-100 (catalog X-100, Sigma–Aldrich), and labeled with 100 μL click reaction mix. The click reaction mix was prepared using BTTAA-based CuAAC Cell Reaction Buffer (catalog CLK-073, Jena Bioscience) and AZDye647 Picolyl Azide (catalog 1300-1, Click Chemistry Tools, Scottsdale, AZ, USA), and was constituted by 67.8 μL Na-Phosphate reaction buffer (pH 7.0), 0.2 μL AZDye647 Picolyl Azide stock solution (1 mM in PBS), 22 μL CuSO4:BTTAA premix, and 10 μL Na-Ascorbate stock solution (1 M in ddH2O). The CuSO4:BTTAA premix was prepared using CuSO4 stock solution (100 mM in ddH2O) and BTTAA stock solution (50 mM in ddH2O) in 1:10 ratio.
After labeling, the MLE12 and AT2 cells were analyzed respectively by the channels for PE and Alexa Fluor 647, using a BD LSRII flow cytometer. The phospholipid synthesis activity was quantified using the differences of the geometric mean fluorescence intensity (MFI) between the positive and the negative controls. The data were analyzed using FlowJo analytical software (version 10) (BD Biosciences).
Lung function measurements
The alveolar mechanic properties were measured using a flexiVentTM (SCIREQ, Montreal, Canada)69. After anesthesia induction using intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine, the murine trachea was cannulated at the site close to the cricoid cartilage, using a 18-gauge needle provided by SCIREQ. The dynamic tube calibration of the flexiVentTM was first completed using the flexiWareTM software, and the mouse was then connected to the Y-tube of the flexiVentTM for lung function measurements. Broadband forced oscillations were selected for measuring alveolar tissue damping and elastance, and the data were retrieved from flexiWareTM software after the measurements were finished.
Bronchoalveolar lavage of mouse lungs
After the mice were euthanized, the trachea was intubated with a 20-gauge catheter (Terumo, Tokyo, Japan). Bronchoalveolar lavage of murine lungs was then performed with 0.7 mL ice-cold PBS for three times (a total of 2.1 mL). The BALF samples were then centrifuged at 500 × g for 5 min at 4 °C. The supernatant was then collected, aliquoted, and stored in a −80 °C freezer. The protein level in BALF was measured using BCA protein assay kit (Thermo Fisher Scientific). The cell pellets were resuspended, and the cell numbers were counted using a hemocytometer.
Bronchoalveolar lavage of human lungs
For patients with ARDS, bronchoalveolar lavage was performed to the lobe most severely affected, based on the results from chest image studies. For the control participants, bronchoalveolar lavage was performed principally to the right middle lobe. Sedative and anesthetic agents were administered based on the clinical standards before bronchoscopy. After bronchial wedging, lavage with 50 mL warm normal saline was performed for four times (a total 200 mL)70. BALF was manually recovered by syringes and was sent to clinical laboratories for microbiology, cytology, and flow cytometric analyses, based on clinical demands. The remaining BALF sample was sent to the research laboratory immediately on ice. After centrifugation at 300 × g for 10 min under 4 °C, 1 mL supernatant was aliquoted and stored in a −80 °C freezer before analyses.
Flow cytometric analyses and sorting of immune cells in BALF
For flow cytometric analysis of immune cells in BALF71, phenol red-free RPMI1640 medium (catalog 11835030, Gibco) instead of PBS was used for lavage. After centrifugation, the cells were resuspended using MACS buffer containing anti-CD16/CD32 antibodies (1:100; catalog 553142, BD Biosciences), and were incubated on ice for 10 min. Surface markers were stained using anti-CD45.2-FITC (1:100; catalog 553772, BD Biosciences), anti-CD11c-PE-Cy7 (1:80; catalog 25-0114-81, eBioscience), anti-Ly6G-Alexa Fluor 700 (1:200; catalog 127621, BioLegend), anti-CD3-PE-Cy5 (1:200; catalog 555276, BD Biosciences), and anti-SiglecF-PE (1:100; catalog 552126, BD Biosciences). After staining on ice for 30 min, the cells were centrifuged and washed. DAPI (0.1 μg/mL; catalog D1306, Invitrogen) was then added as the viability staining, and the cells were analyzed using a BD LSRII flow cytometer. The data were analyzed using FlowJo analytical software. To isolate alveolar macrophages (AMs) for transcriptomic analyses, BALF samples were obtained at baseline and at 4 h after LPS instillation for ALI induction. Alveolar macrophages were then labeled with anti-CD45.2-FITC, anti-CD11c-PE-Cy7, and anti-SiglecF-PE, and were sorted using a BD FACSAriaTM III flow cytometer (Supplementary Fig. 16). For isolating alveolar neutrophils for qPCRs at 24 h after LPS instillation, anti-CD45.2-FITC and anti-Ly6G-Alexa Fluor 700 were used to label neutrophils (Supplementary Fig. 16), which were then sorted into TRIzol Reagent (Invitrogen) for RNA extraction.
Measuring cytokine levels in BALF samples
After centrifugation at 10,000 × g for 10 min at 4 °C, 50 μL supernatant of murine and human BALF samples were used for measuring cytokine and chemokine levels with MILLIPLEX® mouse cytokine/chemokine magnetic bead panel (catalog MCYTOMAG-70K, Merk Millipore) and human cytokine/chemokine/growth factor panel A (catalog HCYTA-60K, Merk Millipore), respectively. The standards for the calibration curves were prepared using PBS, and the assays were performed according to the manufacturer’s instructions. The measured result was designated as 0 pg/mL if it was less than the detection limit. The levels of the cytokine/chemokine in the BALF sample were determined by dividing the measured results by two.
Viability of AT2 cells
To determine the viability of AT2 cells in ALI, Cpt1aiΔAT2/tdTomato−AT2 and controltdTomato−AT2 mice were instilled with LPS. At 24 h after instillation, the single-cell suspension of murine lungs was prepared and was stained using 0.1 μg/mL DAPI (Invitrogen). Viability of tdTomato-positive AT2 cells was measured using a BD FACSAriaTM III flow cytometer, and dead cells were determined based on the positivity of DAPI staining. The results were analyzed using FlowJo analytical software (version 10).
Measuring mitochondrial ROS in AT2 cells
After hemolysis using ACK lysing buffer (catalog A10492-01, Gibco, Waltham, MA, USA), single-cell suspension of murine lungs was incubated in MACS buffer containing anti-CD16/CD32 antibodies (1:100, BD Bioscience) on ice for 10 min. The cells were then stained using anti-CD45.2-FITC (1:100, BD Bioscience) and anti-EpCAM-PE-Cy7 (1:150; catalog 25-5791-80, eBioscience) on ice for 30 min. After centrifugation and wash, the cells were incubated in staining buffer constituted by phenol red-free RPMI1640 medium (Gibco) supplemented with 5 μM MitoSOXTM Mitochondrial Superoxide Indicator (catalog M36008, Thermo Fisher Scientific) at 37 °C for 30 min. After centrifugation and wash, the cells were stained with 0.1 μg/mL DAPI (Invitrogen) and were analyzed using a BD LSRII flow cytometer. The data were analyzed using FlowJo analytical software (version 10), and mitochondrial ROS of AT2 cells were determined by the geometric MFI of mitoSOX staining in the CD45−EpCAM+ cell population.
Transcriptomic analyses
RNA extraction of AT2 cells was performed using TRIzol Reagent (Invitrogen), and RNA from AT2 cells in a mouse formed one sample. DNase digestion (catalog 79254, Qiagen, Hilden, Germany) and RNA clean-up were then performed using the RNeasy Mini kit (catalog 74104, Qiagen). The quality and integrity of total RNA were estimated using the Qubit 3.0 fluorometer (Thermo Fisher Scientific) and a Qsep100 system (BiOptic Inc., New Taipei Taiwan), respectively. The cDNA sequencing libraries were prepared using the SureSelect RNA Library Preparation Kit (catalog G9691A, Agilent Technologies), based on the manufacturer’s instructions. The quality of the cDNA libraries was determined using a Qsep100 system, and the sequencing was performed by Welgene company (Taipei, Taiwan), using an Ilumina NovaSeqTM 6000 sequencer (Illumina, San Diego, CA, USA). For transcriptomic analyses, AMs in BALF sorted from two mice were pooled into one sample, and 150 cells were used as the input for library construction using the Takara SMART-Seq® Stranded kit (catalog 63442, Takara Bio USA, Mountain View, CA, USA), according to the manufacturer’s protocol. The fragment size and the concentration of the library were evaluated using a Qsep100 and a Qubit 3.0 fluorometer, respectively. The library was sequenced by the Taiwan Genomic Industry Alliance (TGIA) company (Taipei, Taiwan), using an Ilumina NovaSeqTM 6000 sequencer.
For analyzing RNAseq data, the cutadapt (v3.5)(https://github.com/marcelm/cutadapt) was applied to remove adapter sequences and to filter out the low-quality reads. The resulting clean reads were then aligned to the mouse genome assembly (GRCm38.p6) by using STAR (v2.7.8a) (https://github.com/alexdobin/STAR) with two-pass mode. To quantify gene expression, a gene count matrix was generated based on the GENCODE vM25 gene annotation using featureCounts (v2.0.1) (http://subread.sourceforge.net). To account for differences in library size between the samples, the trimmed mean of M values (TMM) normalization method implemented by edegR (https://bioconductor.org/) was applied, and the abundance of transcripts was measured with transcripts per million (TPM). For identifying differentially expressed genes, the procedures implemented by the limma package (https://bioconductor.org/) were employed. The p values were computed using linear modeling and empirical Bayes moderation and were adjusted for multiple comparisons through the Benjamini–Hochberg procedure. Over-representation analyses of the differentially expressed genes were performed using the R package clusterProfiler (https://bioconductor.org/). The analyses focused on Gene Ontology (GO) Biological Process sets were obtained from MSigDB (v7.0, C5). The significantly enriched GO sets (adjusted p < 0.05) were utilized to construct association networks using enrichmentMap (https://apps.cytoscape.org/apps/enrichmentmap), and networks were visualized by Cytoscape (version 3.7.0) (https://cytoscape.org/). Heat maps were plotted using the pheatmap package (https://cran.r-project.org/web/packages/pheatmap/), based on the z scores calculated using the gene expressions by log2TPM. The upstream regulator analysis was performed using Ingenuity Pathway Analysis (IPA) (Qiagen). To evaluate the transcriptomic signature in AT2 cells in ARDS, human lung scRNAseq datasets from people with fatal COVID-19 (GSE171524, n = 20 participants; GSE171668, n = 24 participants) and the control (GSE171524, n = 7 participants) were downloaded from Gene Expression Omnibus (Supplementary Fig. 3)28,29. Data pre-processing, including data integration, feature selection, normalization, and data reduction was conducted using the Scanpy (v1.9.0)72. Cell type annotation of each cell was defined by the original works28,29.
Measuring lipid metabolites in BALF samples
To measure PC (catalog ab83377) and PG (catalog ab273288) contents in BALF samples using the commercial fluorometric assay kits (Abcam), 200 μL BALF was mixed with 750 μL 1:2 chloroform:methanol solution for lipid extraction. After vortexing for 1 min, 250 μL chloroform was added to the solution, which was then vortexed for 30 s. The solution was then added with 250 μL ddH2O. After vortex for 30 min, the solution was centrifuged at 1500 × g for 10 min under room temperature. The upper phase was removed gently, and the lower phase was washed using 500 μL 1:1:0.9 chloroform: methanol: ddH2O solution. After vortex for 30 s, the solution was centrifuged again at 1500 × g for 10 min under room temperature. The lower phase was transferred to another Eppendorf tube and was dried by nitrogen. Following resuspension of the extracts using 110 μL PG or PC assay buffer (Abcam), 50 μL solution was used for measurements of PG or PC, and another 50 μL solution was used for background control. The assays were then performed based on the manufacturer’s protocol. To measure free fatty acid contents in BALF samples, 50 μL BALF was used for commercial fluorometric assay (catalog ab65341, Abcam), and the measurements were performed according to the manufacturer’s instructions.
Measuring intracellular TG contents
Intracellular TG contents were measured using commercial TG assay kit (catalog ab65336, Abcam), according to manufacturer’s instructions. After isolation, 1 × 106 AT2 cells were snap-frozen using liquid nitrogen. The pellets were homogenized in 250 μL 5% NP-40 in ddH2O through vortex, and the samples were then heated to 80 °C~100 °C till the solution turned cloudy. The above homogenization and heating process was repeated twice, and the samples were then centrifuged at 21,500 × g for 2 min. The supernatant was obtained for TG measurements through colorimetric assay using a SpectraMax iD3 plate reader (Molecular Devices).
Stable isotope resolved metabolomic
For 13C labeling of metabolites, 0.5 × 106 MLE12 cells were added to a 10-cm dish. After culture for 2 days, the medium was replaced with assay buffer constituted by XF Base medium (Agilent Technologies) containing 10 mM [U−13C] D-glucose (catalog HY-B0389A, MedChemExpress, Monmouth Junction, NJ, USA) and 2 mM glutamine. After incubation at 37 °C for 30 min, the assay buffer was removed, and the extraction was performed according to previous studies73,74. Cells were washed twice with PBS, and once with cold ddH2O. Metabolic quenching was performed using liquid nitrogen, and 1 mL 80% cold methanol was added to each dish. Cells were collected by scraping on dry ice. The dish was then washed twice using 80% methanol, and the recovered solution was pooled together with the cell suspension. After vortex and sonication for 5 min, the cell suspension was centrifuged at 15,000 × g under 4 °C for 5 min, and the supernatant was collected. Evaporation using a speed vacuum was performed at 4 °C, and 200 μL reconstitution solvent (acetonitrile:methanol:water = 4:4:2) was added. After sonication for 5 min, the solution was centrifuged at 15,000 × g under 4 °C for 5 min, and 135 μL supernatant was transferred into a vial. After addition of 15 μL 5 mM medronic acid, the solution was used for analyzing 13C-labeled metabolites through UHPLC-MS/MS (Supplementary Fig. 8a).
Isotopic labeled metabolites were measured using an Agilent 1260 Infinity II UHPLC system (Agilent Technologies) equipped with a binary solvent pump, an autosampler, a sample reservoir, and a column oven. The coupled mass spectrometer was an Agilent 6545XT QTOF with a Jet Stream electrospray ionization source (Agilent Technologies). An ACQUITY BEH Amide Column (2.1 mm × 100 mm, 1.7 µm; catalog 186004801, Waters Corporation, Milford, MA, USA) was performed for the analyte separation. The mobile phase A consisted of 15 mM ammonium acetate and 0.3% ammonia solution (38%) in water. The mobile phase B consisted of 15 mM ammonium acetate and 0.3% ammonia solution (38%) in 90% acetonitrile. The gradient with a flow rate of 0.3 mL min−1 consisted of 0–8 min, 90–50% mobile phase B; 8–10 min, 50% mobile phase B; 10–11 min, 50–90% mobile phase mobile phase B; and 11–20 min, 90% mobile phase B. The autosampler and the column oven were maintained at 4 °C and 40 °C, respectively. The injection volume was 5 μL. Both negative (−) and positive (+) electrospray ionization modes were utilized with the following parameters: dry gas temperature, 280 °C; dry gas flow rate, 8 L min−1; nebulizer pressure, 45 psi; sheath gas temperature, 325 °C; sheath gas flow rate, 10 L min−1; capillary voltage, 3500 V; nozzle voltage, 1000 V; and fragmentor voltage, 120 V. The mass scan range was 50–1000 m/z for positive mode and 50–1100 m/z for negative mode. The acquisition rate was 2 Hz for both modes.
Data from UHPLC-QTOF mass spectrometry were analyzed using Agilent MassHunter Workstation Profinder Software (Version B.10.1) with an in-house database established by Agilent Personal Compound Database and Library. The results exported by Profinder were corrected for the natural distribution of 13C.
Metabolomic profiling of BALF samples
Metabolomic profiling was performed to measure amino acids and metabolites of glycolysis and TCA cycle in BALF samples, which were extracted with 400 μL of acetonitrile/methanol/water (v/v/v = 2:2:1). The extraction was performed by shaking at 1000 rpm for 5 min using a Geno/Grinder 2010, and the samples were then centrifuged using Eppendorf Centrifuge 5810 R at 15,000 × g for 5 min at 4 °C. The supernatant was filtered with 0.2 μm Ministart RC 4 filter, and 90 μL of filtered supernatant was taken and mixed with 10 μL of 5 mM medronic acid. The mixture was then subjected to liquid chromatography-triple quadrupole mass spectrometry analysis, using an Agilent 1290 UHPLC system coupled with an Agilent 6460 triple quadrupole mass spectrometer (Agilent Technologies). The separation was performed using an ACQUITY BEH Amide Column (2.1 mm × 100 mm, 1.7 µm; Waters Corporation), and the column was thermostated at 40 °C during analysis. The mobile phase was constituted by solvent A, 15 mM ammonium acetate with 0.114% NH4OH in water (3 mL of 38% NH4OH in 1000 mL solvent), and solvent B, 15 mM ammonium acetate with 0.114% NH4OH in 90% acetonitrile. For analyzing metabolites of glycolysis and TCA cycle, a gradient elution was used: 0–8 min, 90–50% solvent B, 8–10 min, 50% solvent B, 10–11 min, 50–90% solvent B, 11–16 min, 90% solvent B. For analyzing amino acids, a gradient elution was used: 0–8 min, 85–50% solvent B, 8–10 min, 50% solvent B, 10–10.5 min, 50–85% solvent B, 10.5–17 min, 85% solvent B. The flow rate was 0.3 mL min−1. The injection volume was 5 μL. The negative (−) and positive (+) electrospray ionization modes were utilized with the following parameters: 325 °C (−) or 300 °C (+) for drying gas temperature, 7 L min−1 (−) or 8 L min−1 (+) for drying gas flow, 45 psi or 40 psi for nebulizer pressure, 325 °C (−/+) for sheath gas temperature, 11 L min−1 (−) or 12 L min−1 (+) for sheath gas flow rate, and 3500 V (−/+) for capillary voltage. Nozzle voltage was set at 500 V (−/+). The mass spectrometer was configured in multiple reaction monitoring mode. All the peaks were integrated with MassHunter Quantitative Analysis software.
Lipidomic analyses
For analysis of intracellular acylcarnitine and carnitine levels, 2 × 106 AT2 cells were used for extraction with 500 μL iced methanol. The extraction was performed by shaking at 1000 rpm for 2 min using a Geno/Grinder 2010 (SPEX SamplePrep., Metuchen, NJ, US), and the samples were then put on ice for 5 min. After the addition of 500 μL iced water into the sample mixture, shaking in the Geno/Grinder2010 with the aforementioned parameters was performed again. After on ice for 5 min, the extract was centrifuged using Eppendorf Centrifuge 5810 R at 15,000 × g for 5 min at 4 °C. The supernatant was filtered with 0.2 μm Ministart RC 4 filter (Sartorius, Goettingen, Germany) and subjected to LC–MS analysis. Acylcarnitine profiling in AT2 cells was performed using an Agilent 1290 UHPLC system coupled with an Agilent 6540 QTOF (Agilent Technologies). Injection of 2 μL sample into an Acquity HSS T3 column (2.1 × 100 mm, 1.8 μm) (catalog 186003539, Waters, Milford, MA) was performed, and the analytical column was maintained at 40 °C. The mobile phase was composed of solvent A (water/0.1% formic acid) and solvent B (acetonitrile/0.1% formic acid). The gradient elution program was as follows: 0–1.5 min: 2% B; 1.5–9 min: linear gradient from 2 to 50% B; 9–14 min: linear gradient from 50 to 95% B; and 3 min maintenance in 95% B. The flow rate was 300 μL/min. A Jet Stream electrospray ionization source was used for sample ionization. The following parameters were used throughout the analysis: 325 °C gas temperature, 8 L/min gas flow, 40 psi nebulizer, 325 °C sheath gas temperature, 10 L/min sheath gas flow, 4000 V in positive mode for capillary voltage, and 120 V fragmentation voltage. The mass scan range and acquisition rate were m/z 50–1700 and 2 Hz, respectively. Acylcarnitines were identified using the in-house library with accurate mass and retention times.
For PC, PG, and LPG measurements in BALF and AT2 cell samples were extracted using a modified Folch method. In brief, 100 μL aliquot of BALF or dried AT2 cell samples (2 × 106 cells per sample) were added with deionized water to a total 200 μL solution. The solution was then added with 20 μL of 2,6-di-t-butyl-p-hydroxytoluene (200 mg/mL in methanol). After the addition of 1000 μL homogenization solvent (methanol:chloroform = 1:2), extraction was performed at 1000 rpm for 20 min using a Geno/Grinder 2010. The extract was then centrifuged at 15,000 × g for 5 min at 4 °C. The lower layer of samples was collected. The remaining solution was extracted again. The extracts were pooled together and were then dried with nitrogen. Reconstitution was performed by using 100 μL methanol for the extracts of BALF and using 150 μL methanol for the extracts of AT2 cells. The reconstituted solution was shaken with Geno/Grinder 2010 at 1000 rpm for 2 min and was then centrifuged at 15,000 × g for 5 min at 4 °C. The supernatant was filtered with a 0.2 mm Ministart RC 4 filter and was subjected to the LC–MS analysis for PC, as well as LC–MS/MS analysis for PG and LPG.
PC profiling was performed using an Agilent 1290 UHPLC system coupled with an Agilent 6540 QTOF. An Agilent ZORBAX Eclipse Plus C18 column (2.1 × 100 mm, 1.8 μm; catalog 959758-902) was used for the separation. The sample reservoir and the column oven were maintained at 4 °C and 55 °C, respectively. The injection volume was 5 μL. The mobile phase was composed of solvent A (water/0.1% formic acid/10 mM ammonium acetate) and solvent B (methanol: isopropanol: water = 60: 40: 1/0.1% formic acid/10 mM ammonium acetate). The gradient elution program was as follows: 0–2 min: 35–80% B; 2–7 min: 80–100% B; 7–17 min: 100% B; and 3 min post time. The flow rate was 350 μL/min. A Jet Stream electrospray ionization source was used for sample ionization. The MS parameters were set as follows: 350 °C gas temperature, 7 L/min gas flow, 45 psi nebulizer, 350 °C sheath gas temperature, 11 L/min sheath gas flow, 4000 V in positive mode for capillary voltage, and 120 V fragmentation voltage. The mass scan range and acquisition rate were m/z 100–1600 and 2 Hz, respectively. PC was identified using the in-house library with accurate mass and retention times.
PG and LPG profiling were performed using an Agilent 1290 UHPLC system coupled with an Agilent 6495 C triple quadrupole system (Agilent Technologies). An Agilent ZORBAX Eclipse Plus C18 (2.1 × 100 mm, 1.8 μm) column (Agilent Technologies) was used for the separation. The sample reservoir and the column oven were maintained at 4 °C and 55 °C, respectively. The injection volume was 5 μL. The mobile phase was composed of solvent A (water: acetonitrile = 6: 4/0.1% formic acid/10 mM ammonium acetate) and solvent B (isopropanol: acetonitrile = 9: 1/0.1% formic acid/10 mM ammonium acetate). The gradient elution program was as follows: 0–1 min: 10% B; 1–1.5 min: 10–40% B; 1.5–6 min: 40–70% B; 6–16 min: 70–90% B; 16–18 min: 90–92% B; 18–18.5 min: 92–100% B; 18.5–20.5 min: 100% B; and 2.5 min post time. The flow rate was 350 μL/min. A Jet Stream electrospray ionization source was used for sample ionization. The MS parameters were set as follows: 250 °C gas temperature, 11 L/min gas flow, 25 psi nebulizer, 350 °C sheath gas temperature, 11 L/min sheath gas flow, 3500 V in the positive mode, and 4000 V in the negative mode for capillary voltage, and 166 V fragmentation voltage. The iFunnel positive high- and low-pressure RF were set to 150 V and 60 V, respectively. For the negative mode, the high- and low-pressure RF values were 150 V and 60 V, respectively. Dynamic multiple reaction monitoring was applied for the analysis. The PG- and LPG-specific fragments were identified with a neutral loss of m/z 172 for hydrogen adduct, m/z 189 for ammonium adduct, and m/z 194 for sodium adduct in the positive mode. The fatty acid side chains of PG species were identified in the negative mode.
Data from UHPLC-QTOF mass spectrometry were analyzed by using MassHunter Workstation Profinder Software Version B.08.00 (Agilent Technologies). MassHunter Workstation Quantitative Analysis Software Version B.07.01 (Agilent Technologies, Santa Clara, CA, USA) was used for peak integration of the data from UHPLC-triple quadrupole mass spectrometry.
Murine lung histology
The methods about sample preparations for murine lung histology have been published before8. After euthanasia, inflation of murine lungs was performed using 1.2 mL 4% PFA. The lungs were excised and then fixed in 4% PFA for 24 h at 4 °C. The lobes of murine lungs were separated after fixation, and placed in tissue cassettes (Sakura Finetek USA, Torrance, CA, USA). The samples were immersed in 70% ethanol at 4 °C and were sent to Biotools company (New Taipei, Taiwan) for processing, paraffin embedding, and hematoxylin and eosin staining. Whole slide scan images were then acquired and examined using the CaseViewer software (version 2.4, 3DHISTECH, Budapest, Hungary).
Immunofluorescent staining of murine lungs
Murine lung cryosections, which were prepared as previously published8, were used for immunofluorescent staining. After euthanasia, murine lungs were inflated using 1.5 mL solution formed by the optimum cutting temperature (OCT) formulations (Sakura Finetek USA) and PBS in 2:1 ratio. The lungs were then excised, and fixed in 4% PFA for 24 h at 4 °C. After fixation, the lobes were separated, and dehydrated in 30% sucrose solution for 24 h at 4 °C. After dehydration, the lobes were placed in a cryomold (Sakura Finetek USA), and were embedded by OCT formulations. The samples were then froze at −80 °C, and 30 μm-thick cryosections were retrieved on silane-coated slides immediately before immunofluorescent staining. The methods for blocking, permeation, and staining were described in the section about immunofluorescent staining using human surgical lung samples. The staining was performed using primary antibodies against CXCL1 (1:100; catalog MAB4532-SP, R&D systems), and CXCL2 (1:100; catalog AF-452-SP, R&D systems), and CD45 (conjugated to Alexa Fluor 647, 1:100; catalog 103123, BioLegend), and secondary antibodies against goat IgG (conjugated to Alexa Fluor 488, 1:500; catalog A11055, Thermo Fisher Scientific), and rabbit IgG (conjugated to Alexa Fluor 488, 1:500; catalog A-11008, Thermo Fisher Scientific). The images were acquired by confocal microscopy.
Confocal microscopy
Confocal microscopy was performed in the Microscopy Core Facility, Department of Medical Research, National Taiwan University Hospital, using a Zeiss LSM 880 laser scanning microscope (Oberkochen, Germany). To identify the cell origins of CXCL1 and CXCL2 in lung cryosections from murine LPS-induced ALI model, a 40×/1.3 oil immersed objective was used to obtain ~8.5 μm z stack images. The fluorophores were excited with a 405 nm laser diode (Hoechst 33342), a 488 nm argon laser (Alexa Fluor 488), a 561 nm diode-pumped solid-state laser (tdTomato), and a 633 nm HeNe laser (Alexa Fluor 647). The image was processed using ZEN 2.3 SP1 FP1 (black edition) software (version 14.0.12.201).
Bone marrow-derived neutrophil isolation and experiments
After euthanasia using CO2, the murine femurs and tibias were dissected, and bone marrow flushed using PBS. The marrow cells were then centrifuged at 300 × g for 10 min under 4 °C. The cell pellet was then resuspended using 10 mL PBS and was filtered through a 40 μm strainer (BD Biosciences). After centrifugation at 300 × g for 10 min under 4 °C, the cells were resuspended using MACS buffer. Neutrophils were then negatively selected using the mouse neutrophil isolation kit (catalog 130-097-658, Miltenyi Biotec), through the MACS separation columns. The isolated neutrophils were then resuspended in serum-free DMEM added with 1% penicillin/streptomycin, and were used for in vitro experiments. To investigate the effect of CXCL2 to neutrophil activation, 4 × 105 cells per well were added to a 96-well plate and were activated using 2 nM TNFα (catalog 410-MT-010, R&D systems), along with various concentrations (0, 20, or 40 nM) of CXCL2 (catalog 250-15, PeproTech, Cranbury, NJ, USA). After stimulation for six hours, the neutrophils from each well were used for RNA extraction and qPCR analyses of Cxcl2 and Tbp.
Statistical analysis
Data are represented as outlined in the figure legends and table footnotes. For continuous variables, unpaired Student’s t-tests or Mann–Whitney U tests were used for the comparison between the two groups, as appropriate. The comparison of discrete variables was performed using Pearson’s χ2 test. One-way ANOVA with Bonferroni correction was used for multi-group comparison of continuous variables. The detailed statistical analyses for RNAseq data were described in the method for transcriptomic analyses. A two-sided p-value < 0.05 was statistically significant. All analyses were performed using GraphPad Prism version 10.0.0 (GraphPad Software, San Diego, CA, USA) or SPSS version 18.0.0 (IBM, Armonk, NY, USA).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
RNA sequencing data are available under the accession number GSE224354 and GSE255949 in Gene Expression Omnibus (GEO). Source data underlying Figs. 1b, c, e, f, 2a, c–h, 3a–k, 4b, c, e, g, 5b–h, 6b, d, e, h–l, 7g–k, 8e–m, and Supplementary Figs. 4a–d, f, g, 6b, 7b–i, 8b, c, 9, 10a, 11b, 12b, 13, 15b–m, 19, 20c, 23a–d, 24a, and 4a are provided with this paper as the Source Data file. All the microscopy images in the figures and Supplementary Figs., including Figs. 1a, 7a, and Supplementary Figs. 1, 2, 10b, 21, and 22a, have been deposited in Mendeley online data repository (https://doi.org/10.17632/343cym3z8p.1). Requests for resources reagents and materials should be directed to the lead contact, Kuei-Pin Chung (gbchung@ntu.edu.tw). All the materials and MS-associated raw data generated in this study are available from the lead contact on reasonable request. Source data are provided with this paper.
References
Desai, T. J., Brownfield, D. G. & Krasnow, M. A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190–194 (2014).
Whitsett, J. A. & Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 16, 27–35 (2015).
Treutlein, B. et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014).
Fan, L. C. et al. Alveolar type II epithelial cell FASN maintains lipid homeostasis in experimental COPD. JCI Insight 8, e163403 (2023).
Whitsett, J. A., Wert, S. E. & Weaver, T. E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 61, 105–119 (2010).
Bridges, J. P. et al. LPCAT1 regulates surfactant phospholipid synthesis and is required for transitioning to air breathing in mice. J. Clin. Invest. 120, 1736–1748 (2010).
Tian, Y., Zhou, R., Rehg, J. E. & Jackowski, S. Role of phosphocholine cytidylyltransferase alpha in lung development. Mol. Cell Biol. 27, 975–982 (2007).
Chung, K. P. et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 10, 3390 (2019).
Shin, H. et al. Overexpression of fatty acid synthase attenuates bleomycin induced lung fibrosis by restoring mitochondrial dysfunction in mice. Sci. Rep. 13, 9044 (2023).
Sunaga, H. et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat. Commun. 4, 2563 (2013).
Plataki, M. et al. Fatty acid synthase downregulation contributes to acute lung injury in murine diet-induced obesity. JCI Insight 5, e127823 (2019).
Eaton, S., Bartlett, K. & Pourfarzam, M. Mammalian mitochondrial beta-oxidation. Biochem. J. 320, 345–357 (1996).
Kurtz, D. M. et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl Acad. Sci. USA 95, 15592–15597 (1998).
Goetzman, E. S. et al. Long-chain acyl-CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J. Biol. Chem. 289, 10668–10679 (2014).
Otsubo, C. et al. Long-chain acylcarnitines reduce lung function by inhibiting pulmonary surfactant. J. Biol. Chem. 290, 23897–23904 (2015).
Pham, L. et al. Neutrophil trafficking to the site of infection requires Cpt1a-dependent fatty acid beta-oxidation. Commun. Biol. 5, 1366 (2022).
Shinde, A. et al. Increased mortality from influenza infection in long-chain acyl-CoA dehydrogenase knockout mice. Biochem. Biophys. Res. Commun. 497, 700–704 (2018).
Schoors, S. et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197 (2015).
Wong, B. W. et al. The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 542, 49–54 (2017).
Briant, L. J. B. et al. CPT1a-dependent long-chain fatty acid oxidation contributes to maintaining glucagon secretion from pancreatic islets. Cell Rep. 23, 3300–3311 (2018).
Mihaylova, M. M. et al. Fasting activates fatty acid oxidation to enhance intestinal stem cell function during homeostasis and aging. Cell Stem Cell 22, 769–778 e764 (2018).
Knobloch, M. et al. A fatty acid oxidation-dependent metabolic shift regulates adult neural stem cell activity. Cell Rep. 20, 2144–2155 (2017).
Cui, H. et al. Impairment of fatty acid oxidation in alveolar epithelial cells mediates acute lung injury. Am. J. Respir. Cell Mol. Biol. 60, 167–178 (2019).
Siempos, I. I. et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight 3, e97102 (2018).
Bos, L. D. J. & Ware, L. B. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet 400, 1145–1156 (2022).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Gonzalez, R. F., Allen, L., Gonzales, L., Ballard, P. L. & Dobbs, L. G. HTII-280, a biomarker specific to the apical plasma membrane of human lung alveolar type II cells. J. Histochem. Cytochem. 58, 891–901 (2010).
Delorey, T. M. et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595, 107–113 (2021).
Melms, J. C. et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119 (2021).
Cook, G. A., King, M. T. & Veech, R. L. Ketogenesis and malonyl coenzyme A content of isolated rat hepatocytes. J. Biol. Chem. 253, 2529–2531 (1978).
Hao, F. et al. Butyrate enhances CPT1A activity to promote fatty acid oxidation and iTreg differentiation. Proc. Natl Acad. Sci. USA 118, e2014681118 (2021).
Morant-Ferrando, B. et al. Fatty acid oxidation organizes mitochondrial supercomplexes to sustain astrocytic ROS and cognition. Nat. Metab. 5, 1290–1302 (2023).
Yang, C. et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 56, 414–424 (2014).
Izumi, Y., Benz, A. M., Zorumski, C. F. & Olney, J. W. Effects of lactate and pyruvate on glucose deprivation in rat hippocampal slices. Neuroreport 5, 617–620 (1994).
Hammoud, S. et al. Tubular CPT1A deletion minimally affects aging and chronic kidney injury. JCI Insight 9, e171961 (2024).
Jao, C. Y., Roth, M., Welti, R. & Salic, A. Metabolic labeling and direct imaging of choline phospholipids in vivo. Proc. Natl Acad. Sci. USA 106, 15332–15337 (2009).
Matute-Bello, G., Frevert, C. W. & Martin, T. R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L379–L399 (2008).
Mould, K. J. et al. Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am. J. Respir. Cell Mol. Biol. 57, 294–306 (2017).
Girbl, T. et al. Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity 49, 1062–1076 e1066 (2018).
Roy, R. M. et al. Lactate produced by alveolar type II cells suppresses inflammatory alveolar macrophages in acute lung injury. FASEB J. 37, e23316 (2023).
Zhang, Y. et al. L-lysine ameliorates sepsis-induced acute lung injury in a lipopolysaccharide-induced mouse model. Biomed. Pharmacother. 118, 109307 (2019).
Calkins, C. M. et al. L-arginine attenuates lipopolysaccharide-induced lung chemokine production. Am. J. Physiol. Lung Cell Mol. Physiol. 280, L400–L408 (2001).
Leemans, J. C., Heikens, M., van Kessel, K. P., Florquin, S. & van der Poll, T. Lipoteichoic acid and peptidoglycan from Staphylococcus aureus synergistically induce neutrophil influx into the lungs of mice. Clin. Diagn. Lab Immunol. 10, 950–953 (2003).
Hou, F., Xiao, K., Tang, L. & Xie, L. Diversity of macrophages in lung homeostasis and diseases. Front. Immunol. 12, 753940 (2021).
Kuronuma, K. et al. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J. Biol. Chem. 284, 25488–25500 (2009).
Kinniry, P. et al. KL4-surfactant prevents hyperoxic and LPS-induced lung injury in mice. Pediatr. Pulmonol. 41, 916–928 (2006).
Arbibe, L. et al. Generation of lyso-phospholipids from surfactant in acute lung injury is mediated by type-II phospholipase A2 and inhibited by a direct surfactant protein A-phospholipase A2 protein interaction. J. Clin. Invest. 102, 1152–1160 (1998).
Seeds, M. C. et al. Secretory phospholipase A2-mediated depletion of phosphatidylglycerol in early acute respiratory distress syndrome. Am. J. Med. Sci. 343, 446–451 (2012).
Tojo, K. et al. Early alveolar epithelial cell necrosis is a potential driver of COVID-19-induced acute respiratory distress syndrome. iScience 26, 105748 (2023).
Jiang, D. et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 11, 1173–1179 (2005).
Li, J. L. et al. Neutrophils self-regulate immune complex-mediated cutaneous inflammation through CXCL2. J. Invest. Dermatol. 136, 416–424 (2016).
Lentini, G. et al. Neutrophils enhance their own influx to sites of bacterial infection via endosomal TLR-dependent Cxcl2 production. J. Immunol. 204, 660–670 (2020).
Lee, K., Kerner, J. & Hoppel, C. L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 286, 25655–25662 (2011).
Liu, S. et al. Cis-acting lnc-Cxcl2 restrains neutrophil-mediated lung inflammation by inhibiting epithelial cell CXCL2 expression in virus infection. Proc. Natl Acad. Sci. USA 118, e2108276118 (2021).
Soehnlein, O., Lindbom, L. & Weber, C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 114, 4613–4623 (2009).
Guillon, A. et al. Pneumonia recovery reprograms the alveolar macrophage pool. JCI Insight 5, e133042 (2020).
Robinson, M. J. & Krasnodembskaya, A. D. Therapeutic targeting of metabolic alterations in acute respiratory distress syndrome. Eur. Respir. Rev. 29, 200114 (2020).
Sinha, P. et al. Latent class analysis of ARDS subphenotypes: a secondary analysis of the statins for acutely injured lungs from sepsis (SAILS) study. Intensive Care Med. 44, 1859–1869 (2018).
Folkesson, H. G. & Matthay, M. A. Alveolar epithelial ion and fluid transport: recent progress. Am. J. Respir. Cell Mol. Biol. 35, 10–19 (2006).
Reutershan, J. et al. Critical role of endothelial CXCR2 in LPS-induced neutrophil migration into the lung. J. Clin. Invest. 116, 695–702 (2006).
Flodby, P. et al. Knockout mice reveal a major role for alveolar epithelial type I cells in alveolar fluid clearance. Am. J. Respir. Cell Mol. Biol. 55, 395–406 (2016).
Matute-Bello, G. et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 44, 725–738 (2011).
Frasch, S. C. et al. Lysophospholipids of different classes mobilize neutrophil secretory vesicles and induce redundant signaling through G2A. J. Immunol. 178, 6540–6548 (2007).
Grasselli, G. et al. ESICM guidelines on acute respiratory distress syndrome: definition, phenotyping and respiratory support strategies. Intensive Care Med. 49, 27–759 (2023).
Force, A. D. T. et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA 307, 2526–2533 (2012).
Agudelo, C. W. et al. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (COPD). PLoS ONE 15, e0228279 (2020).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
McGovern, T. K., Robichaud, A., Fereydoonzad, L., Schuessler, T. F. & Martin, J. G. Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J. Vis. Exp. 15, e50172 (2013).
Wells, J. M. et al. Safety and tolerability of comprehensive research bronchoscopy in chronic obstructive pulmonary disease. Results from the SPIROMICS Bronchoscopy Substudy. Ann. Am. Thorac. Soc. 16, 439–446 (2019).
Misharin, A. V., Morales-Nebreda, L., Mutlu, G. M., Budinger, G. R. & Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 49, 503–510 (2013).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Lorenz, M. A., Burant, C. F. & Kennedy, R. T. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal. Chem. 83, 3406–3414 (2011).
DeWaal, D. et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 9, 446 (2018).
Acknowledgements
We thank the National Laboratory Animal Center (NLAC), NARLabs, Taiwan, and the Laboratory Animal Center, National Taiwan University College of Medicine, for animal husbandry. We thank the technical services provided by the Transgenic Mouse Models Core Facility of the National Core Facility for Biopharmaceuticals, National Science and Technology Council, Taiwan, and the Gene Knockout Mouse Core Laboratory of the National Taiwan University Center of Genomic and Precision Medicine. We thank the Metabolomics Core Lab, National Taiwan University Centers of Genomic and Precision Medicine, for helping with lipidomic analyses. We thank Dr. Guan-Yuan Chen, Department of Forensic Medicine, National Taiwan University, for helping with lipid extraction. We thank Dr. Po-Han Chuang, Graduate Institute of Clinical Medicine, National Taiwan University College of Medicine, for helping with the animal experiments. We thank the Bond Biotech company for helping with immunohistochemical staining of CPT1a and LCAD and the Department of Pathology (NTUH) for preparing tissue sections of surgical samples and immunohistochemical staining of CK. We thank the Biotools company (New Taipei, Taiwan) for processing murine lung samples and staining murine lung sections. We thank the Microscopy Core Facility in the Department of Medical Research, National Taiwan University Hospital (NTUH), for helping with cryosection and confocal microscopy. We thank the 7th Core Laboratory (NTUH) for helping with extracellular flux analyses, and we also thank the 3rd Core Laboratory for helping with flow cytometric sorting and analyses. We are grateful for the technical support provided by the Sequencing and Biochemistry Core, Department of Medical Research (NTUH), and for the sequencing services provided by Welgene Biotech (Taipei, Taiwan) and the Taiwan Genomic Industry Alliance (TGIA) Company (Taipei, Taiwan). We thank Mr. Shan-An Chen (Agilent Technology) and the 10th Core Laboratory (NTUH) in setting up the tracing system for 13C-labeled metabolites. We also thank the research nursing staffs at NTUH, NTUH Yunlin Branch, and Fu Jen Catholic University Hospital (New Taipei, Taiwan) who participated in this project and helped with the enrollment of patients in the clinical cohort. This study was supported by the NTUH (109-O04 [K.P.C.], 110-O07 [K.P.C.], 110-S4872 [K.P.C., J.Y.C.], 111-S0075 [K.P.C., J.Y.C.], and 113-S0079 [K.P.C., S.Y.R.]), National Taiwan University School of Pharmacy Endowment Fund in support of the Platform for Clinical Mass Spectrometry and NMR Structure Elucidation (C.H.K.), National Science and Technology Council (MOST-108-2628-B-002-017 [K.P.C.], MOST-109-2628-B-002-044 [K.P.C.], MOST-110-2628-B-002-029 [K.P.C.], MOST-110-2628-B-002-045-MY3 [K.P.C.], MOST-111-2628-B-002-030-MY3 [K.P.C.], MOST-110-2314-B-002-262 [S.Y.R.], and MOST 107-2314-B-002-235-MY3 [Y.F.C.]), National Taiwan University Laurel Research Projects (NTUCDP-112L7745 [K.P.C.] and NTUCDP-112L7746 [K.P.C.]), National Taiwan University New Faculty Grant (110T099 [K.P.C.] and NTU-NFG-110L7422 [K.P.C.]) and the research funding provided by Mr. Barry Lam, the chairman of Quanta Computer Inc (C.J.Y.).
Author information
Authors and Affiliations
Contributions
K.P.C. and C.J.Y. conceived and designed the study; K.P.C., Y.J.C., and Y.H.J. took care of animal husbandry and performed all the experiments; K.P.C. and C.L.H. performed all the data analyses; C.N.C., H.C.K., Y.T.L., and C.H.K. established and performed all metabolomic and lipidomic analyses in this study; Y.L.H. and M.S.H. prepared the pathological sections and interpreted the immunohistochemical staining results; Y.F.C., W.L.L., S.Y.R., and J.Y.C. helped recruitments of the clinical cohort and collected the clinical samples; P.C. and A.M.K.C. shared the key mouse lines in this study; K.N., M.P., S.M.C., A.M.K.C., C.H.K., and C.J.Y. provided critical discussion for data interpretation and revision; K.P.C. wrote the manuscript; M.P., and S.M.C. helped to organize and to edit the manuscript; and all coauthors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
A.M.K.C. is a cofounder and equity stockholder for Proterris, which develops therapeutic uses for CO. A.M.K.C. has a use patent on CO. Additionally, A.M.K.C. has patents in chronic obstructive pulmonary disease (US Patent 10905682). M.P. participates an advisory board meeting for InflaRx. All the other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Laura Herrero, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chung, KP., Cheng, CN., Chen, YJ. et al. Alveolar epithelial cells mitigate neutrophilic inflammation in lung injury through regulating mitochondrial fatty acid oxidation. Nat Commun 15, 7241 (2024). https://doi.org/10.1038/s41467-024-51683-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-51683-1
- Springer Nature Limited