

Abstract
Genomic imprinting is observed in endosperm, a placenta-like seed tissue, where transposable elements (TEs) and repeat-derived small RNAs (sRNAs) mediate epigenetic changes in plants. In imprinting, uniparental gene expression arises due to parent-specific epigenetic marks on one allele but not on the other. The importance of sRNAs and their regulation in endosperm development or in imprinting is poorly understood in crops. Here we show that a previously uncharacterized CLASSY (CLSY)-family chromatin remodeler named OsCLSY3 is essential for rice endosperm development and imprinting, acting as an upstream player in the sRNA pathway. Comparative transcriptome and genetic analysis indicated its endosperm-preferred expression and its likely paternal imprinted nature. These important features are modulated by RNA-directed DNA methylation (RdDM) of tandemly arranged TEs in its promoter. Upon perturbation of OsCLSY3 in transgenic lines, we observe defects in endosperm development and a loss of around 70% of all sRNAs. Interestingly, well-conserved endosperm-specific sRNAs (siren) that are vital for reproductive fitness in angiosperms are also dependent on OsCLSY3. We observed that many imprinted genes and seed development-associated genes are under the control of OsCLSY3. These results support an essential role of OsCLSY3 in rice endosperm development and imprinting, and propose similar regulatory strategies involving CLSY3 homologs among other cereals.
Similar content being viewed by others

Introduction
Seed formation is a major stage in the life cycle of gymnosperms and angiosperms. The embryo is embedded in a nourishment tissue and protected by a seed coat in a typical seed, and such an arrangement protects the embryo from unfavorable environmental conditions and mechanical damage. Unlike gymnosperms, angiosperms seeds evolved a unique nourishment tissue called endosperm which is also a product of fertilization like embryo. Among the two haploid sperm nuclei in the pollen, one fertilizes the egg and the other fertilizes with typically a diploid central cell, in a process called as double fertilization. This results in a diploid embryo and a triploid endosperm1,2.
Apart from the nourishment of the developing embryo, endosperm also senses the environmental parameters such as temperature, nutrient availability, biotic factors, etc. to control seed germination3,4,5,6. Endosperm derived signals such as small molecules, hormones, proteins, peptides, RNAs, etc. play crucial roles in the development of the embryo7. Besides, endosperm is the major source for carbohydrates, proteins and fat for most of the animals, including humans8.
Despite several landmark studies, mechanism of endosperm development is still an obscure area of research8. Multiple studies in model plant Arabidopsis indicated that genetic, hormonal and epigenetic pathways play vital roles in endosperm development. Unique ploidy, genomic imprinting and chromatin organization in endosperm distinguishes it from other plant tissues2,9. Imprinting is important since it regulates endosperm development, providing reproductive fitness to the embryo. A gene is imprinted when one of the parental alleles is partially or completely silenced due to allele-specific epigenetic marks in chromatin and the other allele gets expressed. Among the imprinted genes that are paternally silent but maternally expressed are called maternally expressed imprinted genes (MEGs), and genes that are maternally silent but paternally expressed are referred to as paternally expressed imprinted genes (PEGs)9,10,11,12,13,14.
Epigenetic players such as DNA methylation, demethylation and polycomb group (PcG) of proteins and associated processes regulate genomic imprinting through chromatin modifications in Arabidopsis15,16,17. In agreement with this, mutations in several epigenetic-associated imprinted genes led to defective endosperm and embryos14,18,19,20,21. Surprisingly, across plants, most of the major epigenetic regulators of imprinting themselves were imprinted in endosperm, including FIS2, MEA, VIM5, OsFIE1, OsEMF2a, ZmFIE1, ZmFIE2 genes. Often, tissue- and allele-specific expression of these major players were also tightly regulated by epigenetic pathways10,22,23,24,25,26,27,28,29. Thus, epigenetic pathways seem to play a crucial role in endosperm development by regulating expression of imprinted and development-related genes.
DNA methylation, considered as the primary epigenetic mark, is a major contributor to genomic imprinting in plants30,31. The cytosine methylation of DNA is peculiar in plants as it is observed in CG, CHG and CHH contexts (where H corresponds to A, T, or C). Multiple DNA methyltransferases encoded by plants establish and maintain methylation marks. There is a division of labor to maintain these marks, the CG methylation is maintained by METHYLTRANSFERASE1 (MET1) while CHG methylation requires CHROMOMETHYLASE3 (CMT3). The CHH methylation is regulated by a de novo DNA methyltransferase named DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) which is guided by RdDM. Another methyltransferase named CMT2 also maintains CHH methylation in specific loci32.
A plant-specific DNA-dependent RNA polymerase IV (Pol IV) generates small (∼30–40 nt) transcripts predominantly from TEs and repeats to initiate RdDM. These RNAs get converted into double-stranded (ds) forms by RNA DEPENDENT RNA POLYMERASE2 (RDR2). The ds substrates are processed into 23–24 nt sRNA duplexes by DICER-LIKE3 (DCL3). The 24 nt sRNAs are preferentially loaded into ARGONAUTE4/6/9 (AGO4/6/9) to guide DRM2 to methylate TEs and repeats by associating with long non-coding transcripts (150–200 nt) generated by another plant-specific polymerase named Pol V. Pol IV is specifically recruited to TEs and repeats by a family of SNF2 chromatin remodelers named CLASSY (CLSY) along with SAWADEE HOMEODOMAIN HOMOLOG1 (SHH1) proteins. The tissue-specific expression of CLSYs regulates tissue-specific DNA methylation in Arabidopsis33,34,35,36,37,38.
Although RdDM is known to regulate imprinting in Arabidopsis39, there seems to be some variations in the way it contributes to genomic imprinting across plants. For example, RdDM pathway derived 24 nt sRNAs are majorly maternally biased in the young seeds of Arabidopsis40,41 and these sRNAs regulate imprinting and seed development42,43. However, paternal genome also contributes substantial amount of sRNAs in endosperm44. In maize and Brassica rapa, 24 nt sRNAs were majorly maternally biased in endosperm45,46 whereas in rice, 24 nt sRNAs were derived from both parental genomes14,27,47. Unlike other tissues, a major portion of 24 nt sRNAs were majorly derived from genic regions and not from TEs in rice endosperm47. The same study found a unique class of highly expressing sRNA loci in rice endosperm named siRNA endosperm-specific (siren) loci. These loci overlapped with genic and inter-genic regions47. Multiple studies have also revealed the presence of siren loci in B. rapa and Arabidopsis, both in endosperm and ovule tissues. The siren derived sRNAs not just induced DNA methylation46,48, but also regulated expression of adjacent genes. Siren loci derived sRNAs can also trigger DNA methylation in trans at specific protein-coding genes48,49, indicating their versatile nature, functions of which is not well understood. In rice, maize and Arabidopsis, many imprinted sRNA loci are located proximal to the imprinted genes. Surprisingly, silenced alleles of many imprinted genes generate imprinted sRNAs14,27,44,45,47.
The early stages of endosperm development are very similar between cereals like rice and dicots like Arabidopsis. However, differences are observed in the later stages of development. After the cellularization, endosperm among cereals differentiates into two types of tissues—the outer aleurone layer and the inner starchy endosperm. The cereal endosperm rapidly accumulates large amounts of storage material which nourishes the embryo during embryogenesis and seed germination. In Arabidopsis, endosperm provides nourishment only during embryogenesis. How such variations in development are imposed and maintained in plants are unknown50,51. Such a variation holds huge commercial and agronomic value. Few imprinting-associated genes were found conserved between dicots and monocots14,25,27,47,52,53,54, while, the number of conserved imprinted genes were comparatively higher among monocots25,26,54. Cereals seem to have many different regulators for endosperm development; however, their contributions are unknown.
Here, we used a transcriptome screen to identify epigenetic players of endosperm development in rice. A previously unannotated ortholog of Arabidopsis CLSY3 was identified. This gene is majorly expressed in endosperm and regulated key TE-derived sRNAs, siren loci and imprinted sRNA loci. Using genetic and molecular methods including knockdown (kd) and knockout (KO) lines, we show that rice CLSY3-dependent loci regulate several imprinted genes and seed development-related genes. Mutation in OsCLSY3 negatively affected endosperm development, whereas its overexpression (OE) led to larger seeds with defective cellularization. OsCLSY3 predominantly bound to long terminal repeat (LTR) TEs in the genome as seen in ChIP-analysis performed using two different epitope tags. We also identified that OsCLSY3 itself is a MEG and its silencing in vegetative tissues was maintained through an RdDM loop operating at TEs in its promoter. These results indicated RdDM-controlled tissue-specific roles of a MEG chromatin regulator OsCLSY3 in rice endosperm development.
Results
OsCLSY3 is an endosperm-preferred imprinted gene
To understand key regulators of endosperm development in rice, we performed a tissue-specific transcriptome analysis using rice seeds. We isolated rice embryo as well as endosperm (20–25 days after pollination—DAP), and performed transcriptomic analysis with biological replicates using an elite indica rice variety Pusa Basmati-1 (henceforth PB1) as described in “Methods” section (Fig. 1A and Supplementary Fig. 1A). More than 90% of the intact reads of about 30 million reads/library were mapped to rice genome (Supplementary Table 1). Well-known tissue-specific marker genes55 that express either in embryo, such as OsH1, OsARF2, OsRS2, OsGRF4, or in endosperm, such as OsFIE1, OsNF-YC1128,56, showed expected patterns, indicating that the tissues taken for analysis were free from cross-contamination (Supplementary Fig. 1B, C). The green (non-seed) tissue contamination, considered as a major problem in seed transcriptomes, was not observed in embryo and endosperm transcriptomes, since expression of green tissue-specific genes such as Os12g0516000 and Os09g0537700 (OsRRP) were not detected (Supplementary Fig. 1D)57. To find the endosperm and embryo-preferred genes, we considered log2 2-fold upregulated and downregulated genes, respectively, in endosperm when compared to embryo for further analysis. Around 3607 transcripts (3459 annotated) showed embryo-preferred expression, while transcripts that expressed highly in endosperm were around 3686 (3367 annotated) (Supplementary Data 1 and 2). From the gene ontology (GO) analysis, we observed embryo-preferred genes were majorly involved in hormonal pathways, transcription-related functions, development-related, while endosperm-preferred genes were related to carbohydrate, protein metabolism and transport-related pathways (Supplementary Fig. 1E, F). The genes that showed endosperm-preferred expression in dicots such as AP2, WRKY, bHLH, MADS box, NAC transcription factors behaved similarly in rice58,59,60. We also observed multiple carbohydrate storage, starch branching and sugar metabolism-related genes such as OsSGL, OsGBP, SBDCP1, and OsFLO2 were uniquely expressed in cereal endosperm (Supplementary Data 1 and 2).

A Schematic showing EM (embryo) and EN (endosperm) tissues taken for transcriptome analysis. B Venn diagram showing overlap between epigenetic genes, EN-preferred genes and all known imprinted genes. C Heatmap depicting tissue-specific expression of epigenetic genes. Root (GSE166669), seedling (GSE229604), VL vegetative leaf (GSE229604), FL flag leaf (GSE111472), YP young panicle (GSE180457), MP mature panicle (GSE107903), AN anther (GSE180457), Y-EN young EN (15 days after pollination—DAP), M-EN mature EN (25 DAP) (GSE229961). Row Z-score was plotted. D Phylogenetic tree for DRD1 family proteins in Arabidopsis, rice and maize. 1000 bootstrap replications. E Domain architecture of CLSY3 proteins (predicted). F Expression of rice CLSYs across tissues (RNA-seq). G RT-qPCR analysis of OsCLSY3 and OsCLSY4 across different tissues. OsActin served as an internal control. Data represent means ± standard error (SE). n = 3. Experiment was repeated at least twice with independent samples and consistent results were obtained. H Vector map of P: OsCLSY3: GUS, and GUS expression pattern across reproductive tissues. Y-F1—spikelet before anthesis, Y-F2—spikelet 1 d before pollination, OV—unfertilized ovule. Scale bar (SB)—1 mm. I Scheme depicting WP and PB1 cross, and the SNPs identified in CDS of OsARF22 (PEG), OsCLSY3 (putative MEG) and OsSHH1 (BiG). Punnett square showing possible EN genotypes. Transcript contribution from the WWP genotype seeds is shown for imprinted genes. Stacked barplots showing theoretical and observed transcript contribution of three genes in WWP EN. Dotted lines indicate the theoretical percentage of maternal and paternal SNPs. Source data are provided as a Source Data file.
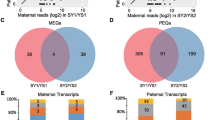
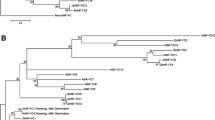
We explored if the differential expression of genes were due to epigenetic players, chromatin modifiers, transcription factors and associated co-factors. To find possible endosperm-preferred epigenetic regulators, we compared expression of 159 epigenetic genes in endosperm61,62,63,64,65. Among these, 21 were highly expressed in endosperm (Fig. 1B). We observed that candidates such as SDG714, OsAGO12, OsMORC6, ENL-1 like chromatin remodelers, several methyltransferases, JMJ genes and previously studied PcG complex-related genes showed endosperm-preferred expression (Supplementary Data 1 and 2). Many epigenetic genes themselves were imprinted and they can regulate endosperm development and imprinting of other genes10,21,22,40,66,67,68. To find endosperm-preferred imprinted epigenetic genes, we overlapped endosperm-preferred genes with 635 known imprinted genes14,27,52,53 and 159 epigenetic genes (Supplementary Data 3). We found three epigenetic genes, i.e., OsFIE1, OsJMJ706 and an unannotated gene Os02g0650800 (LOC_Os02g43460) that were imprinted as well as highly expressed in rice endosperm (Fig. 1B). The roles of OsFIE1 in endosperm development28,29,67,68,69, and OsJMJ706 in flower development are well known70,71. However, identity and function of Os02g0650800, which is a majorly endosperm-preferred gene that is listed as an imprinted gene in two published datasets was unknown (Fig. 1C). The gene was grouped in DRD1 family of SNF2 domain containing chromatin remodeler in rice (CHR740)72. A phylogenetic analysis indicated that CHR740 was close to Arabidopsis CLSY3 (Fig. 1D) and matched previous phylogenetic studies73 (Supplementary Table 2). The gene Os02g0650800 (LOC_Os02g43460) was recently named as FEL174. The CHR740 had 29.3% amino acid identity, 44.5% amino acid similarity with Arabidopsis CLSY3 and both having N-terminal Intrinsically disordered region (IDR), C-terminal SNF2 domains as predicted in InterProScan and Prosite tools75,76 (Fig. 1E). IDRs are crucial for many chromatin associated epigenetic regulators77. Monocots such as rice and maize seem to have three CLSY genes unlike four CLSYs found in Arabidopsis (Fig. 1D)72,73,78. We found that maize ortholog of OsCLSY3 GRMZM2G178435 (RMRL1), is also a MEG25 and it is closely related to OsCLSY3 (45.3% amino acid identity and 57.5% amino acid similarity). We noted that rice CHR722 (Os07g0692600/LOC_Os07g49210/FEL2) and CHR742 (Os05g0392400/LOC_Os05g32610/FEM2) were similar to Arabidopsis CLSY1 and CLSY4, respectively (Fig.1D). The CHR722 was already denoted as OsCLSY1, and also as where it was implicated in anaerobic germination and seedling growth79 and the same gene also named as FEL2 in a recent report74. In Arabidopsis, CLSYs have tissue-specific expression, while CLSY1, CLSY2 are majorly expressed in leaf and flower buds, whereas CLSY3, CLSY4 are majorly expressed in unfertilized ovules37. We observed that OsCLSY3 and OsCLSY1 are majorly expressed in endosperm and embryos, respectively in PB1. However, unlike Arabidopsis, OsCLSY4/FEM2 is expressed ubiquitously (Fig.1F and Supplementary Fig. 2A, B). The expression patterns of CLSY genes in maize also matched the same pattern as in rice, indicating that monocot CLSYs have similar expression patterns (Supplementary Fig. 2C). RT-qPCR analysis across different rice tissues further confirmed spatiotemporal expression of CLSYs (Fig. 1G and Supplementary Fig. 2D). In indica rice PB1, using RNA-seq and RT-qPCR analysis, we found that OsCLSY3 and OsCLSY4 were expressed in different stages of endosperm unlike a previous report from japonica rice74. In order to further clarify the tissue-specific expression of OsCLSY3, we generated a β-glucuronidase (GUS) reporter construct driven by 1.1 kb promoter of OsCLSY3 (Supplementary Fig. 2E). We observed that its expression was restricted in the unfertilized ovule, specific-anther stages and all tissues within the endosperm, but not in embryo (Fig. 1H).
To investigate imprinting status of OsCLSY3 further, we identified single nucleotide polymorphisms (SNPs) in the OsCLSY3 CDS of different rice varieties. We performed crossing with indica rice variety Whiteponni (WP) as a maternal and PB1 as a paternal parent. Endosperm of F1 plants was genotyped and imprinting status of OsCLSY3 (10 DAP) was tested (Supplementary Fig. 2F). We used OsSHH1 as a bi-allelic gene (BiG) and a published PEG OsARF22 as controls27. We observed that for OsCLSY3, 86.1% of transcripts came from the maternal genome when compared to OsSHH1 (BiG) which showed 65.3%. As expected in the case for OsARF22, a PEG, 86.4% of transcripts were from the paternal genome (Fig. 1I and Supplementary Fig. 2F). These analyses demonstrated that OsCLSY3 is a MEG in rice.
DNA methylation at MITE TEs controls expression of OsCLSY3
Since MEGs in rice are majorly regulated by DNA methylation at their proximal regions, we probed DNA methylation status of OsCLSY3 promoter to understand the basis for its endosperm-preferred expression. Two heavily methylated Miniature Inverted TE repeats (MITE)-like regions, one similar to Ditto element, and another to Tourist element, were observed 600 bp upstream of OsCLSY3 transcription start site. To investigate the role of methylation in OsCLSY3 regulation, PB1 seeds were germinated in MS media and transferred to DNA methyltransferase blocker 5-aza-2′-deoxycytidine (AZA) containing media, as described27 (Supplementary Fig. 3A). We observed ectopic expression of OsFIE1, a well-known endosperm-specific MEG as well as OsCLSY3 in a dose-dependent manner (Supplementary Fig. 3B). Ubiquitously expressed OsCLSY4 was found unaltered upon AZA treatment, indicating that DNA methylation regulated the expression of OsCLSY3 but not of OsCLSY4 (Supplementary Fig. 3B). In leaf, the OsCLSY3 promoter (2 kb) was heavily methylated majorly at CHH sites when compared to the OsCLSY4 promoter (Supplementary Fig. 3C). Since TEs and repeats at the gene promoter can regulate tissue-specific DNA methylation16,31, we measured DNA methylation levels of those MITEs by targeted bisulfite (BS)-PCR (Supplementary Table 1). DNA methylation at MITE TEs was higher in vegetative leaf and panicle tissues when compared to endosperm (Supplementary Fig. 3D). This result was in agreement with published embryo and endosperm DNA methylation datasets52, specific to OsCLSY3 promoter and not OsCLSY4 promoter (Supplementary Fig. 3E). These results supported the antagonistic relation between DNA methylation at OsCLSY3 promoter and its expression across tissues.
Further, to study the role of MITE TEs in context of transcriptional regulation of OsCLSY3, we generated transgenic GUS reporter plants, with GUS gene driven by wild type (PB1) OsCLSY3 promoter (1.1 kb) (Fig. 1H) or by OsCLSY3 promoter with deleted MITE TEs. Removal of TEs significantly elevated GUS expression which further suggested that DNA methylation repressed OsCLSY3 expression via MITE TEs (Supplementary Fig. 3F). Further, the GUS expression was increased upon AZA treatment, supporting our previous observations (Supplementary Fig. 3G, H). We also generated a transgenic line in which GUS gene is driven by CLSY3 promoter having a single MITE (Tourist MITE). The plant showed higher GUS expression when compared to native OsCLSY3 promoter with two MITEs (Supplementary Fig. 3F). Upon AZA treatment, the GUS expression was also higher in these transgenic plants (Supplementary Fig. 3H, I). Short TEs near to gene are regulated by the RdDM pathway when cytosine methylation marks are majorly at CHH contexts80. Since MITEs were methylated densely at CHH sites, we quantified the sRNA levels across different tissues from previously published datasets81,82. We found that 24 nt sRNAs at the MITEs TEs were more abundant in embryo and flag leaf when compared to endosperm in our sRNA datasets (Supplementary Fig. 3J) and in northern analysis (Supplementary Fig. 3K). The sRNA level variation across tissues also suggested a clear correlation between sRNAs and DNA methylation at the OsCLSY3 promoter.
DNA methylation at MITE TEs is regulated through RdDM
If the RdDM pathway is involved in regulation of DNA methylation of MITE TEs, signatures of perturbation of this regulation must be seen across mutants in the RdDM pathway. Abundance of MITE derived sRNAs in osnrpd1-kd panicle81 were drastically reduced in comparison to PB1 (Fig. 2A, B). Since sRNAs that establish DNA methylation are loaded into AGO4 in Arabidopsis83, we investigated available rice AGO RNA Immunoprecipitation (RIP) datasets84 and found that MITE derived sRNAs were enriched in OsAGO4a and OsAGO4b (Fig. 2C). Correspondingly, OsCLSY3 mRNA expression was high in osnrpd1-kd young panicle and anther tissues (Fig. 2D). We also measured sRNA and DNA methylation levels using published datasets of several RdDM mutants64,85,86. We found that RdDM mutants in rice such as osrdr2/fem1 and osnrp(d/e)2 showed reduction of MITE derived sRNAs in the OsCLSY3 promoter, whereas, as expected, MITE derived sRNAs were unaltered in osnrpe1 (Fig. 2E and Supplementary Table 3). Correspondingly, there was higher expression of OsCLSY3 in some mutants such as osdrm2 and osnrpd1-kd RNA-seq datasets that are publicly available (Supplementary Fig. 4A and Supplementary Table 3). However, similar increase in OsCLSY4 mRNA was not observed in these mutants (Supplementary Fig. 4B). Since Arabidopsis CLSYs recruit Pol IV at specific TEs and repeats36, we hypothesized that MITE TEs might be regulated by another member of the CLSY family. OsCLSY4 was found majorly expressed in vegetative tissues unlike OsCLSY3 (Fig. 1F, G). We hypothesized that OsCLSY4 might be recruiting Pol IV to the MITE regions in the OsCLSY3 promoter in vegetative tissues. To check this, we generated clsy4-kd plants by artificial miRNA (amiR) strategy87,88,89 and compared silencing of MITEs through sRNAs. Independent amiR-expressing transgenic lines had reduced expression of OsCLSY4 (Supplementary Fig. 4C, D). In clsy4-kd leaf, we observed a dose-dependent reduction of MITE sRNAs (Fig. 2F). Also, we observed drastic reduction in MITE derived sRNAs in clsy4-kd similar to osnrpd1-kd panicle, indicating that OsCLSY4 is likely recruiting Pol IV into OsCLSY3 promoter (Fig. 2F). Using bisulfite sequencing (BS-PCR), we found a reduction of DNA methylation at the OsCLSY3 promoter in leaf tissues of clsy4-kd, which indicated that OsCLSY4 controls expression of OsCLSY3 via RdDM (Fig. 2F). Also, OsCLSY3 level was elevated in 5-day old clsy4-kd seedling tissues (Supplementary Fig. 4E). The osclsy4-kd plants generated in this study showed various agronomic phenotypes such as reduced height of plants, increased tillering and less grain filling matching previous observations in fem2 (osclsy4) mutant in japonica rice74 (Supplementary Fig. 4F–H). However, we also found osclsy4-kd plants had smaller endosperm a phenotype that was not explored in fem2 mutant74 (Supplementary Fig. 4F). We also quantified DNA methylation levels in the MITE region in several known RdDM mutant datasets86,90. We observed, similar to sRNAs, methylation was also reduced in osrdr2/fem1 mutant in rice (Fig. 2G). Among datasets derived from several DNA methyltransferase mutants in rice86 (Supplementary Table 3), only in osdrm2 and its other combinations, a total abolishment of DNA methylation at OsCLSY3 promoter was observed (Fig. 2H). All these results conclusively demonstrate that the RdDM pathway regulates expression of OsCLSY3.

A Integrated Genomics Viewer (IGV) screenshots showing OsCLSY3 promoter derived sRNA levels (16–35 nt) in osnrpd1-kd panicle. B Northern blot showing sRNA levels in osnrpd1-kd panicle. C IGV screenshots showing enrichment of OsCLSY3 promoter derived sRNAs (16–35 nt) in RIP of different AGOs in rice (GSE18250 and GSE20748). D Barplots showing OsCLSY3 level in osnrpd1-kd panicles (top) and osnrpd1-kd anther (bottom). OsActin served as internal control. Data represents mean ± SE of 2 independent biological replicates with 3 technical replicates each (n = 6). Comparisons were made with paired two-tailed Student’s t-tests (P < 0.05 considered as significant). E IGV screenshots showing status of MITE derived sRNAs among RdDM mutants of rice (GSE130166, GSE158709, Nip—Japonica cultivar Nipponbare) (PRJNA758109, Hwa—Japonica cultivar Hwayoung). F Northern blots showing MITE derived sRNA level in osclsy4-kd and osnrpd1-kd (left panels) and analysis of DNA methylation in OsCLSY3 promoter in osclsy4-kd leaf by BS-PCR (right panel). U6 served as loading control. G IGV screenshots showing methylation status of 800 bp MITE TE region in OsCLSY3 promoter in osrdr2 seedlings (GSE130168). H IGV screenshot showing methylation status in leaves of ddccc mutant (GSE138705). ddccc is osdrm2/osdrm1/oscmt2a/oscmt2b/oscmt3 (Nip—Japonica cultivar Nipponbare). Source data are provided as a Source Data file. (B), (D) and (F) were repeated at least twice with consistent results.
OsCLSY3 knockout (KO) plants exhibited sterility
While OsCLSY3 is an endosperm-preferred imprinted gene in monocots such as rice and maize25,27,53, in Arabidopsis its homolog is not an imprinted gene58,91. However, the RdDM pathway derived sRNAs regulate endosperm development in Arabidopsis43. Since role of CLSY3 in the context of rice endosperm development is not explored, we targeted OsCLSY3 with CRISPR-Cas9 gRNA against a unique region of first exon of OsCLSY3 and generated KO plants. Presence of intact T-DNA was confirmed by junction-fragment southern blot analysis (Supplementary Fig. 5A). In all these plants, the OsCLSY3 gene was edited as desired (Supplementary Fig. 5B). Vegetative growth of KO plants was unaffected (Supplementary Fig. 5C), however, three mutants (KO #1, #4, #5) showed strong phenotypes in panicles including complete sterility, indicating that OsCLSY3 plays an essential role in reproduction (Supplementary Fig. 5D). Other edited mutants in which defects were not observed, had either one or two amino acids deletions without altering the protein significantly. A KO of a homolog of OsCLSY3 in japonica rice named FEL1 did not show adverse phenotype74. This variation in phenotypes between subspecies of rice might be due to the sequence difference in the N-terminal end where OsCLSY3 and FEL1 are quite dissimilar, or due to the difference in editing and the nature of the truncated protein generated in these experiments. The nrpd1 mutant of rice and Capsella showed very low yield due to drastic pollen defects81,92. In order to check if KO plants show pollen sterility, we performed pollen viability assay and found non-viable pollen grains in KO plants (Supplementary Fig. 5E). A drastic reduction in number and morphologically defective, prematurely dead pollens were observed in KO lines (Supplementary Fig. 5F, G). There were very few endosperms developing in KO seeds and they showed severe morphological defects. The endosperms were partially filled and had brown spots on the surface (Supplementary Fig. 5H). However, none of the KO plants displayed growth defects in vegetative stages, indicating that the role of OsCLSY3 is restricted to specific tissues such as pollens and endosperm (Supplementary Fig. 5I). All these results collectively suggest that OsCLSY3 is crucial for overall reproductive development.
Knockdown (kd) and overexpression of OsCLSY3 (OE) led to yield-related phenotypes
Since KO plants were sterile, we planned to kd the gene through amiR strategy87,88,89. We generated transgenic plants with two amiRs driven by constitutive promoters in one T-DNA which was named as osclsy3-kd2 (double amiR) (Supplementary Fig. 6A, B). To silence OsCLSY3 in a dose-dependent manner, we also generated transgenics expressing single amiR (Supplementary Fig. 6A, B). Integration of T-DNA in the genome was verified by southern blot (Supplementary Fig. 6C). We obtained a total of 7 transgenic plants (lines 1 and 5–10) with double amiRs (osclsy3-kd2) (Fig. 3A and Supplementary Fig. 6D) and 8 plants (lines 1–8) with single amiR (osclsy3-kd1) (Fig. 3A). We selected two different osclsy3-kd lines for phenotyping and RT-qPCR (osclsy3-kd1 plant #1 was denoted as osclsy3-kd1, and osclsy3-kd2 plant #5 as osclsy3-kd2, respectively). About 76% and 60% reduction in OsCLSY3 transcripts were observed in osclsy3-kd2 and osclsy3-kd1 endosperm tissues, respectively (Fig. 3B). Both osclsy3-kd lines did not show abnormalities in vegetative tissues (Fig. 3C, E), but they had smaller seeds when compared to PB1 (Fig. 3D). The osclsy3-kd had reduced seed length, seed width and weight, indicating a crucial role of OsCLSY3 in endosperm development (Fig. 3F). We obtained one transgenic line (osclsy3-kd2 #2) in which T-DNA was intact but amiR was completely silenced (Fig. 3A and Supplementary Fig. 6C). We observed that seeds of this line were similar to PB1 (Supplementary Fig. 6E). The osclsy3-kd plants displayed minor alterations in primary root length in the second generation, probably due to the change in germination resulting from abnormal endosperms (Supplementary Fig. 6F, H). To investigate the tissue-specific role of OsCLSY3 further, we generated OE plants driven by a constitutive promoter (Supplementary Fig. 6G). The single copy OE plants (Fig. 3G and Supplementary Fig. 6G, I), showed robust vegetative growth and had bigger seeds when compared to PB1 (Fig. 3H–K). We observed that grain length and grain width were also increased in OE plants (Fig. 3F). However, the grain filling rate was significantly reduced probably due to trade-off phenotype (Fig. 3L). To study the internal morphology of osclsy3-kd and OE seeds, we sectioned endosperm and observed chalkiness, a signature of altered cellularization, in OE endosperms (Supplementary Fig. 6J). In KO, osclsy3-kd and OE lines, endosperm filling and cellularization timing defects were observed when we performed detailed histochemistry and scanning electron microscope (SEM) analysis (Supplementary Fig. 7A–D). The chalkiness is considered as a poor agronomic quality of the grains93,94. In OsCLSY3 OE seedlings, we observed shorter roots which was exactly the opposite of that we observed in osclsy3-kd seedlings (Supplementary Fig. 6F). These phenotypes were in agreement with perturbed endosperm development, since this tissue plays important roles in germination and seedling development4,95. Our germination assay revealed slower seed germination in OE when compared to PB1 and osclsy3-kd (Fig. 3M, N). These results collectively suggest that proper spatiotemporal expression of OsCLSY3 is crucial for normal development in rice.

A Northern blots showing expression of amiRs. B Barplot showing levels of OsCLSY3 expression in osclsy3-kd EN. OsActin served as internal control. Data represents mean ± SE of 2 independent transgenic lines with 3 technical replicates each. Experiments repeated more than twice. Comparisons were made with paired two-tailed Student’s t-tests (P < 0.05 was considered significant). C Images showing morphology of osclsy3-kd plants, panicles and seeds when compared to equally grown PB1. D Seed morphology of osclsy3-kd with and without de-husking. E Boxplots showing height and panicle length of osclsy3-kd plants. Boxes show median values and interquartile range. Whiskers show minimum and maximum values, data points were represented as round dots. Two-tailed Student’s t-test (P < 0.05 was considered significant). F Boxplots showing five seeds length, width and weight in osclsy3-kd. G Barplot showing expression of OsCLSY3 in OE plant leaves. OsActin served as internal control. Data represents mean ± SE of 2 independent transgenic lines with 3 technical replicates each. Experiments repeated three times. Two-tailed Student’s t-test (P < 0.05 was considered significant). H Images showing spikelet and seed morphology in OE. (SB—1 mm). I Images showing de-husked OE seeds (SB—1 mm). J Images showing morphology of OE plants, panicles and seeds (with and without de-husking) compared to PB1. K Boxplots showing height, flag leaf and panicle length of OE plants. PB1 panicles were the same ones used in 4E. L Boxplot showing percentage of seed filling in OE and osclsy3-kd plants. M Image showing seed germination across OsCLSY3 mis-expression lines. N Percentage of germinated seeds in OsCLSY3 genotypes. Data represents mean ± SE of 4 biological replicates with n = 5 seeds each, and the assay was repeated twice. Source data are provided as a Source Data file. All experiments were repeated at least twice with consistent results.
Majority of 23–24 nt sRNA producing loci in endosperm were OsCLSY3-dependent
CLSYs in Arabidopsis recruit Pol IV into repeats and transposons, hallmark of which include production of 24 nt sRNAs derived from these regions36,73. To identify endosperm expressing sRNA loci, sRNA sequencing in osclsy3-kd was carried out (osclsy3-kd2). Nearly 90% of about 20 million reads/library mapped to the reference rice genome (Supplementary Table 1). The 23–24 nt sRNA profile was different in endosperm when compared to other tissues as observed previously46,47. Unlike flag leaf and embryo tissues, fewer sRNA loci contributed to the 23–24 nt sRNA pool in endosperm (Fig. 4A). A clear reduction in different size classes of sRNAs was observed in osclsy3-kd endosperm (Fig. 4B and Supplementary Fig. 8A). Principal component analysis (PCA) also indicated that 23–24 nt sRNAs were altered in osclsy3-kd and osnrpd1-kd endosperm tissues (Supplementary Fig. 8B). Further analysis indicated that the sRNAs dependent on OsCLSY3 were also reduced in osnrpd1-kd lines (Fig. 4C). Reduction in 23–24 nt sRNAs was observed chromosome-wide (Fig. 4D). These sRNAs were derived from genic regions as well as from class I (replicates in the genome by copy–paste mechanism) and class II TEs (replicates in genome by cut–paste mechanism) (Fig. 4E). Majority of the 23–24 nt sRNAs which had 5′A were drastically reduced in osclsy3-kd (Supplementary Fig. 8C). To identify CLSY3-dependent sRNA loci, we quantified number of loci present in PB1 and osclsy3-kd endosperm by ShortStack analysis (Supplementary Data 4 and 5). We observed that around 70% of sRNA loci lost 23–24 nt sRNAs in osclsy3-kd, when compared to PB1 in a bedtools based analysis and these were termed CLSY3-dependent sRNA loci (Fig. 4F and Supplementary Data 6). As expected, the CLSY3-dependent sRNA loci were also regulated by Pol IV (Fig. 4C). The bedtools intersect analysis showed the CLSY3-depenedent sRNAs predominantly originated from LTRs, different DNA TEs and genes (Supplementary Fig. 8D). Along with 23–24 nt sRNAs, there was also reduction in 21–22 nt sRNAs derived from the same loci (Supplementary Fig. 8E) as shown previously in Arabidopsis endosperm44. Since osclsy3-kd plants displayed a global reduction of sRNAs, we checked pools of 21–22 nt sRNAs mapped to miRNA encoding loci96,97, and they were not significantly reduced in osclsy3-kd (Fig. 4G). The 23–24 nt CLSY3-dependent sRNAs were derived from both class I (Gypsy, LINE1, SINE, LTR) and class II (MITE, En-Spm) TEs (Fig. 4H and Supplementary Fig. 8F).

A Percentage cumulative sum plots for sRNAs (23–24 nt) in rice tissues. The arrows indicate cumulative percentage of loci which generates 90% of all sRNAs. B Stacked barplot showing abundance of mapped sRNAs (20–25 nt) in PB1 and osclsy3-kd EN. C Boxplot indicating Pol IV dependency at CLSY3-dependent sRNA loci. D IGV screenshots showing levels of sRNAs (23–24 nt) across different chromosomes. E Stacked barplot showing sRNAs (23–24 nt) across class I and class II TEs. F Venn diagram showing sRNA loci across PB1 and osclsy3-kd EN. G Boxplot showing expression of miRNA transcripts in PB1 and osclsy3-kd EN. H Boxplots showing expression of sRNAs derived from class I (bottom) and class II (top) TEs in PB1 and osclsy3-kd EN. I IGV screenshots showing sRNA (sRNA-seq) and CHH methylation (whole-genome methylome) status of hypomethylated loci in PB1 and osclsy3-kd EN. J IGV screenshots showing redistribution of CHH methylation in CLSY3-dependent sRNA loci in PB1 and osclsy3-kd EN. In (C), (G) and (H), boxes show median values and interquartile range. Whiskers show minimum and maximum values excluding outliers. Comparisons were made with two-sided Wilcoxon test (P < 0.01 was considered significant). Source data are provided as a Source Data file.
In order to further understand the role of OsCLSY3 in genome-wide DNA methylation through sRNAs, we performed whole-genome BS sequencing (Supplementary Table 1). The level of DNA methylation in EN tissues was at much lower levels than other reproductive tissues as observed previously in model plants including Arabidopsis98,99. However, a clear reduction in DNA methylation was observed in several loci overlapping with CLSY3-dependent sRNAs (Fig. 4I and Supplementary Fig. 9A). We analyzed DNA methylation at 21,653 CLSY3-dependent loci that lost sRNAs and 2620 CLSY3-dependent loci that gained sRNAs (Supplementary Data 6). After removing non-numerical values, among the 13,259 CLSY3-dependent loci that lost sRNAs, DNA was hypermethylated in 7257 loci and hypomethylated in 4953 loci. Among the 1800 CLSY3-dependent sRNA gained loci, 898 loci showed increased DNA methylation, while 723 loci showed decrease in DNA methylation (Supplementary Data 7). In all these loci, reduction was largely in CHH context. However, as observed previously, there was a clear hypermethylation in the adjacent cytosines next to CLSY3-dependent loci in osclsy3-kd lines (Fig. 4J and Supplementary Fig. 9B). As indicated previously73,74, this hypermethylation was largely over CHH contexts (Supplementary Fig. 9C–E). These are likely due to the redundancy between CLSY members, or overlap and/or competition with other epigenetic information. However, a clear reduction in DNA methylation across 4953 loci that also lost CLSY3-dependent sRNAs in osclsy3-kd indicated regulation of RdDM by OsCLSY3 in endosperm tissues (Supplementary Fig. 9F–H). These results collectively suggest that OsCLSY3 is a major regulator of sRNAs in endosperm and CLSY3-dependent sRNAs are crucial for silencing of specific classes of TEs and repeats through RdDM.
OsCLSY3 preferentially binds to specific motifs in TE-rich regions
In order to understand the role of OsCLSY3 in silencing specific regions in the genome, we performed chromatin immunoprecipitation followed by sequencing (ChIP-seq) in panicle tissue (Supplementary Table 1). We generated transgenic rice lines expressing OsCLSY3 under its own promoter with two different epitope tags (GFP and 2Xmyc) at C-terminal (Supplementary Figs. 6B and 10A). We obtained 8864 peaks with GFP-tagged CLSY3 and 7958 peaks with 2Xmyc tagged CLSY3, respectively (Supplementary Fig. 10B and Supplementary Data 8). There were more than 90% peaks (7115 peaks) overlapping between ChIP-seq datasets with two different tags, indicating a clear enrichment for specific binding regions (Fig. 5A). These 7115 peaks were broad peaks as observed in Arabidopsis CLSY members (Fig. 5B). Among the peaks bound by OsCLSY3, there was a clear enrichment for LTR TE regions (Fig. 5C and Supplementary Fig. 10C). This is striking because thousands of regions are bound by a single CLSY in rice with a preference for TEs (Fig. 5D), while Arabidopsis CLSY proteins seem to be binding to fewer regions in hundreds without a clear preference for genomic features37. In Arabidopsis, CLSY3 bound to a short AT rich motif in <200 loci37. Interestingly, OsCLSY3 bound to specific regions that are enriched with two 29-nt long sequence motifs (Fig. 5E). Rice CLSY3 also had minor preference for a few other TE classes but the number of overlapping peaks were <300, and in those loci CLSY3 bound to longer sequence motifs (Supplementary Fig. 10D). Among 7115 CLSY3-bound peaks, 1398 peaks clearly overlapped with CLSY3-dependent sRNA loci. In these 1398 peaks, sRNA abundance was clearly reduced and the regions were adjacent to, or overlapped with CLSY3-dependent sRNA loci (Fig. 5F, G).

A Venn diagram showing overlap of OsCLSY3 ChIP peaks upon pull-down with two tags. B Metaplots showing OsCLSY3 (7115) and shuffled peaks over enriched regions. Input—sheared DNA from non-transgenic plant. Replicates are almost merged. C Enrichment of OsCLSY3 ChIP-seq peaks across different genomic features. D IGV screenshots showing enrichment of OsCLSY3 over multiple LTR TEs. E Sequence specificity in CLSY3 binding. F IGV screenshots showing overlap of CLSY3-dependent sRNAs in EN over OsCLSY3 binding sites. G Boxplot showing sRNA status at OsCLSY3 ChIP-seq peaks that overlap with CLSY3-dependent sRNA loci (3 kb extended both sides, N-1398). Boxes showing median values and interquartile range. Whiskers show minimum and maximum values, excluding outliers. Comparisons were made with two-sided Wilcoxon test (P < 0.01 was considered significant). Source data are provided as a Source Data file.
OsCLSY3 regulates expression of endosperm-specific sRNAs (siren RNAs) and adjoining genes in endosperm
Since in Arabidopsis ovules, CLSY3 and CLSY4 generate sRNAs from siren loci37,38, we investigated if OsCLSY3 is also involved in the regulation of siren loci (Supplementary Data 9). We measured 23–24 nt sRNAs derived from known rice siren loci52, and found sRNAs from these loci were reduced in osnrpd1-kd endosperm tissues (Fig. 6A and Supplementary Fig. 11A, B). We also found the same pool of siren loci derived sRNAs were reduced in osclsy3-kd endosperm tissues (Fig. 6B–D). As observed previously, siren loci were less expressed in embryo and flag leaf tissues47,52. The sRNAs from siren loci were also reduced in osnrpd1-kd endosperm (Fig. 6D and Supplementary Fig. 11B). According to expression levels of siren loci in osclsy3-kd endosperm, we further categorized siren loci into three groups (Supplementary Data 8). Among all the categories, siren loci derived sRNAs were downregulated in osnrpd1-kd (Supplementary Fig. 11C, D). We found among 797 siren loci in rice, 316 were majorly downregulated in osclsy3-kd and these were likely regulated by OsCLSY3 (category-I). Category-2 loci of about 464 were moderately affected in osclsy3-kd. We also got 17 loci which were upregulated in osclsy3-kd (category-3) (Supplementary Data 9 and Supplementary Fig. 12A, B). In a sRNA northern blot analysis, we found siren sRNAs were reduced in osclsy3-kd and osnrpd1-kd endosperm tissues, indicating a direct role of OsCLSY3 in their accumulation (Fig. 6F). Since siren loci derived sRNAs have the potential to guide DNA methylation at cis or trans loci46,47,48,49, we performed a targeted BS-PCR to understand the impact of DNA methylation upon osclsy3-kd and osnrpd1-kd. The observed reduction of methylation was comparatively more drastic in nrpd1-kd than in osclsy3-kd endosperm, probably due to compensatory effect, likely involving other CLSYs as discussed above (Supplementary Fig. 11E). While we did not find global reduction in DNA methylation when all siren loci were considered (Supplementary Fig. 12C), we observed clear hypomethylation in multiple siren loci (Fig. 6E and Supplementary Fig. 12D, E). Among 316 category-1 siren loci where CLSY3-dependent sRNAs were clearly reduced, DNA hypomethylation was observed in 94 loci, whereas hypermethylation was observed in 134 loci in osclsy3-kd lines. In category-2 siren loci, DNA hypermethylation was observed in 176 loci and hypomethylation was observed in 146 loci. In category-3, among 17 loci, 7 loci were hypomethylated and 6 loci got hypermethylation. Lack of complete correlation between levels of CLSY3-dependent siren RNAs and DNA methylation indicated a possible role for other size classes of sRNAs regulating DNA methylation. A crosstalk with DNA demethylation pathways that might contribute to altered DNA methylation in many loci is also a distinct possibility in osclsy3-kd lines. This is also observed previously in Arabidopsis37. We also observed hypermethylation of DNA methylation in multiple siren loci as discussed earlier73,74. These indicate a partially redundant role for OsCLSYs in regulating siren loci in endosperm tissues. These results collectively suggested that OsCLSY3 regulated a major portion of sRNAs production from the siren loci and contributed to their DNA methylation status.

A Boxplot showing sRNAs (23–24 nt) derived from siren loci in osnrpd1-kd EN (797 loci). ASINH converted RPKM values were used for boxplot. B Boxplot representing status of sRNAs (23–24 nt) from siren loci in osclsy3-kd EN. C Heatmap showing siren sRNAs in osclsy3-kd EN. Row Z-score was plotted. D IGV screenshot showing expression of siren loci in osclsy3-kd and osnrpd1-kd EN. VL vegetative leaf. E IGV screenshots showing expression of siren sRNAs and DNA methylation in osclsy3-kd EN. F Northern blot showing siren sRNAs in RdDM mutants. G Volcano plot representing all DEGs of osclsy3-kd. H Boxplot showing sRNA levels near DEGs. I Correlation plot showing accumulation of CLSY3-dependent sRNAs and mRNA expression of adjacent genes (RPKM). J IGV screenshots showing expression of two selected DEGs and levels of adjacent sRNA loci in osclsy3-kd EN (violet lines mark DNA hypomethylated regions). K Heatmap showing expression of genes adjacent to siren loci (510) and seed development (102) genes. Row Z-score was plotted. In (A), (B) and (H), boxes show median values and interquartile range. Whiskers shows minimum and maximum values, excluding outliers. Comparisons were made with two-sided Wilcoxon test (P < 0.01 was considered significant). Source data are provided as a Source Data file.
Although endosperm development-related phenotypes were not reported in Arabidopsis clsy mutants, we found smaller sized endosperm in osclsy3-kd seeds when compared to PB1 (Fig. 3D). To understand the mechanism that might be operating, a transcriptomic analysis was carried out in osclsy3-kd endosperm tissues (Supplementary Table 1). We shortlisted 2870 differentially expressed genes (DEGs) with log2 1.5-fold upregulation or downregulation (Fig. 6G and Supplementary Data 10, 11). To understand the correlation between levels of CLSY3-dependent sRNAs and DEGs, we compared 23–24 nt sRNA levels ±2 kb windows near DEGs and observed a reduction of sRNAs near the DEGs (Fig. 6H). A strong antagonistic correlation (Pearson’s correlation coefficient: −0.96) between CLSY3-dependent sRNAs and gene expression was observed among thousands of loci, indicating that CLSY3-dependent sRNAs might be regulating their expression (Fig. 6I and Supplementary Data 12). This observation suggested that significant number of the DEGs were regulated by OsCLSY3 in endosperm. Around 24 genes which were upregulated in osclsy3-kd, clearly overlapped with CLSY3-dependent sRNA loci and their DNA methylation levels were reduced in osclsy3-kd (Fig. 6J and Supplementary Data 13). It is possible that CLSY3 also plays a significant role in the expression of the remaining DEGs through less abundant sRNAs or due to altered development of the plants. We also found mis-expression of 58 well-known endosperm development and yield-related genes100,101 having siren loci adjacent to them (±2 kb) in osclsy3-kd lines (Fig. 6K). Around 1258 genes which located next to CLSY3-dependent sRNA loci were significantly mis-expressed in osclsy3-kd (Supplementary Fig. 13A and Supplementary Data 13). Around 12 MADS box genes previously identified as crucial regulators of endosperm development21,102,103,104,105,106 were mis-expressed in osclsy3-kd (Supplementary Fig. 13A). As a control, we analyzed few genes that overlapped with CLSY3-independent sRNA loci. As expected, we observed these genes were unaltered in osclsy3-kd (Supplementary Fig. 13B). In addition, many well-known seed development-related genes such as OsMKKK10, OsFAD2, OsTAR1, OsNF-YB1 known for their role in seed development related to signaling and fat/carbohydrate metabolism were mis-expressed in osclsy3-kd endosperm (Supplementary Fig. 13C). Among these 58 genes were overlapped with CLSY3-dependent sRNA loci indicating a direct role of OsCLSY3 in their regulation (Supplementary Data 13). These results collectively demonstrate that CLSY3-dependent sRNAs direct endosperm development by regulating expression of multiple development, hormone and metabolism-related genes.
OsCLSY3 regulates expression of imprinted genes through imprinted sRNA loci
It was observed that RdDM dependent sRNAs regulated expression of imprinted genes in Arabidopsis43,44, however, it was not known if CLSYs played any role in genomic imprinting. In osclsy3-kd transcriptome, we observed mis-expression of well-established imprinted genes, suggesting a possible role for OsCLSY3 in imprinting (Fig. 7A, B). The detailed analysis further showed 265 genes were upregulated and 44 genes were downregulated in osclsy3-kd endosperm (Supplementary Fig. 14A and Supplementary Data 14). The majority of those imprinted genes were also a part of the DEGs (Fig. 6G). Quantification of total sRNAs derived from gene bodies and 5′ and 3′ regions of 2 kb of all imprinted genes showed a drastic reduction in osclsy3-kd (Fig. 7C). However, many imprinted genes which mis-expressed in osclsy3-kd, did not overlap with CLSY3-dependent sRNA loci. To understand direct role of sRNAs in regulation of imprinted genes, we further focused on the imprinted genes which were proximal to CLSY3-dependent sRNA loci. In Arabidopsis, imprinted sRNAs regulated expression of proximal imprinted genes44. In rice, imprinted sRNA loci were also found adjacent to many imprinted genes, and it was hypothesized previously that these sRNAs might be regulating imprinted genes14,27,47,52. We found among the 15 maternally expressed sRNA loci, around 9 loci were downregulated, whereas all 16 paternally expressed sRNA loci were downregulated in osclsy3-kd endosperm (Fig. 7D). As expected, imprinted sRNA loci were also downregulated in osnrpd1-kd similar to osclsy3-kd (Supplementary Fig. 14B, C and Supplementary Data 15). Interestingly, all the 20 imprinted genes identified in rice that have antagonistic expression pattern with imprinted sRNAs, showed upregulation in osclsy3-kd lines (Fig. 7E and Supplementary Data 16). Out of those, nine imprinted genes clearly showed significant upregulation in osclsy3-kd when compared to PB1, correlating with reduced DNA methylation in many of them (Fig. 7F). While DNA methylation across all imprinted genes was largely unchanged (Supplementary Fig. 14D), in the above examples, there was a clear reduction in DNA methylation. Measurement of DNA methylation level by targeted BS-PCR indicated a reduction of methylation in osclsy3-kd as well as in osnrpd1-kd endosperm (Supplementary Fig. 15A). In agreement with this, DNA methylation by quantitative chop-qPCR showed decrease in DNA methylation in osclsy3-kd in selected imprinted genes (Fig. 7F). In this experiment, OsActin and another control locus (Chr5:2048475–2049215) did not show any significant change as expected (Fig. 7F and Supplementary Fig. 15C). We further verified our observation by whole-genome methylation analysis. The majorly of the loci which were used in targeted BS-PCR and quantitative chop-qPCR showed decreased DNA methylation in osclsy3-kd (Fig. 7G and Supplementary Fig. 15B, C). In case of five imprinted genes, we observed a decrease in DNA methylation in CHH context (Fig. 7H and Supplementary Figs. 15D, 16A). However, for four imprinted genes, we observed either redistribution or increase DNA methylation in CHH context (Supplementary Fig. 16B). These results collectively suggested that OsCLSY3 mediated sRNA-directed DNA methylation contributed to the expression of some imprinted genes.

A Heatmap showing expression of all imprinted genes in osclsy3-kd EN (635 genes). B Boxplot representing expression of imprinted genes in osclsy3-kd EN. ASINH converted RPKM values were used for boxplot. C Boxplots showing abundance of CLSY3-dependent sRNAs in the promoter, terminator and gene body of imprinted genes. D Heatmaps showing expression of sRNAs in imprinted sRNA loci. Row Z-score was plotted. E Heatmap representing expression of imprinted genes proximal to imprinted sRNA loci. F Chop-qPCR showing methylation levels in six imprinted genes. Data represent means ± SE, n = 3. Experiments repeated twice with consistent results. G IGV screenshots showing the sRNA and DNA methylation status of two significantly changed imprinted sRNA loci used in (F). H IGV screenshots showing DNA methylation, sRNA and mRNA levels of two selected imprinted genes (violet lines mark hypomethylated regions). In (B) and (C), boxes show median values and interquartile range. Whiskers shows minimum and maximum values, excluding outliers. Comparisons were made with two-sided Wilcoxon test (P < 0.01 was considered significant). Source data are provided as a Source Data file.
Discussion
OsCLSY3 is essential for endosperm development in rice
Endosperm is unusual because it is a triploid tissue with more accessible chromatin where TEs and gene regulation is vastly different from other tissues including embryo8,107. Development of endosperm is unique and regulated by multiple genes that are further regulated by epigenetic pathways9,30. Only in endosperm tissue, genomic imprinting, a key process for its development is observed. In agreement with all these, several DNA methylation and histone modification pathway players such as—FIE, MEA, NRPD in Arabidopsis and OsFIE1, OsEMF2a, ZmFIE1 in monocots, were identified as crucial players for endosperm development and imprinting21,23,28,29,66,67,69,108. Interestingly, many of these genes were also found imprinted in endosperm where they were preferably expressed. Along with DNA methylation, DNA demethylation pathway players, such as demethylase DME in Arabidopsis and ROS1a in rice central cells, were also implicated in imprinting across plants15,109,110. sRNAs are crucial players in plant epigenetics and their expression drastically increases in reproductive tissues81,111. Unlike Arabidopsis, other dicots like B. rapa and Capsella RdDM mutants showed pollen and seed abnormalities40,92,112. In rice too, RdDM pathway genes such as Pol IV (OsNRPD1), OsRDR2/FEM1, Pol V (OsNRPE1) regulate important agronomic traits such as panicle development, seed setting, pollen development, etc.63,64,81,85,90. The guides for sRNA production in plants are a family of chromatin remodelers named CLSYs that regulate DNA methylation in a tissue- and locus-specific manner36,37.
In rice, CLSYs are also showed tissue-specific expression. We and a previous report found OsCLSY4/FEM2 expressed ubiquitously74 indicating it is a major CLSY, while OsCLSY3 in indica line PB1 majorly expressed in endosperm and specific-reproductive tissues. We also found endosperm-related abnormalities in osclsy3-kd as well as in osclsy4-kd lines in indica rice. This indicates OsCLSY4 is also important for rice endosperm and seed development. It would be interesting to identify how redundant and non-redundant functions of these CLSY proteins promote endosperm development, its timing as well as genomic imprinting. Since single mutants in japonica rice in fel1 were not studied74, it is difficult to understand if there is a difference in expression patterns and functions of OsCLSY members between indica and japonica lines.
There are multiple reports suggesting how CLSY can show natural variation between closely related rice lines and between Arabidopsis ecotypes. In Arabidopsis, using genome-wide association study, a natural genetic variation of CLSY1 having D538E amino acid change was found associated with reduced lateral roots under the low K+ conditions in specific ecotypes113. It has been elegantly proposed that such variations in epigenetic players including CLSYs might help in adaptation to diverse environmental conditions38. In agreement with these, variable phenotypes were also reported in a few RdDM pathway mutants between japonica rice varieties85,90.
Using genetic, genomic and molecular approaches, we identified OsCLSY3, a previously unannotated, imprinted and tissue-specifically expressed gene as a critical player in these processes. An ortholog of this gene is imprinted and majorly expressed in maize endosperm, indicating that monocot endosperms that are preserved during seed development, also have atypical regulatory layers. Unlike Arabidopsis, OsCLSY3 bound to thousands of specific sequence motifs of 20–50 nt length predominantly from LTR TE regions. Among 7115 CLSY3 peaks, only 1398 peaks were overlapped with CLSY3-dependent sRNA loci, probably because of different tissues used for analysis. The strong phenotypes observed in the seeds of OsCLSY3 mis-expression lines clearly indicate the importance of OsCLSY3 in seed development (Fig. 3D, H and Supplementary Fig. 5H). Our results conclusively suggest that OsCLSY3 is an upstream regulator, essential for reproduction and endosperm development (Fig. 8).

A Tissue-preferred expression of OsCLSY3 regulated by the RdDM pathway through MITE TEs present in its promoter. In vegetative tissues, OsCLSY4 recruits Pol IV into OsCLSY3 promoter and methylates MITE TEs via 24 nt sRNAs (left panel). In endosperm, OsROS1a induces demethylation of the maternal alleles of OsCLSY3 promoter leading to its expression (right panel). Here, OsCLSY4 is not very active to induce silencing in TEs. B OsCLSY3 regulated expression of TE and repeat-derived sRNAs, siren loci and imprinted sRNAs in endosperm. Those sRNAs directed DNA methylation and induced associated epigenetic regulations that are crucial for proper development. CLSY3-dependent sRNAs regulated expression of imprinted and seed development-related genes, thereby contributing to endosperm size, cellularization and its quality. Roles of all indicated genes on OsCLSY3-related regulation, except those with question marks, were delineated in this study. Filled and unfilled lollypops indicate methylated and unmethylated DNA, respectively.
The RdDM pathway regulates tissue-specific expression of OsCLSY3 via TEs
One of the most striking features of a few epigenetic regulators of imprinting operating in endosperm is their imprinted nature. Imprinting and expression of such players were tightly regulated by epigenetic pathways, usually involving TEs or repeats. To maintain the genome integrity, plants were thought to have evolved silencing involving various epigenetic layers114, including silencing mechanisms that have the ability to spread or influence neighboring genes65. Several such examples exist, for example, in Arabidopsis, reproductive-tissue-specific and paternally imprinted FWA gene was regulated by SINE retroelements located in its promoter. Here, DNA methylation marks silence the gene in vegetative tissue and paternal allele in endosperm16,115. In Arabidopsis, a balance between DNA methylation and demethylation was regulated by a TE located in the 5′ flanking sequences of demethylase gene ROS1116. Also, DNA binding sites of imprinted gene PHE1 have TEs of RC/Helitron type117. The paternally imprinted gene ALLANTOINASE (ALN) is a negative regulator of seed dormancy in Arabidopsis, and it is regulated by a DNA transposon named POGO present in its promoter4. In rice too, a CACTA DNA transposon derived miRNA820 negatively regulated DNA methyltransferase OsDRM2 by PTGS118. Rice tillering is regulated by the RdDM pathway through MITEs elements present at the OsmiRNA156j and D14 gene119. A stowaway-like MITE embedded in the 3′ UTR of Ghd2 (a CONSTANS-like gene) regulates its expression through RdDM120. A PcG complex MEG gene MEA in Arabidopsis is itself silenced by PcG complex in vegetative stages121. All these studies suggest a strong feedback loop for effective control of a critical process such as endosperm development, acting as a negative deterrent. Our finding of imprinted nature of OsCLSY3 through two tandem MITE TEs, a Ditto element and a Tourist element, supports this idea. Those TEs were methylated in vegetative tissues by the RdDM pathway where another CLSY namely OsCLSY4 recruited Pol IV to regulate its expression. The presence of MITEs as regulatory elements of a crucial gene such as OsCLSY3 is not entirely surprising. After all, MITEs are the largest family of TEs in the rice genome, located close to more than 23,000 genes (nearly 58% of all genes)14. Most of these MITEs are targets of RdDM and are methylated in most tissues99. In endosperm, methylation at TEs occupying the OsCLSY3 promoter was likely abolished due to ROS1a-dependent hypomethylation, similar to other genes under its control110. The expression level of OsCLSY3 appears to be vital in determining endosperm size, its morphology and functions. OE of OsCLSY3 produced a larger endosperm with chalkiness (Supplementary Fig. 6J) implicating its role in cellularization. The chalkiness is an indication of starch and protein synthesis and storage defect in endosperm, suggesting that OsCLSY3 is important for maintaining nutrient quality. In monocot seeds, endosperm plays an important role in seed germination as seen in mis-expression lines of OsCLSY3, where seed germination rate and initial root development were affected (Supplementary Fig. 6F, H).
OsCLSY3 regulates siren loci
In endosperm of flowering plants, fewer sRNA loci closer to genes contribute a large number of sRNAs in endosperm tissues46,47. Such sRNAs, named siren, are a conserved feature of Angiosperms, but they are not identical sequence-wise45,46,47,48. The siren loci derived sRNAs direct DNA methylation of genes by RdDM46. Recently, studies showed that siren sRNAs can also methylate protein-coding genes in trans48. In our study, we showed OsCLSY3 is the major regulator of siren loci in rice. We also observed siren loci derived sRNAs guide DNA methylation which regulates expression of proximal genes (Fig. 6J). The involvement of OsCLSY3, likely with other CLSYs, in the generation of siren loci is in complete agreement with its larger role as a key regulator of endosperm development. Our whole-genome DNA methylation analysis also suggested redundancy in the functions of CLSYs as observed previously in Arabidopsis and recently in rice38,73,74.
OsCLSY3 regulates expression of many imprinted genes and seed development-related genes
In Arabidopsis, rice and maize, previous studies indicated that imprinted sRNA loci were located near the silenced allele of few imprinted genes14,27,40,44,45,47,52. Due to these observations, it was suspected that imprinted sRNAs might be regulating expression of imprinted genes44. Same study also documented perturbation of maternal and paternal sRNA ratios in endosperm that can shift the mRNA ratio generated from parental genomes44,122. Unlike Arabidopsis, where Pol IV appears to be a PEG, Pol IV is not an imprinted gene in rice endosperm probably because OsCLSY3 is a MEG acting as an upstream player. We observed that OsCLSY3 regulates expression of many imprinted sRNA loci as well as some adjoining imprinted genes (Fig. 7D, E). In rice, OsCLSY3 might be regulating transcription of genes via sRNAs as observed in the case of nrpd1 mutant in Arabidopsis.
In several examples, CLSY3-dependent sRNAs regulated genes via DNA methylation. Several genes also had expression changes independent of DNA methylation or sRNAs. Such variations might be due to alterations in other epigenetics marks, altered chromatin state or due to mis-regulation of regulatory modules. We observed several auxin, brassinosteroid signaling pathway genes such as TUD1, OsDWARF/BRD1, BRD2, OsGSK3 which were well documented as important players in grain size123,124,125,126, were mis-expressed in osclsy3-kd lines. Grain development-related MAPK pathway genes127,128 and transcription factors (TF) such as OsMADS6, OsMADS29, OsWRKY53 which were known for starch synthesis, proper cellularization, cell size regulation and programmed cell death also mis-regulated in osclsy3-kd lines103,129,130,131,132 (Supplementary Data 13). Multiple starch, protein, lipid metabolism-related genes such as OsSGL, RSS, OsSWEET11, OsFAD2, OsLHT1, OsGAD3 that were important for synthesis and their transport, were mis-regulated in osclsy3-kd, further indicating a critical role of OsCLSY3133,134,135,136,137. Since size, quality and nutritive aspects of rice endosperm, a major staple food for the majority, is under OsCLSY3 control, this gene can be an important candidate to improve nutritional benefits of rice grains.
Methods
Rice transformation and plant growth
For generating rice transgenic plants, Agrobacterium-mediated transformation was performed as described previously138,139. About 21-day-old embryogenic calli were used from indica variety rice (Oryza sativa indica) PB1. Calli were infected with freshly grown Agrobacterium tumefaciens strain LBA4404 (pSB1), where pSB1 carries extra copies of vir genes, carrying the designated binary plasmid. The regenerated transgenic plants were maintained in a growth chamber at 23 °C with 16h/8h light/dark cycle at 70% RH. The plants were then transferred to a greenhouse.
Vector design and construction
To generate OE plants, the OsCLSY3 gene (Os02g0650800) (5.4 kb) was amplified using appropriate primers (Supplementary Table 4) from genomic DNA. The full-length gene was cloned under maize ubiquitin promoter (P: ZmUbi) in pCAMBIA1300 with hygromycin phosphotransferase (hph) gene as a selection marker. The 3xFLAG epitope tag was added to N-terminal of OsCLSY3. Two osclsy3-kd lines were generated using amiR strategy. The amiRs were designed using the WMD3 web tool87,88, with the stringent criteria for robust amiR generation as described89. The amiR-precursors were synthesized by GeneArt (Thermo Fisher). The amiR sequences were cloned into pCAMBIA1300 under P: ZmUbi and into pRT100 vector under CaMV 35S (P:35S) promoter. For constructing a double amiR containing binary vector, an amiR cassette containing pRT100 was cloned into a binary vector that already had P: ZmUbi driven amiR. The KO lines were generated using CRISPR-Cas9 strategy. Guide RNA was designed using CRISPR-PLANT (http://omap.org/crispr/index.html). Guide RNA targeting a unique region of OsCLSY3 gene at first exon (5′-GUCUUCCTCCCGGCUCUCCU-3′) was cloned into pRGEB32 vector (Addgene #63142)138. All the constructs were verified by restriction analysis and/or Sanger sequencing. Constructs were mobilized into Agrobacterium strain LBA4404 (pSB1), and the mobilization was verified by PCR analysis.
Genetic crosses
To understand the imprinting status of the OsCLSY3 gene, two rice varieties—WP and PB1 were crossed. The hybrid was confirmed by Sanger sequencing of known SNP containing regions. Endosperm from the hybrid plant was collected and dissected into embryo and endosperm. Embryo region was used for the DNA isolation and endosperm was used for RNA isolation. After validation of the genotype, cDNA was synthesized from 1.5 µg of total RNA using Thermo Scientific RevertAid First Strand cDNA Synthesis Kit as per manufacturer’s instructions, and regions of interest were amplified. The amplicons were purified and deep sequenced on NovaSeq 6000 (2 × 100 bp mode). The obtained reads were adapter trimmed using cutadapt139 and aligned to genes by using CRISPResso2140.
5-Aza-2′-deoxycytidine (AZA) treatment
After 3 days of germination in half-strength MS media, seedlings were placed in MS media containing 35 and 70 mg/l Aza27. DMSO was used as control. After 7 days, seedlings were collected for RNA extraction or GUS staining.
Phenotyping of transgenic plants
Phenotypes of transgenic plants, such as plant height, leaf length, panicle length, were measured using (n > 6) mature plants grown for 4 months along with appropriate control plants. Other details including replicates are mentioned in the figure legends. Images of rice spikelets and seeds from PB1, osclsy3-kd and OE lines were obtained using Lecia S8APO stereomicroscope and Nikon camera. For statistical analysis, paired t-test was used.
RNA extraction and RT-qPCR
Total RNA extraction from rice tissues was performed using TRIzol® Reagent (Invitrogen) as per manufacturer instructions. For endosperm tissue, RNA isolation was performed as described earlier141. RT-qPCR was performed for expression of CLSYs and other RdDM pathway-related genes. First-strand cDNA was synthesized from 1.5 µg of total RNA using Thermo Scientific RevertAid First Strand cDNA Synthesis Kit as per manufacturer instructions, and the qPCR was carried out with Solis Biodyne—5x HOT Firepol Evagreen qPCR Master Mix. OsActin (Os03g0718100) and Glyceraldehyde-3-phosphate dehydrogenase (OsGAPDH) (Os04g0486600) were used as internal control. RT-qPCRs were performed at least three times using the BioRad CFX system. Primers used for expression analysis are listed in Supplementary Table 4. The data were plotted using the GraphPad Prism software (version 8).
sRNA northern hybridization
About 8–15 µg of total RNA was used for sRNA northern as described earlier142,143. Hybond N+ (GE Healthcare) membranes were stripped and used for multiple hybridizations. Hybridization was done at 35 °C. The blot was exposed to a phosphor imager screen and re-probed with controls. Typhoon scanner (GE healthcare) was used to detect the hybridization signal. Details of the DNA oligonucleotide probes are provided in Supplementary Table 4.
Southern hybridization
Junction-fragment southern analysis was performed as described earlier144,145. Total DNA was extracted from equally grown control and transgenic plants using CTAB method146. For the probe, hph gene was amplified from binary plasmid (~1 kb). Probe was labeled with [α-32P] dCTP (BRIT, India) using the Rediprime random DNA labeling system (GE Healthcare). The prehybridization, hybridization and subsequent washes were performed at 65 °C. Details of the probe that was used in this study are provided in Supplementary Table 4.
DNA methylation analyses
Total DNA was isolated from endosperm using CTAB method146. Equal amount of DNA was sheared to produce 350 bp fragments using ultrasonication (Covaris). Bisulfite conversion was performed using EZ DNA Methylation-Gold Kit as per manufacturer’s instructions. The libraries were constructed using the IDT xGen™ Methyl-Seq Lib Prep (Catalog no—10009860) as per manufacturer’s instructions and sequenced in NovaSeq 6000 (2 × 150 bp mode). The obtained reads were quality checked and trimmed using Trimmomatic147. The reads were aligned to IRGSP1.0 genome using Bismark aligner tool with default parameters148. DNA methylation status was extracted and coverage reports were generated using Bismark tools. The results were analyzed and plotted using the ViewBS methylation package149.
For the targeted BS sequencing, total DNA was isolated from different tissues using CTAB method. About 200–400 ng of DNA was treated using EZ DNA Methylation-Gold Kit (Zymo Research). The bisulfite treated DNA was used as the template for PCR. Amplification of targeted regions performed by JumpStart™ Taq DNA Polymerase (Sigma). The PCR products were deep sequenced in paired end mode (100 bp) on a Hiseq2500 platform. The obtained reads were quality checked and trimmed using cutadapt139 and aligned to create genome (target sites) using Bismark aligner tool with default parameters148. The obtained results are analyzed using methylation package ViewBS149. Primers used for analysis are provided in Supplementary Table 4.
sRNA library preparation and differential expression analyses
sRNA libraries were prepared as described previously81. The obtained reads were quality checked and trimmed by UEA sRNA Workbench150. The filtered sRNAs were classified into 21–22 nt and 23–24 nt reads, aligned using Bowtie151 -v 1 -m 100 -y -a --best --strata and sRNAs loci were identified using ShortStack152 with following parameters: --nohp --mmap f --mismatches 1 -mincov 2rpmm followed by bedtools intersect. For quantifying the sRNA expression from transposons, siren loci, etc., bedtools multicov153,154 was used to obtain raw abundance and then normalized to RPKM values. These values were plotted as boxplots using custom R scripts in ggplot2155. The Venn diagrams were generated by intervene online tools156. During the generation of Venn diagram, 48 CLSY3-dependent sRNA loci and 3 siren loci were merged by the tool, and this resulted in altered number of a few overlapping loci between features/samples while representing them in Venn diagram when compared to individual lists of loci. For siren loci, all loci were divided into three major categories. The category-1 listed the loci which were more than −1 log2 fold downregulated in osclsy3-kd. The category-2 listed the loci which were mis-regulated in the range of −1 to +1 log2 fold and caterogy-3 had more than +1 log2 fold upregulated loci.
RNA-seq and analyses
RNA-seq was performed using 20-day-old endosperm tissues. Poly(A) enrichment was done before library preparation. Library preparation was done with NEBNext® Ultra™ II Directional RNA Library Prep kit (E7765L) as per manufacturer’s instructions. The obtained libraries were sequenced in paired end mode (100 bp) on the Illumina Hiseq2500 platform. The obtained reads were adapter trimmed using Trimmomatic147. The reads were aligned to IRGSP1.0 genome using HISAT2157 with default parameters. Cufflinks were used to perform DEGs analyses and statistical testing158. Volcano plots were generated for DEGs using custom R scripts with the p value cutoff of <0.05 and absolute log2 (fold change) expression cutoff of more than 1.5. For quantifying the expression of genes, bedtools multicov153,154,159 was used to obtain raw abundance and then normalized to RPKM values. These values were plotted as boxplots and heatmaps using custom R scripts in ggplot2155.
AGO-IP data analyses
Previously published, AGO-IP datasets of various AGOs from rice were processed in the same way as the sRNA datasets, mapping to the IRGSP1.0 genome153,154.
ChIP-seq and its analyses
ChIP-seq was performed as described previously81,160. Briefly, around 1 g of panicles before anthesis were collected and crosslinked with 1% formaldehyde. Equal amount of isolated nuclei were lysed and sheared using ultrasonication (Covaris) until DNA fragments in 350 bp range were obtained. The sheared chromatin was incubated overnight with appropriate antibodies conjugated with protein G dynabeads (Thermo Fisher) at 4 °C. The beads were pre-bound with 4–5 µg of antibodies (Myc-Abcam ab9106 and GFP-Sigma G1544) before incubation with the sheard chromatin. After overnight incubation, washes, elution, de-crosslinking and purification were performed as described earlier81. Purified IP products were used for library preparation using NEBNext® Ultra™ II DNA Library Prep Kit using purification beads (NEB, E7103L) as per manufacturer’s protocol. The libraries were sequenced on NovaSeq 6000 platform (2 × 100 bp mode).
The adapter of the obtained reads was trimmed using cutadapt139 and aligned to IRGSP1.0 genome using Bowtie -v 3 -k 1 -y -a -- best --strata151. The PCR duplicates were removed before further analysis. The aligned bam files were compared with input DNA using deepTools161,162. The peak calling was performed using MACS2163. The coverage signal metaplots were plotted using plotprofile. The number of peaks overlapping with different genomic features were counted by bedtools intersect154.
Chop-qPCR
The chop-qPCR performed with 20 days endosperm DNA of osclsy3-kd and PB1 as described85. Total DNA was isolated by CTAB method and 500 ng DNA digested with NlaIII (10 K units/ml) restriction enzyme for 2 h. The digested DNA was used as template for qPCR. Equally treated DNA, without any enzyme was used as mock. Target loci as well as OsActin with NlaIII restriction site, was used as a control. qPCRs were performed at least twice using BioRad CFX system. Primers used for chop-qPCR are provided in Supplementary Table 4.
GO analysis
The GO analysis was performed using ShinyGO v0.75 platform164. Genes IDs were used from RAPDB. The biological processes with FDR cutoff of p value—0.05.
GUS histochemical assay
Histochemical assay was performed as described165. Plant tissues were collected in 50 mM phosphate buffer that contained 1% Triton-X 100 and incubated for 3 h at 37 °C. Explants were transferred into X-Gluc staining solution (1 mM) and vacuum infiltrated for 15 min. Next, the explants were incubated at 37 °C for 16 h. Further, explants were washed once with double distilled water followed by 70% ethanol and then transferred to acetone: methanol mix (1:3) in rotospin at 4 °C for 1 h to remove chlorophyll. After chlorophyll removal, explants were imaged by Nikon camera or Leica S8APO stereomicroscope.
Histochemistry of endosperm
Seeds were harvested (10 DAP) and fixed under vacuum in 4% PFA solution at 4 °C overnight and sliced to 60 μm sections by vibratome166. The slices were stained in 1% w/v iodine and 1% w/v potassium iodide. The extra stain was removed with 1X PBS wash and imaged under microscope (OLYMPUS BX43). For manual sectioning, 15 DAP endosperms (4% PFA fixed) were dehydrated through an ethanol series. The seeds were cut by razor blade and stained using 1% w/v iodine and 1% w/v potassium iodide. Sections were imaged under Leica S8APO stereomicroscope.
SEM imaging
For SEM imaging, rice spikelets were collected just before flowering and endosperms were collected (15 DAP). Samples were fixed in 16% formaldehyde, 25% glutaraldehyde and 0.2 M cacodylate buffer for 12–16 h. The samples were rinsed with double distilled water and dehydrated in series of ethanol and dried in critical point drying (CPD, Leica EM CPD300), gold coated, and the images were obtained using a Carl Zeiss scanning EM at an accelerating voltage of 2–4 kV as described before81,167. After CPD, endosperms were cut by a sharp razor blade.
Germination assay
The germination assay was performed as described previously168. Total 20 seeds of different genotypes (5 seeds/replicate) were imbibed on the wet filter paper in 5 cm diameter petri-plates. In all the plates, 4 ml of single distilled water was used to wet the filter papers and seeds were germinated at 25 °C for 6 days in the dark. Experiments were repeated twice independently.
Pollen staining
Pollen viability test was performed using I2–KI staining solution containing 0.2% (w/v) I2 and 2% (w/v) KI as described earlier167,169,170. Anthers from six spikelets of mature panicles were collected in 200 µl of solution 1 day before the fertilization. Pollen grains were released in the solution by mechanical shearing. After 10 min, viable pollen grains were counted under the bright-field microscope (OLYMPUS BX43). Round and dark blue stained pollen grains were considered as viable, while very light blue and distorted pollens were considered as non-viable81,171.
Statistics and reproducibility
No data were excluded from the analysis. All northern blots were repeated twice with biological replicate samples. All SEM images were performed at least twice with different samples. Statistical analysis was performed using two-tailed paired Student’s t-test or two-sided Wilcoxon test to determine differences between two groups. Statistical analyses were performed using excel and R studio. Details such as the number of replicates, the level of significance and sample sizes for RT-qPCR, RNA analysis, sequencing, phenotyping were mentioned in the corresponding figure legends, text and Supplementary Data. A full list of primers, probes, sequences and other details are available in Supplementary Tables.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the raw and processed sequencing data generated in this study have been submitted to NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) and are accessible under accession number GSE229961. Other publicly available data used in this study can be found under GSE180457, GSE138705, GSE130168, GSE158709, PRJNA758109, GSE130122, GSE138705, GSE18251, GSE20748. The reads were aligned to IRGSP1.0 genome. Source data are provided with this paper.
References
Pires, N. D. Seed evolution: parental conflicts in a multi-generational household. Biomol. Concepts 5, 71–86 (2014).
Baroux, C., Spillane, C. & Grossniklaus, U. Evolutionary origins of the endosperm in flowering plants. Genome Biol. 3, reviews1026 (2002).
Chahtane, H. et al. The plant pathogen Pseudomonas aeruginosa triggers a DELLA-dependent seed germination arrest in Arabidopsis. Elife 7, e37082 (2018).
Iwasaki, M., Hyvärinen, L., Piskurewicz, U. & Lopez-Molina, L. Non-canonical RNA-directed DNA methylation participates in maternal and environmental control of seed dormancy. Elife 8, e37434 (2019).
De Giorgi, J. et al. The Arabidopsis mature endosperm promotes seedling cuticle formation via release of sulfated peptides. Dev. Cell 56, 3066–3081.e5 (2021).
Iwasaki, M., Penfield, S. & Lopez-Molina, L. Parental and environmental control of seed dormancy in Arabidopsis thaliana. Annu. Rev. Plant Biol. 73, 355–378 (2022).
Doll, N. M. et al. A two-way molecular dialogue between embryo and endosperm is required for seed development. Science 367, 431–435 (2020).
Li, J. & Berger, F. Endosperm: food for humankind and fodder for scientific discoveries. New Phytol. 195, 290–305 (2012).
Gehring, M. Genomic imprinting: insights from plants. Annu. Rev. Genet. 47, 187–208 (2013).
Kiyosue, T. et al. Control of fertilization-independent endosperm development by the MEDEA polycomb gene in Arabidopsis. Proc. Natl. Acad. Sci. USA 96, 4186–4191 (1999).
Kradolfer, D., Wolff, P., Jiang, H., Siretskiy, A. & Köhler, C. An imprinted gene underlies postzygotic reproductive isolation in Arabidopsis thaliana. Dev. Cell 26, 525–535 (2013).
Gutierrez-Marcos, J. F., Pennington, P. D., Costa, L. M. & Dickinson, H. G. Imprinting in the endosperm: a possible role in preventing wide hybridization. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 1105–1111 (2003).
Kinoshita, T. Reproductive barrier and genomic imprinting in the endosperm of flowering plants. Genes Genet. Syst. 82, 177–186 (2007).
Yuan, J. et al. Both maternally and paternally imprinted genes regulate seed development in rice. New Phytol. 216, 373–387 (2017).
Gehring, M. et al. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation Cell. Cell 124, 495–506 (2006).
Kinoshita, T. et al. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation Science. Science 303, 521–523 (2004).
Batista, R. A. & Köhler, C. Genomic imprinting in plants-revisiting existing models. Genes Dev. 34, 24–36 (2020).
Ishikawa, R. et al. Rice interspecies hybrids show precocious or delayed developmental transitions in the endosperm without change to the rate of syncytial nuclear division. Plant J. 65, 798–806 (2011).
Huang, F. et al. Mutants in the imprinted PICKLE RELATED 2 gene suppress seed abortion of fertilization independent seed class mutants and paternal excess interploidy crosses in Arabidopsis. Plant J. 90, 383–395 (2017).
Zhu, H. et al. DNA demethylase ROS1 negatively regulates the imprinting of DOGL4 and seed dormancy in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 115, E9962–E9970 (2018).
Cheng, X. et al. The maternally expressed polycomb group gene OsEMF2a is essential for endosperm cellularization and imprinting in rice. Plant Commun. 2, 100092 (2021).
Luo, M. et al. Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 96, 296–301 (1999).
Grossniklaus, U., Vielle-Calzada, J. P., Hoeppner, M. A. & Gagliano, W. B. Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science 280, 446–450 (1998).
Ohad, N. et al. A mutation that allows endosperm development without fertilization. Proc. Natl. Acad. Sci. USA 93, 5319–5324 (1996).
Waters, A. J. et al. Comprehensive analysis of imprinted genes in maize reveals allelic variation for imprinting and limited conservation with other species. Proc. Natl. Acad. Sci. USA 110, 19639–19644 (2013).
Anderson, S. N., Zhou, P., Higgins, K., Brandvain, Y. & Springer, N. M. Widespread imprinting of transposable elements and variable genes in the maize endosperm. PLoS Genet. 17, e1009491 (2021).
Chen, C. et al. Characterization of imprinted genes in rice reveals conservation of regulation and imprinting with other plant species. Plant Physiol. 177, 1754–1771 (2018).
Luo, M., Platten, D., Chaudhury, A., Peacock, W. J. & Dennis, E. S. Expression, imprinting, and evolution of rice homologs of the polycomb group genes. Mol. Plant 2, 711–723 (2009).
Cheng, X. et al. Functional divergence of two duplicated fertilization independent endosperm genes in rice with respect to seed development. Plant J. 104, 124–137 (2020).
Köhler, C. & Weinhofer-Molisch, I. Mechanisms and evolution of genomic imprinting in plants. Heredity 105, 57–63 (2010).
Satyaki, P. R. V. & Gehring, M. DNA methylation and imprinting in plants: machinery and mechanisms. Crit. Rev. Biochem. Mol. Biol. 52, 163–175 (2017).
Stroud, H. et al. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 21, 64–72 (2014).
Smith, L. M. et al. An SNF2 protein associated with nuclear RNA silencing and the spread of a silencing signal between cells in Arabidopsis. Plant Cell 19, 1507–1521 (2007).
Zhang, H. et al. DTF1 is a core component of RNA-directed DNA methylation and may assist in the recruitment of Pol IV. Proc. Natl. Acad. Sci. USA 110, 8290–8295 (2013).
Law, J. A., Vashisht, A. A., Wohlschlegel, J. A. & Jacobsen, S. E. SHH1, a homeodomain protein required for DNA methylation, as well as RDR2, RDM4, and chromatin remodeling factors, associate with RNA polymerase IV. PLoS Genet. 7, e1002195 (2011).
Zhou, M., Palanca, A. M. S. & Law, J. A. Locus-specific control of the de novo DNA methylation pathway in Arabidopsis by the CLASSY family. Nat. Genet. 50, 865–873 (2018).
Zhou, M. et al. The CLASSY family controls tissue-specific DNA methylation patterns in Arabidopsis. Nat. Commun. 13, 244 (2022).
Martins, L. M. & Law, J. A. Moving targets: mechanisms regulating siRNA production and DNA methylation during plant development. Curr. Opin. Plant Biol. 75, 102435 (2023).
Vu, T. M. et al. RNA-directed DNA methylation regulates parental genomic imprinting at several loci in Arabidopsis. Development 140, 2953–2960 (2013).
Mosher, R. A. et al. Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature 460, 283–286 (2009).
Mosher, R. A. Maternal control of Pol IV-dependent siRNAs in Arabidopsis endosperm. New Phytol. 186, 358–364 (2010).
Lu, J., Zhang, C., Baulcombe, D. C. & Chen, Z. J. Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl. Acad. Sci. USA 109, 5529–5534 (2012).
Kirkbride, R. C. et al. Maternal small RNAs mediate spatial-temporal regulation of gene expression, imprinting, and seed development in Arabidopsis. Proc. Natl. Acad. Sci. USA 116, 2761–2766 (2019).
Erdmann, R. M., Satyaki, P. R. V., Klosinska, M. & Gehring, M. A small RNA pathway mediates allelic dosage in endosperm. Cell Rep. 21, 3364–3372 (2017).
Xin, M. et al. Dynamic parent-of-origin effects on small interfering RNA expression in the developing maize endosperm. BMC Plant Biol. 14, 192 (2014).
Grover, J. W. et al. Abundant expression of maternal siRNAs is a conserved feature of seed development. Proc. Natl. Acad. Sci. USA 117, 15305–15315 (2020).
Rodrigues, J. A. et al. Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc. Natl. Acad. Sci. USA 110, 7934–7939 (2013).
Burgess, D., Chow, H. T., Grover, J. W., Freeling, M. & Mosher, R. A. Ovule siRNAs methylate protein-coding genes in trans. Plant Cell 34, 3647–3664 (2022).
Long, J. et al. Nurse cell–derived small RNAs define paternal epigenetic inheritance in Arabidopsis. Science 373, eabh0556 (2021).
Olsen, O.-A. The modular control of cereal endosperm development. Trends Plant Sci. 25, 279–290 (2020).
Liu, J., Wu, M.-W. & Liu, C.-M. Cereal endosperms: development and storage product accumulation. Annu. Rev. Plant Biol. 73, 255–291 (2022).
Rodrigues, J. A. et al. Divergence among rice cultivars reveals roles for transposition and epimutation in ongoing evolution of genomic imprinting. Proc. Natl. Acad. Sci. USA 118, e2104445118 (2021).
Luo, M. et al. A genome-wide survey of imprinted genes in rice seeds reveals imprinting primarily occurs in the endosperm. PLoS Genet. 7, e1002125 (2011).
Waters, A. J. et al. Parent-of-origin effects on gene expression and DNA methylation in the maize endosperm. Plant Cell 23, 4221–4233 (2011).
Sato, Y. et al. A rice homeobox gene, OSH1, is expressed before organ differentiation in a specific region during early embryogenesis. Proc. Natl. Acad. Sci. USA 93, 8117–8122 (1996).
Zhiguo, E. et al. A group of nuclear factor Y transcription factors are sub-functionalized during endosperm development in monocots. J. Exp. Bot. 69, 2495–2510 (2018).
Li, H. et al. Isolation of five rice nonendosperm tissue-expressed promoters and evaluation of their activities in transgenic rice. Plant Biotechnol. J. 16, 1138–1147 (2018).
Hsieh, T.-F. et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 108, 1755–1762 (2011).
Le, B. H. et al. Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. USA 107, 8063–8070 (2010).
Mahto, A., Mathew, I. E. & Agarwal, P. Decoding the transcriptome of rice seed during development. in Advances in Seed Biology (InTech, 2017).
Chen, X. & Zhou, D.-X. Rice epigenomics and epigenetics: challenges and opportunities. Curr. Opin. Plant Biol. 16, 164–169 (2013).
Shi, J., Dong, A. & Shen, W.-H. Epigenetic regulation of rice flowering and reproduction. Front. Plant Sci. 5, 803 (2014).
Higo, A. et al. DNA methylation is reconfigured at the onset of reproduction in rice shoot apical meristem. Nat. Commun. 11, 4079 (2020).
Wang, L. et al. Reinforcement of CHH methylation through RNA-directed DNA methylation ensures sexual reproduction in rice. Plant Physiol. 188, 1189–1209 (2022).
Deng, X., Song, X., Wei, L., Liu, C. & Cao, X. Epigenetic regulation and epigenomic landscape in rice. Natl. Sci. Rev. 3, 309–327 (2016).
Köhler, C., Page, D. R., Gagliardini, V. & Grossniklaus, U. The Arabidopsis thaliana MEDEA polycomb group protein controls expression of PHERES1 by parental imprinting. Nat. Genet. 37, 28–30 (2005).
Zhang, L. et al. Identification and characterization of an epi-allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 24, 4407–4421 (2012).
Dhatt, B. K. et al. Allelic variation in rice fertilization independent endosperm 1 contributes to grain width under high night temperature stress. New Phytol. 229, 335–350 (2021).
Huang, X. et al. Imprinted gene OsFIE1 modulates rice seed development by influencing nutrient metabolism and modifying genome H3K27me3. Plant J. 87, 305–317 (2016).
Sun, Q. & Zhou, D.-X. Rice jmjC domain-containing gene JMJ706 encodes H3K9 demethylase required for floral organ development. Proc. Natl. Acad. Sci. USA 105, 13679–13684 (2008).
Liu, H. et al. OsmiR396d-regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiol. 165, 160–174 (2014).
Hu, Y. et al. Analysis of rice Snf2 family proteins and their potential roles in epigenetic regulation. Plant Physiol. Biochem. 70, 33–42 (2013).
Yang, D.-L. et al. Four putative SWI2/SNF2 chromatin remodelers have dual roles in regulating DNA methylation in Arabidopsis. Cell Discov. 4, 55 (2018).
Xu, D., Zeng, L., Wang, L. & Yang, D.-L. Rice requires a chromatin remodeler for polymerase IV-small interfering RNA production and genomic immunity. Plant Physiol. https://doi.org/10.1093/plphys/kiad624 (2023).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Sigrist, C. J. A. et al. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 3, 265–274 (2002).
Musselman, C. A. & Kutateladze, T. G. Characterization of functional disordered regions within chromatin-associated proteins. iScience 24, 102070 (2021).
Hale, C. J., Stonaker, J. L., Gross, S. M. & Hollick, J. B. A novel Snf2 protein maintains trans-generational regulatory states established by paramutation in maize. PLoS Biol. 5, e275 (2007).
Castano-Duque, L., Ghosal, S., Quilloy, F. A., Mitchell-Olds, T. & Dixit, S. An epigenetic pathway in rice connects genetic variation to anaerobic germination and seedling establishment. Plant Physiol. 186, 1042–1059 (2021).
Matzke, M. A., Kanno, T. & Matzke, A. J. M. RNA-directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plants. Annu. Rev. Plant Biol. 66, 243–267 (2015).
Hari Sundar G, V. et al. Plant polymerase IV sensitizes chromatin through histone modifications to preclude spread of silencing into protein-coding domains. Genome Res. https://doi.org/10.1101/gr.277353.122 (2023).
Swetha, C. et al. Major domestication-related phenotypes in Indica rice are due to loss of miRNA-mediated laccase silencing. Plant Cell 30, 2649–2662 (2018).
Zilberman, D., Cao, X. & Jacobsen, S. E. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299, 716–719 (2003).
Wu, L. et al. Rice MicroRNA effector complexes and targets. Plant Cell 21, 3421–3435 (2009).
Chakraborty, T., Trujillo, J. T., Kendall, T. & Mosher, R. A. A null allele of the pol IV second subunit impacts stature and reproductive development in Oryza sativa. Plant J. 111, 748–755 (2022).
Hu, D. et al. Multiplex CRISPR-Cas9 editing of DNA methyltransferases in rice uncovers a class of non-CG methylation specific for GC-rich regions. Plant Cell 33, 2950–2964 (2021).
Ossowski, S., Schwab, R. & Weigel, D. Gene silencing in plants using artificial microRNAs and other small RNAs. Plant J. 53, 674–690 (2008).
Warthmann, N., Chen, H., Ossowski, S., Weigel, D. & Hervé, P. Highly specific gene silencing by artificial miRNAs in rice. PLoS ONE 3, e1829 (2008).
Narjala, A., Nair, A., Tirumalai, V., Hari Sundar, G. V. & Shivaprasad, P. V. A conserved sequence signature is essential for robust plant miRNA biogenesis. Nucleic Acids Res. 48, 3103–3118 (2020).
Zheng, K. et al. The effect of RNA polymerase V on 24-nt siRNA accumulation depends on DNA methylation contexts and histone modifications in rice. Proc. Natl. Acad. Sci. USA 118, e2100709118 (2021).
Wolff, P., Jiang, H., Wang, G., Santos-González, J. & Köhler, C. Paternally expressed imprinted genes establish postzygotic hybridization barriers in Arabidopsis thaliana. Elife 4, e10074 (2015).
Wang, Z. et al. Polymerase IV plays a crucial role in pollen development in Capsella. Plant Cell 32, 950–966 (2020).
Wada, H. et al. Multiple strategies for heat adaptation to prevent chalkiness in the rice endosperm. J. Exp. Bot. 70, 1299–1311 (2019).
An, L. et al. Embryo-endosperm interaction and its agronomic relevance to rice quality. Front. Plant Sci. 11, 587641 (2020).
Yan, D., Duermeyer, L., Leoveanu, C. & Nambara, E. The functions of the endosperm during seed germination. Plant Cell Physiol. 55, 1521–1533 (2014).
Griffiths-Jones, S., Saini, H. K., van Dongen, S. & Enright, A. J. miRBase: tools for microRNA genomics. Nucleic Acids Res. 36, D154–D158 (2008).
Anushree, N. & Shivaprasad, P. V. Regulation of plant miRNA biogenesis. In Proc. Indian National Science Academy (A Phys. Sci.) 84, 439–453 (2018).
Hsieh, T.-F. et al. Genome-wide demethylation of Arabidopsis endosperm. Science 324, 1451–1454 (2009).
Zemach, A. et al. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 107, 18729–18734 (2010).
Li, P. et al. Genes and their molecular functions determining seed structure, components, and quality of rice. Rice 15, 18 (2022).
Li, N., Xu, R. & Li, Y. Molecular networks of seed size control in plants. Annu. Rev. Plant Biol. 70, 435–463 (2019).
Arora, R. et al. MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8, 242 (2007).
Zhang, J., Nallamilli, B. R., Mujahid, H. & Peng, Z. OsMADS6 plays an essential role in endosperm nutrient accumulation and is subject to epigenetic regulation in rice (Oryza sativa). Plant J. 64, 604–617 (2010).
Paul, P. et al. MADS78 and MADS79 are essential regulators of early seed development in rice. Plant Physiol. 182, 933–948 (2020).
Chen, C. et al. Heat stress yields a unique MADS box transcription factor in determining seed size and thermal sensitivity. Plant Physiol. 171, 606–622 (2016).
Zhang, H., Xu, H., Feng, M. & Zhu, Y. Suppression of OsMADS7 in rice endosperm stabilizes amylose content under high temperature stress. Plant Biotechnol. J. 16, 18–26 (2018).
Jiang, H. & Köhler, C. Evolution, function, and regulation of genomic imprinting in plant seed development. J. Exp. Bot. 63, 4713–4722 (2012).
Tonosaki, K. et al. Mutation of the imprinted gene OsEMF2a induces autonomous endosperm development and delayed cellularization in rice. Plant Cell 33, 85–103 (2021).
Xiao, W. et al. Imprinting of the MEA Polycomb gene is controlled by antagonism between MET1 methyltransferase and DME glycosylase. Dev. Cell 5, 891–901 (2003).
Kim, M. Y. et al. DNA demethylation by ROS1a in rice vegetative cells promotes methylation in sperm. Proc. Natl. Acad. Sci. USA 116, 9652–9657 (2019).
Chow, H. T. & Mosher, R. A. Small RNA-mediated DNA methylation during plant reproduction. Plant Cell 35, 1787–1800 (2023).
Grover, J. W. et al. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 94, 575–582 (2018).
Shahzad, Z., Eaglesfield, R., Carr, C. & Amtmann, A. Cryptic variation in RNA-directed DNA-methylation controls lateral root development when auxin signalling is perturbed. Nat. Commun. 11, 218 (2020).
Quesneville, H. Twenty years of transposable element analysis in the Arabidopsis thaliana genome. Mob. DNA 11, 28 (2020).
Kinoshita, Y. et al. Control of FWA gene silencing in Arabidopsis thaliana by SINE-related direct repeats. Plant J. 49, 38–45 (2007).
Williams, B. P., Pignatta, D., Henikoff, S. & Gehring, M. Methylation-sensitive expression of a DNA demethylase gene serves as an epigenetic rheostat. PLoS Genet. 11, e1005142 (2015).
Batista, R. A. et al. The MADS-box transcription factor PHERES1 controls imprinting in the endosperm by binding to domesticated transposons. Elife 8, e50541 (2019).
Nosaka, M. et al. Role of transposon-derived small RNAs in the interplay between genomes and parasitic DNA in rice. PLoS Genet. 8, e1002953 (2012).
Xu, L. et al. Regulation of rice tillering by RNA-directed DNA methylation at miniature inverted-repeat transposable elements. Mol. Plant 13, 851–863 (2020).
Shen, J. et al. Translational repression by a miniature inverted-repeat transposable element in the 3′ untranslated region. Nat. Commun. 8, 14651 (2017).
Jullien, P. E., Katz, A., Oliva, M., Ohad, N. & Berger, F. Polycomb group complexes self-regulate imprinting of the polycomb group gene MEDEA in Arabidopsis. Curr. Biol. 16, 486–492 (2006).
Satyaki, P. R. V. & Gehring, M. RNA Pol IV induces antagonistic parent-of-origin effects on Arabidopsis endosperm. PLoS Biol. 20, e3001602 (2022).
Hu, X. et al. The U-box E3 ubiquitin ligase TUD1 functions with a heterotrimeric G α subunit to regulate brassinosteroid-mediated growth in rice. PLoS Genet. 9, e1003391 (2013).
Gao, X. et al. Rice qGL3/OsPPKL1 functions with the GSK3/SHAGGY-like kinase OsGSK3 to modulate brassinosteroid signaling. Plant Cell 31, 1077–1093 (2019).
Qin, R. et al. LTBSG1, a new allele of BRD2, regulates panicle and grain development in rice by brassinosteroid biosynthetic pathway. Genes 9, 292 (2018).
Huang, J. et al. Natural variation of the BRD2 allele affects plant height and grain size in rice. Planta 256, 27 (2022).
Liu, Z. et al. OsMKKK70 regulates grain size and leaf angle in rice through the OsMKK4-OsMAPK6-OsWRKY53 signaling pathway. J. Integr. Plant Biol. 63, 2043–2057 (2021).
Xu, R. et al. Control of grain size and weight by the OsMKKK10-OsMKK4-OsMAPK6 signaling pathway in rice. Mol. Plant 11, 860–873 (2018).
Yin, L.-L. & Xue, H.-W. The MADS29 transcription factor regulates the degradation of the nucellus and the nucellar projection during rice seed development. Plant Cell 24, 1049–1065 (2012).
Nayar, S., Sharma, R., Tyagi, A. K. & Kapoor, S. Functional delineation of rice MADS29 reveals its role in embryo and endosperm development by affecting hormone homeostasis. J. Exp. Bot. 64, 4239–4253 (2013).
Nayar, S., Kapoor, M. & Kapoor, S. Post-translational regulation of rice MADS29 function: homodimerization or binary interactions with other seed-expressed MADS proteins modulate its translocation into the nucleus. J. Exp. Bot. 65, 5339–5350 (2014).
Tang, J., Mei, E., He, M., Bu, Q. & Tian, X. Functions of OsWRKY24, OsWRKY70 and OsWRKY53 in regulating grain size in rice. Planta 255, 92 (2022).
Ogawa, D. et al. RSS1 regulates the cell cycle and maintains meristematic activity under stress conditions in rice. Nat. Commun. 2, 278 (2011).
Tiwari, G. J., Liu, Q., Shreshtha, P., Li, Z. & Rahman, S. RNAi-mediated down-regulation of the expression of OsFAD2-1: effect on lipid accumulation and expression of lipid biosynthetic genes in the rice grain. BMC Plant Biol. 16, 1–13 (2016).
Yang, J., Luo, D., Yang, B., Frommer, W. B. & Eom, J.-S. SWEET11 and 15 as key players in seed filling in rice. New Phytol. 218, 604–615 (2018).
Peng, B. et al. Scanning electron microscopic observation on chalkiness of rice mutant OsLHT1 grains. J. Agric. Sci. 14, 54 (2022).
Liu, Z. et al. Transcription factor OsSGL is a regulator of starch synthesis and grain quality in rice. J. Exp. Bot. 73, 3417–3430 (2022).
Xie, K. & Yang, Y. RNA-guided genome editing in plants using a CRISPR-Cas system. Mol. Plant 6, 1975–1983 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011).
Clement, K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226 (2019).
Wang, G., Wang, G., Zhang, X., Wang, F. & Song, R. Isolation of high quality RNA from cereal seeds containing high levels of starch. Phytochem. Anal. 23, 159–163 (2012).
Shivaprasad, P. V., Dunn, R. M., Santos, B. A., Bassett, A. & Baulcombe, D. C. Extraordinary transgressive phenotypes of hybrid tomato are influenced by epigenetics and small silencing RNAs. EMBO J. 31, 257–266 (2012).
Tirumalai, V., Prasad, M. & Shivaprasad, P. V. RNA blot analysis for the detection and quantification of plant microRNAs. J. Vis. Exp. https://doi.org/10.3791/61394 (2020).
Ramanathan, V. & Veluthambi, K. Transfer of non-T-DNA portions of the Agrobacterium tumefaciens Ti plasmid pTiA6 from the left terminus of TL-DNA. Plant Mol. Biol. 28, 1149–1154 (1995).
Vivek Hari Sundar, G. & Shivaprasad, P. V. Investigation of transposon DNA methylation and copy number variation in plants using southern hybridisation. Bio Protoc. 12, e4432 (2022).
Rogers, S. O. & Bendich, A. J. Extraction of total cellular DNA from plants, algae and fungi. in Plant Molecular Biology Manual 183–190 (Springer Netherlands, Dordrecht, 1994).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27, 1571–1572 (2011).
Huang, X., Zhang, S., Li, K., Thimmapuram, J. & Xie, S. ViewBS: a powerful toolkit for visualization of high-throughput bisulfite sequencing data. Bioinformatics 34, 708–709 (2018).
Stocks, M. B. et al. The UEA sRNA Workbench (version 4.4): a comprehensive suite of tools for analyzing miRNAs and sRNAs. Bioinformatics 34, 3382–3384 (2018).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Axtell, M. J. ShortStack: comprehensive annotation and quantification of small RNA genes. RNA 19, 740–751 (2013).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Quinlan, A. R. BEDTools: the Swiss-army tool for genome feature analysis. Curr. Protoc. Bioinform. 47, 11.12.1–34 (2014).
Wickham, H. Ggplot2 (Springer International Publishing, Cham, Switzerland, 2016).
Khan, A. & Mathelier, A. Intervene: a tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 18, 1–8 (2017).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Patwardhan, M. N., Wenger, C. D., Davis, E. S., Phanstiel, D. H. & Bedtoolsr An R package for genomic data analysis and manipulation. J. Open Source Softw. 4, 1742 (2019).
Song, L., Koga, Y. & Ecker, J. R. Profiling of transcription factor binding events by chromatin immunoprecipitation sequencing (ChIP-seq). Curr. Protoc. Plant Biol. 1, 293–306 (2016).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020).
Jefferson, R. A., Kavanagh, T. A. & Bevan, M. W. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907 (1987).
Shariq, M. et al. Adult neural stem cells have latent inflammatory potential that is kept suppressed by Tcf4 to facilitate adult neurogenesis. Sci. Adv. 7, eabf5606 (2021).
Pachamuthu, K. et al. Rice-specific Argonaute 17 controls reproductive growth and yield-associated phenotypes. Plant Mol. Biol. 105, 99–114 (2021).
He, Y. et al. Indole-3-acetate beta-glucosyltransferase OsIAGLU regulates seed vigour through mediating crosstalk between auxin and abscisic acid in rice. Plant Biotechnol. J. 18, 1933–1945 (2020).
Khatun, S. & Flowers, T. J. The estimation of pollen viability in rice. J. Exp. Bot. 46, 151–154 (1995).
Yao, M. et al. Downregulation of OsAGO17 by artificial microRNA causes pollen abortion resulting in the reduction of grain yield in rice. Electron. J. Biotechnol. https://doi.org/10.1016/j.ejbt.2018.07.001 (2018).
Das, S., Swetha, C., Pachamuthu, K., Nair, A. & Shivaprasad, P. V. Loss of function of Oryza sativa Argonaute 18 induces male sterility and reduction in phased small RNAs. Plant Reprod. 33, 59–73 (2020).
Acknowledgements
We thank Prof. K. Veluthambi for Agrobacterium strains, WP, PB1 seeds and binary plasmids. We thank genomics, electron microscopy, CIFF, IT, radiation, greenhouse and lab-kitchen facilities at the NCBS. We acknowledge help from Dr. Anushree Narjala in bioinformatics analysis and Dr. M. Dhandapani for crossing demonstration. We thank all the lab members for discussions and comments. This work was supported by NCBS-TIFR core funding, Government of India. This study was also supported by the Department of Atomic Energy, Government of India, under Project Identification No. RTI 4006 (1303/3/2019/R&D-II/DAE/4749 dated 16.7.2020). These funding agencies did not participate in the designing of experiments, analysis, or interpretation of data, or in writing of the manuscript. We thank anonymous reviewers for their valuable suggestions and insightful comments.
Author information
Authors and Affiliations
Contributions
P.V.S. and A.K.P. designed all experiments and discussed results and wrote the manuscript. P.V.S. obtained funding. A.K.P. performed most of the experiments and bioinformatic analyses. V.H.-S.G. generated osnrpd1-kd datasets and performed bioinformatic analysis. A.B.N. performed embryo and endosperm transcriptome analysis. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pal, A.K., Gandhivel, V.HS., Nambiar, A.B. et al. Upstream regulator of genomic imprinting in rice endosperm is a small RNA-associated chromatin remodeler. Nat Commun 15, 7807 (2024). https://doi.org/10.1038/s41467-024-52239-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52239-z
- Springer Nature Limited