
Abstract
Clinical and neuroanatomical correlates of daytime sleepiness in Parkinson’s disease (PD) remain inconsistent in the literature. Two studies were conducted here. The first evaluated the interrelation between non-motor and motor symptoms, using a principal component analysis, associated with daytime sleepiness in PD. The second identified the neuroanatomical substrates associated with daytime sleepiness in PD using magnetic resonance imaging (MRI). In the first study, 77 participants with PD completed an extensive clinical, cognitive testing and a polysomnographic recording. In the second study, 29 PD participants also underwent MRI acquisition of T1-weighted images. Vertex-based cortical and subcortical surface analysis, deformation-based morphometry, and voxel-based morphometry were performed to assess the association between daytime sleepiness severity and structural brain changes in participants. In both studies, the severity of daytime sleepiness and the presence of excessive daytime sleepiness (EDS; total score >10) were measured using the Epworth Sleepiness Scale. We found that individuals with EDS had a higher score on a component including higher dosage of dopamine receptor agonists, motor symptoms severity, shorter sleep latency, and greater sleep efficiency. Moreover, increased daytime sleepiness severity was associated with a larger surface area in the right insula, contracted surfaces in the right putamen and right lateral amygdala, and a larger surface in the right posterior amygdala. Hence, daytime sleepiness in PD was associated with dopaminergic receptor agonists dosage, motor impairment, and objective sleep measures. Moreover, neuroanatomical changes in cortical and subcortical regions related to vigilance, motor, and emotional states were associated with more severe daytime sleepiness.
Similar content being viewed by others


Introduction
Excessive daytime sleepiness (EDS), a marked inability to remain awake and alert during the day1,2, is a major non-motor symptom affecting 20–60% of adults with Parkinson’s disease (PD)3. While motor and non-motor symptoms in PD tend to coexist and are often related to one another4, much less is known about the interdependence between the symptoms associated with EDS in PD. Nonetheless, EDS has been independently but inconsistently linked to various symptoms in PD, such as more severe depressive and anxiety symptoms, worsening motor impairment, and poor quality of life5,6,7,8,9,10,11,12,13. Measures of global cognitive functioning, executive control and processing speed were also reported to be worse in PD adults with EDS than those without14,15. As for the interdependence of EDS in PD specifically, two studies have evaluated its interrelation with other motor and non-motor symptoms and provided inconsistent results16,17. One study used exploratory factor analysis and found that daytime sleepiness was part of a factor that also included cognitive, autonomic, axial, psychotic and depressive symptoms17. In contrast, the other study, using a principal component analysis, found that EDS formed a unique component unrelated to motor and other non-motor symptoms16. Further-designed studies are required to understand the interrelations between clinical symptoms in PD. Then, determining whether the clinical manifestation of EDS plays a role in these inter-relational associations between other motor and non-motor symptoms in PD would be easier to establish.
The etiology of EDS in PD is complex and multifactorial18,19. EDS in PD is a common adverse effect of dopaminergic therapy (e.g., levodopa or dopaminergic agonists), although some discrepancies in sleep and wakefulness have been shown depending on the dopaminergic agents used18,19,20,21. Some studies also showed that EDS in PD is associated with complaints of nonrestorative nocturnal sleep7,8,10,11. However, most of the studies using nocturnal polysomnography (PSG) showed no group differences nor associations between daytime sleepiness and nighttime sleep efficiency, awakenings, or sleep stages13,22,23,24,25. Furthermore, the neuroanatomical correlates of EDS in PD are still poorly understood. Only few magnetic resonance imaging (MRI) studies using voxel-based morphometry (VBM) evaluated the gray matter alterations associated with EDS in PD and provided inconsistent results26,27,28. Indeed, some studies have shown a widespread gray matter volume reduction in cortical regions27,28, while another have reported increased gray matter volume in the bilateral hippocampal and parahippocampal gyri in PD individuals with EDS compared to those without EDS26. A proposed explanation is that VBM relies solely on a volumetric representation of the brain, which can amplify the impact of partial volume and obscure the detection of slight pathological variations in the cerebral cortex and subcortical surface29. Other techniques, such as vertex-based cortical and subcortical surface analysis and deformation-based morphometry, may provide a more comprehensive and accurate description of the brain abnormalities associated with EDS in PD.
To further explore the correlates of EDS in PD, we performed two studies. In Study 1, we aimed to evaluate the interdependence between clinical symptoms reportedly associated with EDS in PD, and to determine whether the clinical manifestation of EDS could discriminate individuals with PD on the components resulting from this prior assessment. To do this, we evaluated the inter-relations between clinical variables (cognition, neuropsychiatric, motor, dopaminergic medication, sleep-related complaints of insomnia, and PSG sleep measures) using a data-driven approach with a principal component analysis (PCA). Then, we compared PD participants with and without EDS on the resulting components of the PCA. In Study 2, we assessed the neuroanatomical substrates underlying daytime sleepiness in PD using surface-, volume- and deformation-based MRI analyses.
Results
The flowchart for both studies is presented in Fig. 1.

For Study 1, 95 participants met the inclusion criteria. Of these, 18 were excluded: dementia (n = 4) or PSG date > 6 months from the clinical visit (n = 14). Then, 77 participants were included in the analyses (25 with and 52 without EDS). For Study 2, 43 participants met the inclusion criteria. Of these, 14 were excluded: dementia (n = 4) and MRI date > 6 months from the clinical visit (n = 10). Then, 29 participants were included (11 with and 18 without EDS). PD Parkinson’s disease, PSG polysomnography, MRI magnetic resonance imaging, EDS excessive daytime sleepiness.
In Study 1, individuals with EDS were younger and took a higher dosage of DA receptor agonists (Table 1). No significant between-group differences were found for sex, education, PD duration, disease severity, motor symptom severity, the proportion of individuals with RBD or MCI, MMSE score, LEDD, the proportion of individuals taking DA receptor agonists, antidepressants, other medication, and questionnaires (except ESS). Moreover, the two groups were similar on all PSG variables (Table 2).
In Study 2, individuals with EDS took a higher dosage of DA receptor agonists (Table 3). Age, sex, education, PD duration and disease severity, motor symptoms severity, proportion of individuals with RBD or MCI, MMSE score, LEDD, proportion of individuals taking DA receptor agonists, antidepressant, others medication and questionnaires (except ESS) were similar between groups.
For Study 1, PCA analyses revealed six components from the fourteen variables, and the resulting factor structure explained 68.3% of the variables’ variance. The six components and their loadings are listed in Table 4.
Component 1 accounted for 20.3% of the total variance and included four variables: DA receptor agonists dosage, UPDRS-III, sleep latency, and sleep efficiency. Component 2 accounted for 13.0% of the total variance and was composed of four variables: UPDRS-III, MMSE, Trail Making Test B, and Bells Test. Component 3 accounted for 11.5% of the total variance and comprised two variables: BDI-II and BAI. Component 4 accounted for 8.0% of the total variance and included three variables: PD duration, N2-N3 sleep, and Bells Test. Component 5 accounted for 7.8% of the total variance and included two variables: ISI and AHI. Component 6 accounted for 7.6% of the total variance and comprised three variables: DA receptor agonists dosage, UPDRS-III, and mean O2 saturation.
A group difference was found for component 1 (Fig. 2). Compared to individuals without EDS (M = −0.21, SD = 0.97), those with EDS (M = 0.43, SD = 0.94) had a higher component 1 score that included higher dosage of DA receptor agonists (LEDD in mg), higher UPDRS-III scores, shorter sleep latency, and greater sleep efficiency. A statistical trend was also observed for component 4. No significant between-group difference was found for components 2, 3, 5, and 6.

The full line in each violin plot represents the median and the dotted line represents the first and the third quartile. *Individuals with EDS had a higher score on component 1 as compared to those without EDS (F = 4.66, p = 0.03, ES = 0.06; PD-nEDS: M = −0.21, SD = 0.97; PD-EDS: M = 0.43, SD = 0.94). A statistical trend was found for component 4 (F = 3.11, p = 0.08, ES = 0.04; PD-nEDS: M = 0.12, SD = 1.01; PD-EDS: M = −0.26, SD = 0.95), whereas no difference was found for components 2 (F = 1.92, p = 0.17, ES = 0.03; PD-nEDS: M = 0.00, SD = 0.91; PD-EDS: M = −0.00, SD = 1.19), 3 (F = 1.17, p = 0.28, ES = 0.02; PD-nEDS: M = −0.12, SD = 0.92; PD-EDS: M = 0.06, SD = 0.74), 5 (F = 0.00, p = 0.99, ES = 0.00; PD-nEDS: M = −0.02, SD = 1.05; PD-EDS: M = 0.05, SD = 0.90), and 6 (F = 1.65, p = 0.20, ES = 0.02; PD-nEDS: M = −0.15, SD = 1.05; PD-EDS: M = 0.32, SD = 0.81). PD Parkinson’s disease, PD-EDS PD with excessive daytime sleepiness, PD-nEDS PD without EDS, AHI apnea-hypopnea index, DA dopaminergic agonist, M mean, SD standard deviation, ES effect sizes.
In Study 2, for vertex-based cortical surface in PD, higher ESS scores were associated with larger surface area in the right anterior insula extending to the medial orbitofrontal cortex (Fig. 3a and Table 5). Moreover, between-group differences revealed that individuals with EDS had larger surface area in the right rostral middle frontal cortex, left supramarginal cortex extending to the posterior insula and superior temporal gyrus and left caudal middle frontal area. Individuals with EDS also had decreased cortical volume in the left parieto-occipital sulcus including the fundus (Fig. 3b and Table 5).

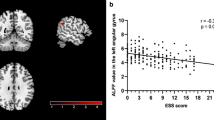
A (i). A higher ESS score was associated with larger surface area in the right anterior insula extending to the medial orbitofrontal cortex. (ii). Individuals with EDS had larger surface area in the right rostral middle frontal cortex, left supramarginal cortex extending to the posterior insula and superior temporal gyrus and left caudal middle frontal area. B Individuals with EDS had decreased cortical volume in the left parieto-occipital sulcus including the fundus. The color bar indicates the logarithmic scale of p values (−log10) for between-group differences, with red-yellow areas representing reductions in the first compared to the last group in the contrast (corrected with Monte Carlo simulation at p < 0.05 with age, sex, and disease duration as covariates as well as total intracranial volume for cortical volume analysis). C A higher ESS score was associated with contraction (red) in the lateral and medial surface of the right putamen, contraction in the posterior surface of the right amygdala, and expansion (blue) in the posterior surface of the right amygdala. ESS Epworth Sleepiness Scale, PD-EDS Parkinson’s disease with excessive daytime sleepiness, PD-nEDS PD without EDS.
As for the vertex-based subcortical shape in PD, higher ESS scores were associated with contraction in the lateral and medial surface of the right putamen (r = −0.41, p = 0.029) and the lateral surface of the right amygdala (r = −0.61, p = 0.001), and expansion in the posterior surface of the right amygdala (r = 0.57, p = 0.001) (Fig. 3c and Table 5). Moreover, individuals with EDS show surface contraction of the right putamen and surface expansion on the right amygdala compared to those without EDS.
No significant association with ESS scores or group differences were found for whole-brain analyses with cortical thickness, VBM or DBM techniques.
Discussion
This article presents two studies conducted to (1) clarify the association between daytime sleepiness and PD after computing a PCA to create components of other clinical motor and non-motor symptoms, and (2) determine the neuroanatomical changes associated with daytime sleepiness in PD. In Study 1, each component obtained from the PCA defined a putative discriminant combination of variables to contrast PD individuals based on EDS (defined as a score >10 on the ESS). The results highlight that the combination of a higher dosage of DA receptor agonists, more severe motor symptoms, shorter sleep latency, and greater sleep efficiency can distinguish PD individuals with EDS from those without. In Study 2, the results show that more severe daytime sleepiness in PD is associated with increased cortical surface area (right anterior insula) and subcortical surface contraction (right putamen and right amygdala). These regions are associated to vigilance, motor, and emotional states. Together, these two studies provide new insight to better understand the complex relationships between daytime sleepiness and the other clinical symptoms, as well as structural brain changes reported in PD.
With the magnitude of symptoms that may or may not be independently involved in daytime sleepiness in PD5,6,7,8,9,10,11,12,13, it becomes necessary to provide an approach allowing both data reduction while also evaluating the interrelations between these symptoms. Here, Study 1 offers a sensitive picture of the relationship between clinical symptoms related to daytime sleepiness in PD. Using a PCA, we propose an alternative strategy to establish the interdependence of clinical factors associated with daytime sleepiness in PD first. Our study highlights the importance of including objective sleep variables to assess the interdependence of clinical symptoms in PD, contrary to similar studies that have solely used questionnaire-based sleep measures16,17. Nocturnal sleep metrics (sleep latency, sleep efficiency, N2-N3 sleep, AHI) and variables associated with parkinsonism (dosage of DA receptor agonists, PD duration, and severity of motor symptoms) were found within most components of the PCA, making them non-negligible features in PD.
The results highlight that PD individuals with EDS differ from those without EDS on a component including higher DA receptor agonists dosage, more severe motor symptoms, shorter sleep latency, and greater sleep efficiency. Of note, Study 1 illustrates that the presence of EDS is significant not only for the component with the greatest variance but also for objective sleep variables (sleep latency and efficiency) that were not previously described by other studies16,17. In Study 1, participants taking a DA receptor agonist were exclusively on pramipexole, which has a high affinity with D2/D3 receptors. This medication has been shown to induce somnolence and daytime sleep episodes in PD19,30. It suggests that a higher dose of pramipexole to treat the worst motor symptoms can also concomitantly shorten sleep latency and increase sleep efficiency due to its sedative nature. A double-blind placebo-controlled study performed in healthy adults also found that pramipexole administration significantly reduced mean sleep latency and increased total sleep duration on the Multiple Sleep Latency Test31. On the other hand, most studies using nocturnal PSG reported no associations between daytime sleepiness and most PSG variables in PD13,22,23,24,25. One of them, however, reported that PD individuals with EDS had shorter sleep latency compared to those without EDS13. While shorter sleep latency and greater sleep efficiency are included in the most discriminative component in Study 1, one must remember that it is the component considering the interrelation between all four variables that differ between PD individuals with and without EDS.
The two PD groups did not differ on neuropsychiatric symptoms including anxiety and depression (component 3), and on motor severity, global cognition, executive control and processing speed (component 2). This result is in line with a previous study in PD that also used a PCA16; they showed that EDS was not interrelated to anxiety, depression, or global cognition16. Moreover, PD individuals with and without EDS did not differ on insomnia severity and AHI (component 5), and on nocturnal oxygen saturation, motor severity and DA receptor agonists dosage (component 6). These results corroborate previous studies, which found no significant association between daytime sleepiness and objective measures related to sleep apnea in PD13,32,33,34.
As for Study 2, we applied surface-based analyses of cortical and subcortical structures to allow a more exhaustive picture of daytime sleepiness’s structural brain correlates in PD. Increased daytime sleepiness was associated with a larger surface area in the right anterior insula extending to the medial orbitofrontal cortex. Between-group comparisons also revealed a larger surface area in PD individuals with EDS in the right rostral middle frontal cortex, left supramarginal cortex extending to the posterior insula and superior temporal gyrus and left caudal middle frontal area, and reduced cortical volume in the left parieto-occipital sulcus. The insula is a cortical region highly interconnected with several cortical (frontal, temporal, parietal, occipital, limbic areas) and subcortical (putamen, thalamus, amygdala, and hippocampus) regions35,36. It plays an important part in multiple brain networks involved in a wide variety of functions including sensorimotor, olfacto-gustatory, socio-emotional, and cognition37. In particular, it is suggested that the insular cortex plays a role in the salience network and is associated with subjective sleepiness38,39,40,41. Thus, the insula is considered as a central hub processing relevant information related to vigilance, motor responses, and emotional states37,40,41,42,43. In PD, neuroimaging studies have revealed functional and structural alterations of the insula which would play a potential role in non-motor symptoms reported in this population40. Interestingly, increased surface area has been reported in isolated REM sleep behavior disorder44, a strong prodromal phenotype associated with the development of PD42, and shown to occur preferentially within regions with a higher expression of genes involved in the inflammatory response43. Increased area may therefore be consequential to ongoing inflammatory response in the brain. Otherwise, local surface area has been hypothesized to reflect underlying white matter fibers45. Increased surface area could result from underlying white matter degeneration since the tension or shrinkage of white matter fibers could lead to deeper sulci and extended cortical surface area45,46. Importantly, previous research reported white matter alterations in PD individuals with EDS, which may underlie anterior insula extending to the medial orbitofrontal cortex. Hence, a previous study in PD individuals with EDS reported bilateral white matter damage (illustrated by increased axial diffusivity) notably in the superior corona radiata and the temporal part of superior longitudinal fascicles26, as these pass underneath the structures shown in Study 2. Microstructural changes of white matter in sleep-related circuits (particularly bilateral fornix and bilateral inferior longitudinal fasciculus) have also been observed in drug-naïve PD individuals with EDS compared to those without EDS47. Future multimodal neuroimaging studies using both vertex based, white-matter integrity metrics, and should validate this result interpretation.
Increased daytime sleepiness were also associated with surface contraction of the right putamen and right lateral amygdala, as well as a surface expansion of the posterior surface of the right amygdala. These alterations were also found in PD individuals with EDS (contraction of right putamen, and expansion of right amygdala) compared to those without EDS. Since the right putamen and amygdala are highly inter-interconnected with the right insula40, these results could reflect alterations in the mesocorticolimbic circuitry in PD individuals with more severe daytime sleepiness. Putamen atrophy is a frequent feature reported in PD48,49. One study, however, reported a localized pallidum and putamen volume hypertrophy, in the left dorsolateral and the right dorsal, respectively, for drug-naïve PD individuals with EDS, and no significant group differences were found in the shape analysis of the other subcortical nuclei50. While the latter is contrary to our results, their participants were drug-naïve and in an early stage of PD. Here, participants in Study 2 were more advanced in their disease progression. Our results imply that putamen and amygdala atrophy could occur as the disease progresses. Accordingly, association of daytime sleepiness with nigrostriatal dopaminergic degeneration, namely for the putamen, was reported in PD individuals at stage 2 of the Hoehn and Yahr scale, but not in PD individuals in stage 1 using SPECT51.
No significant association with daytime sleepiness was found using VBM and DBM techniques. To date, the studies that used VBM to evaluate the structural correlates of sleepiness in PD provided inconsistent results26,27,28. One found a widespread gray matter volume reduction in cortical regions (frontal, temporal, occipital, and limbic), in addition to atrophy of the basal forebrain in PD individuals with EDS as compared to those without EDS and healthy controls27. Another study, with a larger sample size, had similar results28. They identified in PD a loss of integrity and atrophy in the anterior cingulate network that were associated with EDS28. The other study reported increased gray matter volume in the bilateral hippocampal and parahippocampal gyri in PD individuals with EDS compared to those without EDS26. However, most studies did not control for multiple comparisons (only one did28). There has been no study to date evaluating changes in brain morphology related to EDS using DBM in PD.
These two studies have some limitations. Conventional approaches in research usually assess symptoms independently, which provides key information on the bidirectional link between the two variables studied. However, the clinical perspective and reality of PD individuals are rather complex phenomena, and they imply an interplay between clinical symptoms4. A conventional approach infers that symptoms in PD are mutually exclusive and rules out the assumption that many symptoms in PD are interrelated to each other. While the approach with a PCA considers this interrelation between clinical symptoms linked to daytime sleepiness in PD and provides additional clinical information, challenges in interpretation and limitations can arise. Of note, it is essential to mention that the PCA method used in Study 1 is a data-driven approach, and to maintain enough statistical power, we were limited to 14 variables. Furthermore, given the complexity of the protocol (nighttime PSG, neuropsychology, MRI), we cannot exclude bias in selecting our sample (e.g., interest to partake in sleep study due to personal sleep complaints, or participants with advanced PD would be less inclined to embark on our research protocol). Future studies should evaluate whether our conclusions apply to a larger and more representative sample with a wider range of variables. It should also be noted that daytime sleepiness is a subjectively-rated symptom that individuals often underrecognize23; caregiver input would have been valuable but was not always available. Another alternative for future studies would be the inclusion of objective measures that are sensitive to the measurement of EDS in PD. Given the strong association between the use of pramipexole and daytime sleepiness in PD, it could be of interest to evaluate whether the timing of pramipexole intake (e.g. evening administration) is associated with shorter sleep latency/greater sleep efficiency and with daytime sleepiness complaints. Moreover, as this study excluded participants with dementia, the assessment of correlations between cognition and daytime sleepiness is limited to mild cognitive changes. As for Study 2, group analyses were considered complimentary due to the limited sample size. Although participants in Study 2 were carefully selected to ensure minimizing selection biases and controlling for several confounding variables, and being stricter in terms of multiple comparisons, a larger sample could increase the statistical power and allow the identification of other brain region changes related to the severity of daytime sleepiness in PD. Nevertheless, future studies could use the data provided here for a priori power analysis and have an accurate required sample size.
Methods
Participants
PD participants were recruited from the movement disorders clinics of the Centre Hospitalier de l’Université de Montréal and the McGill University Health Centre (Montreal, Canada) as part of a longitudinal study on sleep and cognition in PD. They were consecutive individuals seen at their annual assessment and were referred by a neurologist for this study regardless of sleep complaints including daytime sleepiness. All participants underwent one night in the sleep laboratory at the Centre for Advanced Research in Sleep Medicine of the Centre Intégré universitaire de santé et de services sociaux du Nord-de-l’Île-de-Montréal (CIUSSS-NÎM)—Hôpital du Sacré-Cœur de Montréal. Research protocols were approved by the ethics committee of the CIUSSS-NÎM – Hôpital du Sacré-Cœur de Montréal and by the ethics committee CIUSSS du Centre-Sud-de-l’Île-de-Montréal—Comité d’éthique de la recherche vieillissement-neuroimagerie (Montreal, Quebec). All participants gave their informed written consent to participate.
For Study 1, PD participants underwent multiple clinical visits including a nocturnal PSG, neurological exam, a complete neuropsychological assessment, as well as sleep and mood questionnaires. Participants between 45 and 85 years old with a diagnosis of idiopathic PD confirmed by a movement disorder specialist were included52. Exclusion criteria were: (i) dementia according to the neuropsychological assessment53, or the recommendations of the Movement Disorder Society Task Force for PD54; (ii) a major psychiatric disorder (including bipolar disorder, untreated major depression, schizophrenia) according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision53; (iii) history of stroke, or cerebrovascular disease; and (iv) PSG date > 6 months from the clinical visit. They were taking their usual medication during the study. Antiparkinsonian medication, including dopamine (DA) receptor agonists, was converted into levodopa equivalent daily dose (LEDD mg) according to Tomlinson et al. 55. Motor symptom severity was evaluated by a neurologist in the “on” state using the Unified Parkinson’s Disease Rating Scale, Part III (UPDRS-III)56. The Hoehn and Yahr scale was also used to assess disease severity57.
For Study 2, PD participants underwent the same protocol as Study 1 in addition to an MRI. Similar inclusion and exclusion criteria were retained from Study 1 apart from the PSG date criterion since it was not included in the MRI analyses. All participants of Study 2 also had to have an MRI scan < 6 months from the clinical visit.
Procedure
Polysomnography (PSG)recordings included EEG (10-20 system, referential montage with linked ears), chin electromyogram and left and right electrooculography. Signals were digitalized at a sampling rate of 256 Hz using commercial software (Harmonie, Stellate System). Sleep stages were identified according to the American Academy of Sleep Medicine criteria58. Oral and nasal airflow plus thoracic and abdominal movements were recorded in concomitance with pulse oximetry was performed to measure the respiratory event index. Apneas were defined as an airflow reduction of ≥90% lasting ≥10 s. Hypopneas were defined as an airflow reduction of ≥30% lasting ≥10 s accompanied by either an oxygen desaturation of ≥ 3% or arousal, as recommended58. The diagnostic criteria for REM sleep behavior disorder (RBD) were: REM sleep without atonia defined as tonic EMG activity >30% and/or phasic EMG activity >15% of the total REM sleep time (on 3-s mini-epochs within 30-s REM sleep epochs) and at least one of the following two criteria: 1) a history of undesirable and potentially harmful behaviors during sleep or 2) complex behaviors during REM sleep recorded during the night spent in the laboratory59,60,61.
Both studies offered PD participants a comprehensive neuropsychological assessment measuring five cognitive domains: attention, executive functions, verbal episodic memory and learning, visuospatial, and language abilities (see Table S1). Cognitive status was determined by consensus between the neurologist (R.B.P.) and neuropsychologist (J.F.G.). Mild cognitive impairment (MCI) diagnosis was based on specific criteria detailed elsewhere62,63. The Mini-Mental State Examination (MMSE) was also completed as a measure of global cognitive functioning64. Self-reported questionnaires were used to assess the severity of depressive (Beck Depression Inventory second edition (BDI-II)65), anxiety (Beck Anxiety Inventory (BAI)66), insomnia (Insomnia Severity Index (ISI)67), and apathy (proportion of individuals with a score ≥1 on the UPDRS Part I item 456) symptoms. The Epworth Sleepiness Scale (ESS) was used to evaluate the severity of daytime sleepiness symptoms68.
In Study 2, PD participants were imaged on a Siemens 3 T TrioTIM scanner with a 12-channel head matrix coil at the Institut universitaire de gériatrie de Montréal. T1-weighted images were acquired using an MPRAGE sequence with the following parameters: repetition time 2.3 seconds, echo time 2.91 ms, inversion time 900 milliseconds, 9-degree flip angle, 256 × 240 mm field of view, 256 × 240 matrix resolution (voxel size: 1 × 1 × 1 mm), and 240 Hz/Px bandwidth.
Vertex-based surface analyses were performed to investigate the local changes occurring in the cortical and subcortical surfaces. Cortical surface processing was conducted with FreeSurfer (version 6.0.0) using default parameters, as described previously44,69, which generated cortical surface maps that quantified thickness, surface area, and volume at each vertex. Thickness and surface area maps were smoothed with a filter of full width at half maximum of 20 mm and volume maps of 15 mm. Subcortical surface processing using FSL-FIRST was then performed to study the surface of the left and right putamen, caudate, pallidum, thalamus, hippocampus, amygdala, and nucleus accumbens70. Briefly, the structures were segmented and inflated based on shape and intensity information from 336 manually delineated T1-weighted images70. Surfaces were registered to the MNI152 space and then used to detect between-group differences in surface displacement71.
We additionally performed VBM and DBM in CAT12 version 12.7 to describe the volume- and deformation-based changes found in the gray matter tissue of participants. Normalized modulated images were smoothed using a filter of 8 mm and analyzed using default parameters, as described previously72.
Statistical analyses
All variables were examined for their mean, standard deviation, skewness, and kurtosis. Logarithmic transformations (PSG values when applicable) were applied for variables that were substantially out of a normal distribution (i.e., absolute skew value larger than 2 or an absolute kurtosis larger than 773). Non-parametric equivalent tests (demographic and clinical data) were performed when variables were not normally distributed. Statistical analyses were performed using SPSS (version 29) and Matlab (9.6.0) for multiple imputations processing.
For Study 1, we performed a PCA using varimax rotation. Due to the sample size of Study 1, we limited the number of variables included in the PCA. We thus selected 14 a priori variables based on the literature that were shown to be linked to the presence of EDS in PD and that were available in our study14,15,18,19: (1) PD duration starting from symptoms onset (years); (2) DA receptor agonists dosage converted in LEDD (mg)55; (3) motor symptoms severity: UPDRS-III total score « on »; (4) depressive symptom severity: BDI-II total score; (5) anxiety symptoms severity: BAI total score; (6) insomnia symptom severity: ISI total score; (7) sleep latency (min; log-transformed); (8) sleep efficiency (%); (9) N2 and N3 sleep (%); (10) AHI (log-transformed); (11) mean O2 saturation (%); (12) a global cognitive measure: MMSE raw score; (13) executive control and processing speed measure: Trail Making Test, part B (time); and (14) visuospatial selective attention measure: Bells Test (number of omissions).
We selected the 14 a priori variables to be included in the PCA model. Since this selection showed missing data (e.g., incomplete questionnaires), we proceeded to multiple imputation (MI) targeted for exploratory factor analyses as described in Rubin et al. to optimize the sample in the context of limited statistical power74. Further details are described in Supplementary Material (Table S2). Once the MI procedure had been completed, we repeated our statistical analyzes with imputed data, and the effects remained similar.
For the Principal Component Analysis (PCA), this factor analysis created components of variables that were highly intra-correlated but not inter-correlated from one another. Each variable included in a component has a loading which characterizes the magnitude of covariance observed between the variables within the same component. The number of factors was determined using a minimal eigenvalue of 1. Eigenvalues, used as references, were derived from random data sets resulting from running 1000 iterations.
PD participants were divided into two groups based on the presence or absence of EDS via a score higher than 10 on the ESS (PD with EDS > 10, PD without EDS ≤ 10). To evaluate potential confounding variables such as age, sex, educational level and RBD status, one-way analysis of variance (ANOVA) comparing PD participants with and without EDS on these variables were first computed. Only age differed significantly between the two groups (p < 0.05). Hence, an analysis of covariance (ANCOVAs) was performed on each component using age as a covariable to compare participants with and without EDS. F- and p-values using the significant covariate are reported. Effect sizes (ES) were measured using the partial eta squared. Results were considered significant when p < 0.05.
In Study 2, for cortical surface analysis, general linear models (GLM) were built at each vertex to investigate the association between ESS and thickness, surface area, and volume of the cortical surface, controlling for the effects of age, sex, and PD duration. Since dosage of DA receptor agonists and PD duration were strongly correlated (r = 0.58, p = 0.001), only PD duration was included. Total intracranial volume was also added as a covariate for cortical surface area and volume analyses75. We also performed complementary analyses to investigate the presence of thickness, surface area, and volume differences between PD participants with and without EDS using the same covariates. Clusters were considered significant when p < 0.05 based on a Monte Carlo simulation approach.
For subcortical shape analysis, GLM were also used to study at each vertex the association between ESS and surface displacement in the 14 structures while controlling for age, sex and PD duration. Total intracranial volume was not added as a covariate since surface meshes were registered to the MNI152 space. Non-parametric permutation inference was performed using randomize with 10,000 permutations and a threshold-free cluster enhancement (TCFE) approach76. Clusters were considered significant when p < 0.05 after correction for family-wise error. The subcortical surface differences between groups were also investigated as complementary analyses using the same covariates.
Multiple regressions of ESS on VBM and DBM-derived brain measurements were used to predict whole-brain regional gray matter volume (GMV) using CAT12 version 12.7 for SPM12 under MATLAB R2018b (smoothing at 8 mm). All analyses were corrected for age, sex, and PD duration and total intracranial volume was added for VBM analyses. A TCFE was applied, and statistical significance was set at p < 0.05 corrected for multiple comparisons using false-discovery rate (FDR). Additionally, we performed t-tests for complementary between-group comparisons (PD with and without EDS) using the statistical module in CAT12.
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request by any qualified investigator and in compliance with the specifications of the institutional ethics committees. Note that data sharing was not included in the informed consent signed by the patient, so approval from institutional ethics committees is required.
Code availability
The underlying code for this study is not publicly available but may be made available to qualified researchers upon request to the corresponding author (see data availability).
References
Santamaria, J. How to evaluate excessive daytime sleepiness in Parkinson’s disease. Neurology 63, S21–S23 (2004).
Sateia, M. J. International classification of sleep disorders-third edition: highlights and modifications. Chest 146, 1387–1394 (2014).
Chahine, L. M., Amara, A. W. & Videnovic, A. A systematic review of the literature on disorders of sleep and wakefulness in Parkinson’s disease from 2005 to 2015. Sleep. Med. Rev. 35, 33–50 (2017).
Ehgoetz Martens, K. A. & Shine, J. M. The interactions between non-motor symptoms of Parkinson’s disease. Expert Rev. Neurother. 18, 457–460 (2018).
Braga-Neto, P., da Silva-Junior, F. P., Sueli Monte, F., de Bruin, P. F. & de Bruin, V. M. Snoring and excessive daytime sleepiness in Parkinson’s disease. J. Neurol. Sci. 217, 41–45 (2004).
Xiang, Y. Q. et al. Clinical features and correlates of excessive daytime sleepiness in Parkinson’s disease. Front Neurol. 10, 121 (2019).
Yoo, S. W., Kim, J. S., Oh, Y. S., Ryu, D. W. & Lee, K. S. Excessive daytime sleepiness and its impact on quality of life in de novo Parkinson’s disease. Neurol. Sci. 40, 1151–1156 (2019).
Zhu, K., van Hilten, J. J. & Marinus, J. Course and risk factors for excessive daytime sleepiness in Parkinson’s disease. Parkinsonism Relat. Disord. 24, 34–40 (2016).
Feng, F. et al. Excessive daytime sleepiness in Parkinson’s disease: A systematic review and meta-analysis. Parkinsonism Relat. Disord. 85, 133–140 (2021).
Hoglund, A. et al. A 10-year follow-up of excessive daytime sleepiness in Parkinson’s disease. Parkinsons Dis. 2019, 5708515 (2019).
Amara, A. W. et al. Longitudinal assessment of excessive daytime sleepiness in early Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 88, 653–662 (2017).
Wen, M. C. et al. Neural substrates of excessive daytime sleepiness in early drug naive Parkinson’s disease: A resting state functional MRI study. Parkinsonism Relat. Disord. 24, 63–68 (2016).
Cochen De Cock, V. et al. Daytime sleepiness in Parkinson’s disease: a reappraisal. PLoS ONE 9, e107278 (2014).
Maggi, G., Trojano, L., Barone, P. & Santangelo, G. Sleep disorders and cognitive dysfunctions in Parkinson’s disease: a meta-analytic study. Neuropsychol. Rev. 31, 643–682 (2021).
Jester, D. J., Lee, S., Molinari, V. & Volicer, L. Cognitive deficits in Parkinson’s disease with excessive daytime sleepiness: a systematic review. Aging Ment. Health 24, 1769–1780 (2020).
Hoglund, A., Broman, J. E., Palhagen, S., Fredrikson, S. & Hagell, P. Is excessive daytime sleepiness a separate manifestation in Parkinson’s disease? Acta Neurol. Scand. 132, 97–104 (2015).
van Rooden, S. M., Visser, M., Verbaan, D., Marinus, J. & van Hilten, J. J. Patterns of motor and non-motor features in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 80, 846–850 (2009).
Shen, Y., Huang, J. Y., Li, J. & Liu, C. F. Excessive daytime sleepiness in Parkinson’s disease: clinical implications and management. Chin. Med. J. (Engl.) 131, 974–981 (2018).
Liu, H. et al. Excessive daytime sleepiness in Parkinson’s disease. Nat. Sci. Sleep. 14, 1589–1609 (2022).
Bliwise, D. L. et al. Daytime alertness in Parkinson’s disease: potentially dose-dependent, divergent effects by drug class. Mov. Disord. 27, 1118–1124 (2012).
Scanga, A., Lafontaine, A. L. & Kaminska, M. An overview of the effects of levodopa and dopaminergic agonists on sleep disorders in Parkinson’s disease. J. Clin. Sleep. Med. 19, 1133–1144 (2023).
Liguori, C. et al. Daytime sleepiness may be an independent symptom unrelated to sleep quality in Parkinson’s disease. J. Neurol. 266, 636–641 (2019).
Bargiotas, P., Lachenmayer, M. L., Schreier, D. R., Mathis, J. & Bassetti, C. L. Sleepiness and sleepiness perception in patients with Parkinson’s disease: a clinical and electrophysiological study. Sleep 42, zsz004 (2019).
Stauder, M., Klerman, E. B., Wang, W., Erickson, H. & Videnovic, A. Relationships of self-reported and objective measures of sleep, sleepiness, and sleep quality in Parkinson’s disease. Parkinsonism Relat. Disord. 101, 57–61 (2022).
Schreiner, S. J. et al. Sleep spindle and slow wave activity in Parkinson disease with excessive daytime sleepiness. Sleep 46, zsac165 (2022).
Chondrogiorgi, M. et al. Multimodal imaging evaluation of excessive daytime sleepiness in Parkinson’s disease. Int. J. Neurosci. 126, 422–428 (2016).
Kato, S. et al. Widespread cortical and subcortical brain atrophy in Parkinson’s disease with excessive daytime sleepiness. J. Neurol. 259, 318–326 (2012).
de Schipper, L. J., van der Grond, J., Marinus, J., Henselmans, J. M. L. & van Hilten, J. J. Loss of integrity and atrophy in cingulate structural covariance networks in Parkinson’s disease. Neuroimage Clin. 15, 587–593 (2017).
Hutton, C., Draganski, B., Ashburner, J. & Weiskopf, N. A comparison between voxel-based cortical thickness and voxel-based morphometry in normal aging. Neuroimage 48, 371–380 (2009).
Hauser, R. A., Gauger, L., Anderson, W. M. & Zesiewicz, T. A. Pramipexole-induced somnolence and episodes of daytime sleep. Mov. Disord. 15, 658–663 (2000).
Micallef, J. et al. Antiparkinsonian drug-induced sleepiness: a double-blind placebo-controlled study of L-dopa, bromocriptine and pramipexole in healthy subjects. Br. J. Clin. Pharm. 67, 333–340 (2009).
Neikrug, A. B. et al. Effects of sleep disorders on the non-motor symptoms of Parkinson disease. J. Clin. Sleep. Med. 9, 1119–1129 (2013).
De Cock, V. C. et al. Is obstructive sleep apnea a problem in Parkinson’s disease? Sleep. Med. 11, 247–252 (2010).
Beland, S. G. et al. Observational study of the relation between Parkinson’s disease and sleep apnea. J. Parkinsons Dis. 5, 805–811 (2015).
Ghaziri, J. et al. The corticocortical structural connectivity of the human insula. Cereb. Cortex 27, 1216–1228 (2017).
Ghaziri, J. et al. Subcortical structural connectivity of insular subregions. Sci. Rep. 8, 8596 (2018).
Kurth, F., Zilles, K., Fox, P. T., Laird, A. R. & Eickhoff, S. B. A link between the systems: functional differentiation and integration within the human insula revealed by meta-analysis. Brain Struct. Funct. 214, 519–534 (2010).
Motomura, Y. et al. The role of the thalamus in the neurological mechanism of subjective sleepiness: an fMRI study. Nat. Sci. Sleep. 13, 899–921 (2021).
Sridharan, D., Levitin, D. J. & Menon, V. A critical role for the right fronto-insular cortex in switching between central-executive and default-mode networks. Proc. Natl Acad. Sci. USA 105, 12569–12574 (2008).
Christopher, L., Koshimori, Y., Lang, A. E., Criaud, M. & Strafella, A. P. Uncovering the role of the insula in non-motor symptoms of Parkinson’s disease. Brain 137, 2143–2154 (2014).
Uddin, L. Q., Nomi, J. S., Hebert-Seropian, B., Ghaziri, J. & Boucher, O. Structure and function of the human insula. J. Clin. Neurophysiol. 34, 300–306 (2017).
Fereshtehnejad, S. M. et al. Evolution of prodromal Parkinson’s disease and dementia with Lewy bodies: a prospective study. Brain 142, 2051–2067 (2019).
Rahayel, S. et al. Mitochondrial function-associated genes underlie cortical atrophy in prodromal synucleinopathies. Brain 146, 3301–3318 (2023).
Rahayel, S. et al. Brain atrophy in prodromal synucleinopathy is shaped by structural connectivity and gene expression. Brain 145, 3162–3178 (2022).
Van Essen, D. C. A tension-based theory of morphogenesis and compact wiring in the central nervous system. Nature 385, 313–318 (1997).
Jubault, T. et al. Patterns of cortical thickness and surface area in early Parkinson’s disease. Neuroimage 55, 462–467 (2011).
Ashraf-Ganjouei, A. et al. White matter tract alterations in drug-naive Parkinson’s disease patients with excessive daytime sleepiness. Front Neurol. 10, 378 (2019).
De Micco, R., Russo, A. & Tessitore, A. Structural MRI in idiopathic Parkinson’s disease. Int. Rev. Neurobiol. 141, 405–438 (2018).
Sterling, N. W., Lewis, M. M., Du, G. & Huang, X. Structural imaging and parkinson’s disease: moving toward quantitative markers of disease progression. J. Parkinsons Dis. 6, 557–567 (2016).
Gong, L. et al. Striatum shape hypertrophy in early stage Parkinson’s disease with excessive daytime sleepiness. Front Neurosci. 13, 1353 (2019).
Happe, S. et al. Association of daytime sleepiness with nigrostriatal dopaminergic degeneration in early Parkinson’s disease. J. Neurol. 254, 1037–1043 (2007).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
American Psychiatric Association. Diagnostic and statistical manual of mental disorders (5th ed.). Washington, DC: Author (2013).
Dubois, B. et al. Diagnostic procedures for Parkinson’s disease dementia: recommendations from the movement disorder society task force. Mov. Disord. 22, 2314–2324 (2007).
Tomlinson, C. L. et al. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov. Disord. 25, 2649–2653 (2010).
Goetz, C. G. et al. Movement disorder society-sponsored revision of the unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov. Disord. 23, 2129–2170 (2008).
Hoehn, M. M. & Yahr, M. D. Parkinsonism: onset, progression and mortality. Neurology 17, 427–442 (1967).
Berry, R. B. et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the scoring of sleep and associated events. deliberations of the sleep apnea definitions task force of the American Academy of Sleep Medicine. J. Clin. Sleep. Med. 8, 597–619 (2012).
Montplaisir, J. et al. Polysomnographic diagnosis of idiopathic REM sleep behavior disorder. Mov. Disord. 25, 2044–2051 (2010).
Medicine, A.A.O.S. The International Classification of Sleep Disorders 3rd edn (ICSD-3) (American Academy of Sleep Medicine, 2014).
Cesari, M. et al. Video-polysomnography procedures for diagnosis of rapid eye movement sleep behavior disorder (RBD) and the identification of its prodromal stages: guidelines from the International RBD Study Group. Sleep 45, zsab257 (2022).
Litvan, I. et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov. Disord. 27, 349–356 (2012).
De Roy, J. et al. Detecting the Cognitive Prodrome of Dementia in Parkinson’s Disease. J. Parkinsons Dis. 10, 1033–1046 (2020).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 12, 189–198 (1975).
Beck, A. T., Ward, C. H., Mendelson, M., Mock, J. & Erbaugh, J. An inventory for measuring depression. Arch. Gen. Psychiatry 4, 561–571 (1961).
Beck, A. T., Epstein, N., Brown, G. & Steer, R. A. An inventory for measuring clinical anxiety: psychometric properties. J. Consult Clin. Psychol. 56, 893–897 (1988).
Bastien, C. H., Vallieres, A. & Morin, C. M. Validation of the Insomnia Severity Index as an outcome measure for insomnia research. Sleep. Med. 2, 297–307 (2001).
Johns, M. W. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 14, 540–545 (1991).
Dale, A. M., Fischl, B. & Sereno, M. I. Cortical surface-based analysis. I. Segmentation and surface reconstruction. Neuroimage 9, 179–194 (1999).
Patenaude, B., Smith, S. M., Kennedy, D. N. & Jenkinson, M. A Bayesian model of shape and appearance for subcortical brain segmentation. Neuroimage 56, 907–922 (2011).
Rahayel, S. et al. Subcortical amyloid relates to cortical morphology in cognitively normal individuals. Eur. J. Nucl. Med. Mol. Imaging 46, 2358–2369 (2019).
Rahayel, S. et al. Brain atrophy in Parkinson’s disease with polysomnography-confirmed REM sleep behavior disorder. Sleep 42, zsz062 (2019).
Curran, P. J., West, S. G. & Finch, J. F. The robustness of test statistics to nonnormality and specification error in confirmatory factor analysis. Psychol. Methods 1, 14 (1996).
Rubin, R. Multiple Imputation for Nonresponse in Surveys (John Wiley & Sons, Inc, 2004).
Im, K. et al. Brain size and cortical structure in the adult human brain. Cereb. Cortex 18, 2181–2191 (2008).
Smith, S. M. & Nichols, T. E. Threshold-free cluster enhancement: addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 44, 83–98 (2009).
Acknowledgements
This study was funded by the Canadian Institutes of Health Research (CIHR) and the Fonds de recherche du Québec – Santé (FRQ-S). T.R. received a scholarship from the CIHR and FRQ-S. S.R. received a scholarship from the CIHR. A.B. received a scholarship from the Social Sciences and Humanities Research Council (SSHRC). J.M. holds a Canada Research Chair in Sleep Medicine. J.F.G. holds a Canada Research Chair in Cognitive Decline in Pathological Aging. The data supporting the findings of this study are available within the article and/or its supplementary material.
Author information
Authors and Affiliations
Contributions
T.R.: literature review, analysis and interpretation, writing of the original version; R.B.P: study concept and design, analysis and interpretation, critical revision of the manuscript for important intellectual content; S. R.: analysis and interpretation (Neuroimaging study), critical revision of the manuscript for important intellectual content; V.D.: analysis and interpretation (Neuroimaging study), critical revision of the manuscript for important intellectual content; A.B.: analysis and interpretation (Clinical correlates), critical revision of the manuscript. J.M.L.: analysis and interpretation, critical revision of the manuscript for important intellectual content; J.M: study concept and design, study supervision, critical revision of the manuscript for important intellectual content. J.C: analysis and interpretation, critical revision of the manuscript for important intellectual content. J.F.G: study concept and design, study supervision, analysis and interpretation, critical revision of the manuscript for important intellectual content; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors have no conflicts of interest to report related to the presented work. However, the authors would like to share the following financial and non-financial disclosures: T.R. received PhD scholarships from the Fonds de Recherche du Québec—Santé (FRQ-S) and the Canadian Institutes of Health Research (CIHR). R.B.P. received grants from the CIHR and the National Institutes of Health (NIH)/National Institute on Aging (NIA). He received personal compensation for travel, speaker fees, and consultation from Biotie, Biogen, Boehringer-Ingelheim, Roche, and Teva Neurosciences, outside the submitted work. J.M. received personal compensation for consultancy services from Takeda, Merck, Paldin and Eisai outside the submitted work. S.R. received received postdoctoral a scholarship from the CIHR. A.B. received a scholarship from the Social Sciences and Humanities Research Council (SSHRC). V.D. reports no disclosure. J.M. received grants from the CIHR. He holds a Canada Research Chair in Sleep Medicine. J.M.L. reports no disclosure. J.C. received grants from the CIHR. She received personal compensation for consultancy services from Eisai outside the submitted work. J.F.G. received grants from the CIHR, the FRQ-S, and the NIH/NIA. He holds a Canada Research Chair in Cognitive Decline in Pathological Aging.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosinvil, T., Postuma, R.B., Rahayel, S. et al. Clinical symptoms and neuroanatomical substrates of daytime sleepiness in Parkinson’s disease. npj Parkinsons Dis. 10, 149 (2024). https://doi.org/10.1038/s41531-024-00734-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-024-00734-x
- Springer Nature Limited