
Abstract
Cystic fibrosis (CF) is one of the most common genetic diseases worldwide with high carrier frequencies across different ethnicities. Next generation sequencing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene has proven to be an effective screening tool to determine carrier status with high detection rates. Here, we evaluate the performance of the Swift Biosciences Accel-Amplicon CFTR Capture Panel using CFTR-positive DNA samples. This assay is a one-day protocol that allows for one-tube reaction of 87 amplicons that span all coding regions, 5′ and 3′UTR, as well as four intronic regions. In this study, we provide the FASTQ, BAM, and VCF files on seven unique CFTR-positive samples and one normal control sample (14 samples processed including repeated samples). This method generated sequencing data with high coverage and near 100% on-target reads. We found that coverage depth was correlated with the GC content of each exon. This dataset is instrumental for clinical laboratories that are evaluating this technology as part of their carrier screening program.
Measurement(s) | amplicon sequencing |
Technology Type(s) | DNA sequencing |
Factor Type(s) | CFTR mutations |
Sample Characteristic - Organism | Homo sapiens |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.10060244
Similar content being viewed by others

Background & Summary
Cystic fibrosis (CF) is considered one of the most common genetic diseases, affecting 1 in 2500–3500 live births in Caucasian populations1. Over 1500 mutations have been previously reported in the CFTR gene. Due to the high carrier rates, the American College of Obstetricians and Gynecologists (ACOG) suggests CF carrier testing for all women who are considering pregnancy or are currently pregnant2,3,4. In 2004, the American College of Medical Genetics and Genomics (ACMG) published a guideline on testing 23 CFTR mutations with high carrier frequencies across different ethnicities3. However, to increase the detection rate, it has become a common practice for clinical laboratories to expand the CFTR panel to more than 100 mutations, and even full gene analysis5,6,7.
In the past three decades, the detection of CFTR mutations has evolved through various molecular methods, including reverse dot blot, restriction fragment length polymorphism (RFLP), and Sanger sequencing8,9. The advent of next generation sequencing (NGS) leads to a higher clinical sensitivity by screening more targeted CFTR mutations and sequencing of the exonic gene regions, as well as a higher throughput by multiplexing many samples into one sequencing run10,11. While NGS excels at generating large amount of data, it is time-consuming and less cost-effective for sequencing few targets and low volume of samples. Recently, Swift Biosciences released a pre-designed amplicon/library preparation kit that can amplify the CFTR gene using 87 amplicons in one reaction. Combined with Illumina MiSeq Nano kit v2 (300-cycles), this protocol allows for quick turnaround time, low sample volume, and cost effectiveness.
While a previous study had demonstrated that this method could detect frequent and rare CFTR mutations when compared to other methods, the technical specifications were not analysed12. Here we examine the Accel-Amplicon CFTR Panel using CF-positive samples by assessing the performance of this assay. We processed seven CF-positive samples that represent across the CFTR mutation spectrum (missense, nonsense, splicing and indels), and these mutations are recommended in the ACMG guideline3. The first run included one normal sample and three CF-positive samples, and the second run included all samples from the first run, with additional four CF-positive samples (Table 1).
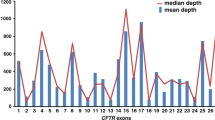
Using the MiSeq Nano v2 kit, the sequencing coverage depth averages for run 1 (four samples) and run 2 (ten samples) are 5753x and 1344x, respectively, with almost 100% of the CFTR target region being more than 20x (Table 1). As expected for amplicon sequencing, 98–99% of sequencing reads are on-target. We analysed the sequencing performance on the exon level. The coding region, 5′UTR and 3′UTR of the CFTR gene has 6123 bp, while the amplicon covers these regions with more than 3000 bp padded region (targeted amplicon size = 9666 bp), with additional amplicons covering four intronic regions (introns 1, 12, 22, and 25) (Table 2). The number of amplicons for each exon correlates with the size of the exons (R2 = 0.9766%) (Fig. 1).

Correlation of amplicon numbers and exon size. The numbers of amplicons for each exon is plotted against the exon size, except intron 1, 12, and 22. A trendline is plotted from the data and R2 is calculated to be 0.9766.
Using the manufacturer’s recommended bioinformatic pipeline, we were able to detect all the mutations in the CF-positive samples. No pathogenic variants were detected in sample 1 (normal control) in both runs. Repeated samples in the inter- and intra-run analyses were found to be concordant (See technical validation section for more details).
Here, we provide the FASTQ files for each of the samples in this validation study. Tables 1 and 2 provide the coverage summary for each sample and each exon. Furthermore, in the method and technical validation section, we describe the steps and quality control (QC) performed to ensure the accuracy and precision of the assay.
To our knowledge, no previous studies have critically evaluated the sequencing performance of the Accel-Amplicon CFTR panel. As analytical performance of the methodology is vital for a clinical test, the data generated in this study can be evaluated by clinical genetic laboratories that are interested in employing the Accel-Amplicon CFTR panel to screen CF carriers. As carrier screening becomes more well-known and consumer demand increases, this method fulfils the need of an affordable and time-sensitive approach to screen CFTR mutations in general population carrier screening with a maximum detection rate.
Methods
Validation samples acquisition and DNA quantification
The following DNA samples (samples 1–3, 5–8) were obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research (see the corresponding Coriell naming convention in Table 3). Sample 4 was acquired from a patient; an informed consent was obtained for research using an IRB protocol (06-004886) at the Center for Applied Genomics at the Children’s Hospital of Philadelphia. The consent agreement states that genotype data may be shared with public data repositories for research purposes, and that the patient’s personal information would be kept private and unidentifiable in any publication or presentation. DNA concentration was calculated using a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, catalogue number Q32851). Samples were diluted down to 5 ng/mL with Pre-PCR TE buffer and a final volume of 10 μL containing 20 ng input DNA was used.
Library preparation
Library preparation was performed using the Accel-Amplicon CFTR panel (Swift Bioscience, catalogue number AL-55048) in accordance with the manufacturer’s protocol. In brief, multiplex PCR was performed on the sample DNA using the reagents provided by the Accel-Amplicon panel kit for 4 cycles of 10 sec at 98 °C, 5 min at 63 °C, 1 min at 65 °C and 22 cycles of 10 sec at 98 °C, 1 min at 64 °C. Size selection and clean-up were performed using SPRIselect beads (Beckman Coulter, catalogue number B23318) with a ratio of 1.2. Indexing sequencing adapters were then ligated to each library at 37 °C for 20 minutes. A second clean-up step was performed using SPRIselect beads at a ratio of 0.85 and rediluted with 20 mL of Post-PCR TE buffer. Quantification of adapted libraries was performed by qPCR using KAPA Library Quantification Kit (KAPA Biosystems, catalogue number 07960140001).
Next-generation sequencing
Illumina MiSeq Nano Reagent Kit V2 was used to sequence the samples (Table 1). The final pooled concentration of 2 nM (5 μL was used) was mixed with 0.2 N NaOH (5 μL). The mixture was then mixed with 990 μL of pre-chilled HT1 to obtained a 10 pM denatured library mixed. No PhiX spike-in was used.
Bioinformatic analysis
Sequencing data was analysed based on the bioinformatic pipeline recommended and provided by Swift Biosciences. In short, adapter-trimmed paired-end FASTQ files were generated by the Illumina MiSeq upon completion of the sequencing run (Note: adapter trimming can be done post FASTQ generation). For each sample, an alignment in Sequence Alignment Map (SAM) format was generated from the pair of FASTQ files using Burrows-Wheeler Aligner (BWA) and hg19 human genome reference. The SAM file was further modified by SAMtools to sort the file by name for Swift primerclip preparation. Due to the presence of synthetic primer sequences at the start or end of reads, the primerclip tool was used to remove these sequences before proceeding with downstream analysis. With both Picard’s AddOrReplaceReadGroups tool and SAMtools, the primer-clipped SAM file was converted to BAM format and an indexed BAM file was generated. Variant calling was performed using GATK HaplotypeCaller. To determine quality metrics at the sample and interval level, Picard’s CollectTargetPcrMetrics was used.
Sanger sequencing
Pathogenic variants were confirmed using Sanger sequencing. PCR was performed using QIAGEN Fast Cycling PCR kit (#203743) with primers flanking the variants of interest (Tables 4 and 5). The PCR conditions were: 5 minutes at 95 °C, 35 cycles [5 seconds at 96 °C, 5 seconds at 58 °C, 40 seconds at 68 °C], 1 minute at 72 °C. PCR products were purified using Applied Biosystems ExoSAP-IT PCR Product Cleanup Reagent (#78201.1.ML). Sequencing reactions were performed using Applied Biosystems BigDye Terminator v1.1 Cycle Sequencing Kit (#4337449), and were purified using Applied Biosystem Centri-Seq. 8-Well Strips (#4367820). Sanger sequencing was performed using Applied Biosystems 3500 Genetic Analyzer (#4440462).
BWA-MEM alignment
bwa mem ${FASTA} ${Sample_ID}_R1.fastq.gz ${Sample_ID}_R2.fastq.gz -U 17 -M -t 32 > ${Sample_ID}_bwa.sam.
SAMtools sort SAM
samtools sort -n ${Sample_ID}_bwa.sam -o ${Sample_ID}_bwa_nsorted.sam.
Primerclip
primerclip Accel-Amplicon_CFTR_masterfile.txt ${Sample_ID}_bwa_nsorted.sam ${Sample_ID}_bwa_primertrimmed.sam.
SAMtools convert SAM to BAM
java -jar picard.jar AddOrReplaceReadGroups I=${Sample_ID}_bwa_primertrimmed.sam O=${Sample_ID}_bwa_primertrimmed.bam SO=coordinate RGID=snpID LB=swift SM=${Sample_ID} PL=illumina PU=miseq VALIDATION_STRINGENCY=STRICT.
samtools index ${Sample_ID}_bwa_primertrimmed.bam ${Sample_ID}_bwa_primertrimmed.bam.bai.
Picard CollectPcrMetrics tool
samtools view -H ${Sample_ID}_bwa_primertrimmed.bam > ${Sample_ID}_bwa_header.txt.
cat ${Sample_ID}_bwa_header.txt cftr_180313_nonmerged_targets_5col.bed > ${Sample_ID}_bwa_fullintervals.
cat ${Sample_ID}_bwa_header.txt cftr_180313_nonmerged_targets_5col.bed > ${Sample_ID}_bwa_noprimerintervals.
java -jar picard.jar CollectTargetedPcrMetrics I=${Sample_ID}_bwa_primertrimmed.bam O=${Sample_ID}_bwa_targetPCRmetrics.txt AI=${Sample_ID}_bwa_fullintervals TI=${Sample_ID}_bwa_noprimerintervals R=${FASTA} PER_TARGET_COVERAGE=${Sample_ID}_bwa_perTargetCov.txt VALIDATION_STRINGENCY=STRICT.
GATK variant calling
java -jar GenomeAnalysisTK.jar -T HaplotypeCaller -R ${FASTA} -I ${Sample_ID}_bwa_primertrimmed.bam -stand_call_conf 20 -stand_emit_conf 20 -mbq 20 -L CFTR_merged_5col.bed -o ${Sample_ID}_bwa_gatkHC.vcf.
Data Records
There are eight unique samples in our cohort. Samples 1–4 were analysed in both runs. Samples 5–8 were analysed in run 2. Sample 4 was run in triplicate in the second run. fastq can be accessed from the Sequence Read Archive (SRA) repository under SRA: SRP19346913. Direct FASTQ files can be downloaded via SRA Toolkit using command line “fastq-dump–split-3 -G SRR#” (Table 5). BAM files can be downloaded at (https://doi.org/10.6084/m9.figshare.11341958.v1), and VCF files can be downloaded at (https://doi.org/10.6084/m9.figshare.10565513.v1)14,15.
Technical Validation
Library quantitation
To evaluate whether the DNA samples were successfully processed using this Swift Accel Amplicon protocol, we used the KAPA Library Quantification Kit to measure the library concentration. During qPCR, primers bound to the Illumina P5 and P7 flow cell oligo sequences and the concentrations of the samples were assessed by measuring the SYBR green fluorescence intensity; this method specifically measures the adapted DNA, excluding any unadapted DNA fragments generated during the PCR step. The concentration of each sample in both runs are listed in Table 6.
Sequencing data assessment
Pooled libraries were sequenced using Illumina MiSeq Nano Reagent Kit V2 kit (300 cycles). The cluster densities for run 1 and 2 were 807 ± 1 k/mm2 and 534 ± 8 k/mm2, with 98.08% and 98.05% of reads of Q30 score or more, respectively (Table 6). Further analyses of the FASTQ files using MultiQC showed that the majority of the base positions had mean quality value of Q38, while the first five bases of reads have lower quality scores (at around Q33) (Fig. 2a). For all FASTQ files, the majority of the reads had quality value of Q38 (Fig. 2b)16. Overall coverage depth of all processed samples is demonstrated in Table 1. As expected, the mean coverage depth in run 1 (5753x) is higher than those of run 2 (1344x), as there are fewer samples pooled into one flow cell in run 1 (Table 1). Moreover, all samples from run 1 have 100% of regions with more than 20x coverage depth (Table 1). For run 2, all samples have less than 20x coverage at the 3′UTR region (chr7:117308320–117308346; CFTR:c.*1158_*1184). This region has no known pathogenic variants described in HGMD or in ClinVar. In addition, samples 5, 6, and 7 have no coverage for two bases in intron 8 (chr7:117188661–117188662; CFTR:c.1210-13_1210-12). This is a common TG repeat deletion that is present in 22.92% of general population according to gnomAD. Next, we assessed the coverage depth per exon, and investigated the inter-exonic depth variability (Tables 2 and 7). We found that the coverage depth was higher as the GC content of the exon was closer to 50% for both runs (Fig. 2c,d). As expected for amplicon sequencing, the majority of sequencing reads (98–99%) were aligned to the targeted regions (See Supplementary File 1 for BED file).

Sequence quality and coverage depth per exon. Sequence quality was assessed using MultiQC. Each green line represents one FASTQ file. (a) Mean quality value across each base position in the read. (b) Number of reads with average quality score. (c,d) For both runs, the coverage depth of exons increases as the GC content approaches 50%.
Assay validation of CF-positive samples
Samples used in this validation study have known pathogenic CFTR mutations (Table 3), and they were used to validate this Swift Accel-Amplicon CFTR Panel for usage in a clinical laboratory setting. Analytical validation is a vital component in the process of launching a clinical genetic test, as it demonstrates the quality and performance of the testing method and the accuracy of the assay result. Here, we evaluate the capability of this assay by assessing the variants that were detected in each sample. As expected, there were no pathogenic variants detected in the control sample (sample 1) for both runs. The pathogenic variants of samples 2–8 were confirmed by the manufacturer-recommended bioinformatic pipeline. These genotypes can be visualized using Integrative Genome Viewer (IGV), and they have also been confirmed using Sanger sequencing (Fig. 3); this yields a 100% sensitivity. Furthermore, samples 1–4 were sequenced in both runs, and sample 4 was sequenced three times in run 2. All results were concordant and matched to the referenced genotypes, hence the repeatability and reproducibility is 100%. Additionally, since there can be non-pathogenic variants in CFTR, we provide a table of all the variants detected in each VCF file for each sample in both run (Online-only Table 1). HGVS nomenclature and GnomAD frequencies for each variant are also listed. Of note, the VCF for sample 1 in run 2 contains a variant that is not present in run 1. This variant is a common two-nucleotide deletion of a TG-repeat stretch in intron 8. This dinucleotide repeat is adjacent to a poly-T stretch that also has common deletions and duplications. This discrepancy may be due to the fact that NGS alignment and annotation tools cannot reliably detect small insertions/deletions at repetitive regions. Sanger sequencing is still the preferred method to reliably detect variants at this repeat.

Variant visualization using IGV and Mutation Surveyor. The variants for each corresponding sample are confirmed by visualizing the BAM files in Integrative Genomic Viewer (IGV). The Sanger sequence traces visualized using MutationSurveyor are also shown for each variant of each sample.
Code availability
Swift Primerclip installation instructions, scripts, and examples can be found at https://github.com/swiftbiosciences/primerclip. Current available methods for downloading the Swift Primerclip tool are a pre-compiled binary for linux on x86_64 and building from source using Haskell-stack build tool. Additional requirements include SAMTools (1.6-2-gf068ac2), Picard Tools (2.1.0), BWA (0.7.17-r1188), GATK (3.5-0-g36282e4), and Java (1.8). Codes and parameters are described as below.
References
Scotet, V. et al. Evidence for decline in the incidence of cystic fibrosis: a 35-year observational study in Brittany, France. Orphanet J Rare Dis 7, 14, https://doi.org/10.1186/1750-1172-7-14 (2012).
Committee on, G. Committee Opinion No. 691: Carrier Screening for Genetic Conditions. Obstet Gynecol 129, e41–e55, https://doi.org/10.1097/AOG.0000000000001952 (2017).
Watson, M. S. et al. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med 6, 387–391, doi:10.109701.GIM.0000139506.11694.7C (2004).
Richards, C. S. et al. Standards and guidelines for CFTR mutation testing. Genet Med 4, 379–391, doi:10.109700125817-200209000-00010 (2002).
Hughes, E. E. et al. Clinical Sensitivity of Cystic Fibrosis Mutation Panels in a Diverse Population. Hum Mutat 37, 201–208, https://doi.org/10.1002/humu.22927 (2016).
Currier, R. J. et al. Genomic sequencing in cystic fibrosis newborn screening: what works best, two-tier predefined CFTR mutation panels or second-tier CFTR panel followed by third-tier sequencing? Genet Med 19, 1159–1163, https://doi.org/10.1038/gim.2017.32 (2017).
Beauchamp, K. A. et al. Sequencing as a first-line methodology for cystic fibrosis carrier screening. Genet Med. https://doi.org/10.1038/s41436-019-0525-y (2019).
Raskin, S., Phillips, J. A. III., Kaplan, G., McClure, M. & Vnencak-Jones, C. Cystic fibrosis genotyping by direct PCR analysis of Guthrie blood spots. PCR Methods Appl 2, 154–156 (1992).
Dooki, M. R., Akhavan-Niaki, H. & Juibary, A. G. Detecting Common CFTR Mutations by Reverse Dot Blot Hybridization Method in Cystic Fibrosis First Report from Northern Iran. Iran J Pediatr 21, 51–57 (2011).
Lucarelli, M. et al. A New Targeted CFTR Mutation Panel Based on Next-Generation Sequencing Technology. J Mol Diagn 19, 788–800, https://doi.org/10.1016/j.jmoldx.2017.06.002 (2017).
Nakano, E. et al. Targeted next-generation sequencing effectively analyzed the cystic fibrosis transmembrane conductance regulator gene in pancreatitis. Dig Dis Sci 60, 1297–1307, https://doi.org/10.1007/s10620-014-3476-9 (2015).
Hendrix, M. M., Foster, S. L. & Cordovado, S. K. Newborn Screening Quality Assurance Program for CFTR Mutation Detection and Gene Sequencing to Identify Cystic Fibrosis. J Inborn Errors Metab Screen 4, https://doi.org/10.1177/2326409816661358 (2016).
NCBI Sequence Read Archive, https://identifiers.org/insdc.sra:SRP193469 (2019).
Vaccaro, C. Performance evaluation of sequencing data using Swift Accel-Amplicon CFTR Panel_BAMs. figshare, https://doi.org/10.6084/m9.figshare.11341958.v1 (2019).
Vaccaro, C. Performance evaluation of sequencing data using Swift Accel-Amplicon CFTR Panel_VCFs. figshare, https://doi.org/10.6084/m9.figshare.10565513.v1 (2019).
Ewels, P., Magnusson, M., Lundin, S. & Kaller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048, https://doi.org/10.1093/bioinformatics/btw354 (2016).
Acknowledgements
Sequencing and analyses in this study were supported by the Center for Applied Genomics at the Children’s Hospital of Philadelphia. The authors thank Drew McUsic for his technical assistance.
Author information
Authors and Affiliations
Contributions
M.L.L. analysed the data, designed and supervised the study, and wrote the manuscript. F.M. performed the experiments and wrote the method session of the manuscript. C.V. and A.W. performed the bioinformatic analysis and wrote the bioinformatic analysis session of the article. A.W. performed the bioinformatic analysis. D.W. analysed the data and wrote the manuscript. T.W. performed the experiments. H.H. and A.S. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Online-only Table
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article

Cite this article
Leung, M.L., Watson, D.J., Vaccaro, C.N. et al. Evaluating sequence data quality from the Swift Accel-Amplicon CFTR Panel. Sci Data 7, 8 (2020). https://doi.org/10.1038/s41597-019-0339-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-019-0339-4
- Springer Nature Limited