
Abstract
Stoliczka’s Asian trident bat (Aselliscus stoliczkanus) is a small-bodied species and very sensitive to climate change. Here, we presented a chromosome-level genome assembly of A. stoliczkanus by combining Illumina sequencing, Nanopore sequencing and high-throughput chromatin conformation capture (Hi-C) sequencing technology. The genome assembly was 2.18 Gb in size with 98.26% of the genome sequences anchored onto 14 autosomes and two sex chromosomes (X and Y). The quality of the genome assembly is very high with a contig and scaffold N50 of 72.98 and 162 Mb, respectively, Benchmarking Universal Single-Copy Orthologs (BUSCO) score of 96.6%, and the consensus quality value (QV) of 47.44. A total of 20,567 genes were predicted and 98.8% of these genes were functionally annotated. Syntenic blocks between A. stoliczkanus and Homo sapiens, together with previous comparative cytogenetic studies, provide valuable foundations for further comparative genomic and cytogenetic studies in mammals. The reference-quality genome of A. stoliczkanus contributes an important resource for conservative genomics and landscape genomics in predicting adaptation and vulnerability to climate change.
Similar content being viewed by others


Background & Summary
Stoliczka’s Asian trident bat (Aselliscus stoliczkanus) is one of the three species in the genus Aselliscus (Hipposideridae)1 and widely distributes in Southeast Asia, including southeastern China, Myanmar, Thailand, Laos, Vietnam, and the Peninsular Malaysia2,3,4,5. It is assessed as least concern (LC) by the International Union for Conservation of Nature (IUCN)2,5. However, as a small-bodied species, A. stoliczkanus is very sensitive to climate change, in particular to humidity6. Thus, although A. stoliczkanus has a wide distribution and is listed as LC on the IUCN Red list, previous field surveys of cave bats revealed that the population size of this species has declined rapidly in recent years7,8. So, A. stoliczkanus has been recognized as near threatened (NT) on China Species Red List9,10. This species can be regarded as a valuable bio-indicator to climate change, as other bat species11,12. However, up to now, no reference-quality genome has been generated for A. stoliczkanus, which is valuable for the assessment of its conservation status and effective protection management13,14.
A. stoliczkanus with diploid chromosome number (2n) of 30 is one of the first bat species whose chromosomes were flow-sorted to be used to generate a whole set of chromosome-specific painting probes15. As far as we know, chromosome-specific paints of A. stoliczkanus have been used in comparative cytogenetic studies on four bat families, including Hipposideridae16, Rhinolophidae15,17,18, Megadermatidae19, and Vespertilionidae20. By integrating comparative chromosomal maps between A. stoliczkanus (Asto) and Myotis myotis (MMY)21, as well as between A. stoliczkanus and Homo sapiens (HSA)15, conserved syntenies of species from 10 bat families have been generated, which have been used to investigate the chromosomal evolution and predict the ancestral karyotype of Chiroptera22,23. Thus, a reference-quality genome of A. stoliczkanus and further whole-genome alignments between A. stoliczkanus and the other two species (MMY and HSA) will be very useful to confirm the genome syntenic blocks identified in previous comparative cytogenetic studies.
In this study, we presented a chromosome-level genome assembly for A. stoliczkanus using a combination of Illumina short-read sequencing (99.76 Gb), Nanopore long-read sequencing (203.07 Gb) and high-throughput chromatin conformation capture (Hi-C) sequencing (222.41 Gb) (Table 1). The final genome assembly size was 2.18 Gb with the contig and scaffold N50 of 72.98 Mb and 162 Mb, respectively (Table 2). Consistent with the karyotype reported in previous studies15,21, the final chromosome-level genome assembly includes 14 autosomes, X and Y chromosome (Figs. 1, 2 and Table 3), containing 99.66% of the total genome assembly. Our genome assembly of A. stolizkanus is comparable to other bat genomes (Fig. 3) and can be reliably used in further comparative genomics. In the genome assembly of A. stoliczkanus we detected 760.02 Mb (34.75% of the genome) repetitive elements (Table 4). After masking repetitive elements, a total of 20,320 protein-coding genes were predicted and 98.8% of them were functionally annotated (Table 5).

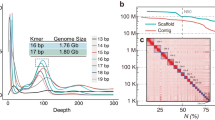
(a) Genomescope profile for 21-mers based on Illumina short-reads. (b) Hi-C contact map for the genome assembly.

Circos showing the genomic structure of A. stolizkanus including chromosome, gene density, repeat density, and GC content (%) from the outer circle to inner one.

The contiguity of the genome assembly revealed by Benchmarking Universal Single-Copy Orthologs (BUSCO) score and N50 of contig and scaffold. Genome assembly of Aselliscus stoliczkanus was shown in red.
By performing genomic synteny analysis, we validated the results of previous comparative cytogenetic studies between A. stoliczkanus and the other two species15,21 (Fig. 4 and Table 3). This consistency also supports the high quality of A. stoliczkanus genome assembly and annotation generated in this study. Our current genomic resource of A. stoliczkanus will be very useful for the assessing of its conservation status and designing effective protection strategies in the future. In addition, syntenic blocks identified between A. stoliczkanus and other species will provide valuable foundations for further comparative genomic and cytogenetic studies in bats and also in mammals.

(a) G-banded karyotype of A. stolizkanus with syntenic blocks between A. stolizkanus (Asto) and two other species (Myotis myotis: MMY; Homo sapiens: HSA) identified in previous comparative cytogenetic studies15. The capital letter “H” in chromosome 14 represents heterochromatin. The diagram for each chromosome is also shown on the left and the black color in the diagram represents the regions in which the GC content of a 1-Mb window is lower than the average GC content of the chromosome. (b) Genomic synteny and collinearity among Asto, MMY and HSA. Chromosomes in the genome assembly of MMY were numbered on the basis of previously published flow karyotype of this species21.
Methods
Sample collection and sequencing
An adult male A. stolizkanus was collected in November 2018 at Xiaogou cave in Yunnan province, China (25°03′16.7″N, 103°22′52.5″E). Bat was euthanized by cervical dislocation and four tissues (muscle, heart, brain, and liver) were sampled with RNase-free tubes. All tissues were frozen immediately in liquid nitrogen and then were stored in a −80 °C freezer. Sampling and tissue collection procedures were approved by the National Animal Research Authority, East China Normal University (approval ID bf20190301).
For genome sequencing, genomic DNA extracted from muscle with DNeasy kits (Qiagen) was used to construct three different sequencing libraries. First, Nanopore long read library (DNA fragment >20 kb) was constructed with the SQK-LSK109 kit (Oxford Nanopore Technologies, UK) and sequenced on a PromethION sequencer (Oxford Nanopore). The quality of Nanopore reads was assessed using Nanoplot v1.40.224 and results have been shown in supplementary Table S1. Then reads were further trimmed by Nanofilt v2.8.024 with default parameters. Second, for genome survey and error correction of Nanopore data, Illumina short read library (DNA fragment ~350 bp) was constructed with the NEBNext Ultra DNA library Pre-Kit and sequenced on the Illumina Novaseq. 6000 platform (pair-end 150 bp). Illumina short reads were assessed and trimmed by fastp v0.23.225 (-q 20 -w 5 -u 40 -n 5). Third, to generate a chromosome-level genome, we created the Hi-C library with the Truseq Nano DNA library Kit and the restriction endonuclease DpnII following procedures described previously26 and sequenced it on the Illumina Hiseq platform (pair-end 150 bp). Hi-C reads were also processed by fastp v0.23.225.
For transcriptome sequencing, the total RNA of each tissue (heart, brain and liver) was extracted using TRIzol (Life Technologies Corp., Carlsbad, CA, USA). A total of three RNA sequencing libraries were constructed using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) and sequenced on the Illumina HiSeq X Ten platform (paired-end 150 bp). RNA-seq reads were trimmed using TRIMMOMATIC v0.3827 with default parameters.
Genome assembly
Jellyfish v2.2.1028 was used to construct the k-mer count histogram (k = 21) based on 99.76 Gb clean short read data. Then, the genome size, heterozygosity and percentage of repeat content were estimated using GenomeScope v2.029. Genomic contig assembly was performed based on 203.07 Gb Nanopore long reads using NextDenovo30 software (https://github.com/nextomics/nextdenovo). Then Nextpolish v1.4.131 was applied to polish the assembly with both Nanopore long reads and Illumina short reads. Redundant contigs were removed using Purge_Dups v1.2.532 with default settings. A total of 222.41 Gb clean Hi-C reads were then mapped to the contig assembly using Juicer v1.633 and chromosome construction was conducted using the 3D-DNA pipeline34 with default settings. We further used Juicebox Assembly Tools35 to manually correct the chromatin contact matrix and built the Hi-C interaction heatmap. The final genome assembly was generated by 3D-DNA post-review pipeline based on the corrected assembly file above.
To evaluate the quality of the genome assembly, we first assessed its integrity using Benchmarking Universal Single-Copy Orthologs (BUSCO v5.2.2)36 with a database of mammals (mammalia_odb10). Second, the Nanopore long reads and Illumina short reads were mapped to the genome assembly using minimap2 v2.24-r112237 and bwa v0.7.17-r118838 with default parameters, respectively. We then estimated the mapping rates using SAMtools v1.16.139. Third, the accuracy of our genome assembly was assessed by calculating the consensus quality (QV) using MERQURY v1.340 based on Illumina short reads and k-mers.
Identification of sex chromosomes
We identified the X chromosome of A. stolizkanus by aligning its genome assembly against the genome of R. ferrumequinum (NCBI accession number: GCF_004115265.2) whose X chromosome had been identified41. Y chromosome of A. stolizkanus was identified by performing blastn searches with Y-linked genes in the mammals (USP9Y and UTY)42.
Repeat annotation
We annotated the repeat sequences in the A. stolizkanus genome using both de novo and homology-based prediction methods. A de novo repeat library was first created using RepeatModeler (https://github.com/Dfam-consortium/RepeatModeler) (the ‘-LTRStruct’ option), which was merged with the bat repeat libraries43, Repbase (http://www.girinst.org/repbase) and Dram database to generate a final custom repeat library. RepeatMasker v4.1.244 was then used to perform repeat sequence annotation with the custom repeat library. A total of 760.02 Mb (34.75% of the genome) repetitive elements were identified, of which 33.12% was transposable elements (TEs), including LINE (19.78%), LTR (5.93%), SINE (2.34%), DNA transposons (4.89%), Rolling-circles (0.13%) and unclassified TEs (0.13%) (Table 4).
Gene annotation
We used three methods to predict protein-coding genes, including ab initio prediction, transcriptome-based prediction and homology-based prediction. The BRAKER2 v2.5.2 pipeline45 was applied to perform ab initio prediction using de novo, homology-based protein and RNA-Seq evidence. For transcriptome-based prediction, a total of 21.3 Gb RNA-seq reads from four tissues were mapped to the genome using HISAT2 v2.2.146 with default parameters and the transcriptome was assembled using STRINGTIE v2.2.147. The open reading frames (ORFs) were predicted by TransDecoder v5.5.0 (https://github.com/TransDecoder/TransDecoder/). Homology-based prediction was performed using GEMOMA v1.9.048,49 based on protein sequences of 11 species including six bats (Rousettus aegyptiacus, Rhinolophus ferrumequinum, Pipistrellus kuhlii, Phyllostomus discolor, Myotis myotis, and Molossus molossus) and five other mammals (Felis catus, Bos taurus, Sus scrofa, Mus musculus, and Homo sapiens). Then, we used EVidenceModeler v2.0.050 to combine genes predicted by the three methods with a weighted consensus (ABINITIO_PREDICTION AUGUSTUS 1, TRANSCRIPT Cufflinks 12, OTHER_PREDICTION GeMoMa 10, OTHER_PREDICTION transdecoder 12). Finally, two rounds of PASA v2.4.151 were conducted to update the EVM result using the transcriptome de novo assembled by Trinity v2.13.252 under the default settings. We performed functional annotation by searching sequences of protein-coding genes against the Uniprot database and nonredundant protein sequence database (NR) using DIAMOND (blastp -e 1e-5)53, and the eggNOG database using EggNOG-mapper54. In addition, INTERPROSCAN55 (-appl Pfam -iprlookup -goterms) was employed to obtain protein domains and motifs and gene ontology (GO).
Genome synteny
Genomic synteny analyses were performed between A. stoliczkanus and two other species (M. myotis and H. sapiens). We first conducted the pairwise alignment of these chromosome-level genomes using LAST software v141056, then we identified and visualized the synteny blocks using MCscan (python version) with default parameters. High collinearity was observed across the three species (Fig. 4b) and synteny blocks identified here could be used to validate the results of previous comparative cytogenetic studies.
Data Records
All raw sequencing data that were used for genome assembly and annotation have been deposited into the National Center for Biotechnology Information (NCBI) with accession number SRR2545963157 and SRR2547626058 for Illumina sequencing data, SRR2547005959 and SRR2547005860 for Nanopore sequencing data, SRR2549003561 for Hi-C sequencing data, SRR2546184762, SRR2546185363, and SRR2546191864 for transcriptome Illumina sequencing data. The final genome assembly and gene annotation results have been deposited in Figshare65 and in the GenBank database of NCBI with accession number JAWWOG00000000066.
Technical Validation
High quality of the genome assembly of A. stoliczkanus was supported by multiple evaluation methods (BUSCO score: 96.6%; QV value: 47.44; mapping rates of Illumina short reads and Nanopore long reads: 99.68% and 99.96%, Table 2). In addition, the final assembled chromosome-level genome (99.66% of the total genome) contained the same number of chromosomes with the karyotype reported previously. Finally, highly homologous genomic segments between A. stoliczkanus and two other species (M. myotis and H. sapiens) revealed by genomic synteny analysis also supported the high quality of the genome assembly and annotation of A. stoliczkanus.
Code availability
All commands and pipelines used in the data processing were all executed according to the manuals and protocols of the corresponding bioinformatics software. If no detailed parameters were provided, default parameters were used. The version of the software has been specified in the Methods section. No custom programming or coding was used.
References
Dobson, G. E. On a new genus and species of Rhinolophidae, with description of a new species of Vesperus, and notes on some other species of insectivorous bats from Persia. J. Asiat. Soc. Bengal. 40, 455–461 (1871).
Bates, P., Bumrungsri, S., Francis, C., Csorba, G. & Furey, N. Aselliscus stoliczkanus. The IUCN Red List of Threatened Species 2008, e.T2155A9300617 (2008).
Francis, C. M. A field guide to the mammals of South-East Asia. Mammalia 73, 78–80 (2009).
Zhang, Z. et al. Variation in Aselliscus stoliczkanus based on morphology and molecular sequence data, with a new record of the genus Aselliscus in China. J. Mammal. 97, 1718–1727 (2016).
Tu, V., Görföl, T., Furey, N. & Csorba, G. Aselliscus stoliczkanus. The IUCN Red List of Threatened Species 2022, e.T214518902A21976509 (2022).
Liu, Y., Wang, Y., Zhang, Z., Bu, Y. & Niu, H. Roost selection and ecology of Stoliczka’s trident bat, Aselliscus stoliczkanus (Hipposideridae, Chiroptera) in China. Mamm. Biol. 95, 143–149 (2019).
Zhang, L., Jones, G. & Zhang, J. Recent surveys of bats (Mammalia: Chiroptera) from China. Acta Chiropterol. 11, 71–88 (2009).
Bu, Y. et al. Geographical distribution, roost selection, and conservation state of cave-dwelling bats in China. Mammalia 79, 409–417 (2015).
Wang, S. & Xie, Y. China Species Red List (Higher Education Press, 2004).
Jiang, Z. China’s Red List of Biodiversity: Vertebrates (Science Press, 2021).
Jones, G., Jacobs, D. S., Kunz, T. H., Willig, M. R. & Racey, P. A. Carpe noctem: the importance of bats as bioindicators. Endanger Species Res. 8, 93–115 (2009).
Festa, F. et al. Bat responses to climate change: a systematic review. Biol. Rev. 98, 19–33 (2023).
Formenti, G. et al. The era of reference genomes in conservation genomics. Trends Ecol. Evol. 37, 197–202 (2022).
Wilder, A. P. et al. The contribution of historical processes to contemporary extinction risk in placental mammals. Science 380, eabn5856 (2023).
Mao, X. et al. Karyotype evolution in Rhinolophus bats (Rhinolophidae, Chiroptera) illuminated by cross-species chromosome painting and G-banding comparison. Chromosome Res. 15, 835–848 (2007).
Mao, X. et al. Karyotypic evolution in family Hipposideridae (Chiroptera, Mammalia) revealed by comparative chromosome painting, G- and C-banding. Zool. Res. 31, 453 (2010).
Volleth, M. et al. Comparative chromosomal studies in Rhinolophus formosae and R. luctus from China and Vietnam: elevation of R. l. lanosus to species rank. Acta Chiropterol. 19, 41–50 (2017).
Volleth, M. et al. Cytogenetic investigations in Bornean Rhinolophoidea revealed cryptic diversity in Rhinolophus sedulus entailing classification of Peninsular Malaysia specimens as a new species. Acta Chiropterol. 23, 1–20 (2021).
Volleth, M. et al. Cytogenetic analyses detect cryptic diversity in Megaderma spasma from Malaysia. Acta Chiropterol. 23, 271–284 (2022).
Kulemzina, A. I. et al. Comparative chromosome painting of four Siberian Vespertilionidae species with Aselliscus stoliczkanus and human probes. Cytogenet. Genome Res. 134, 200–205 (2011).
Ao, L. et al. Karyotypic evolution and phylogenetic relationships in the order Chiroptera as revealed by G-banding comparison and chromosome painting. Chromosome Res. 15, 257–268 (2007).
Mao, X. et al. Comparative cytogenetics of bats (Chiroptera): The prevalence of Robertsonian translocations limits the power of chromosomal characters in resolving interfamily phylogenetic relationships. Chromosome Res. 16, 155–170 (2008).
Sotero-Caio, C. G., Baker, R. J. & Volleth, M. Chromosomal evolution in Chiroptera. Genes 8, 272 (2017).
De Coster, W., D’hert, S., Schultz, D. T., Cruts, M. & Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Belton, J. M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Hu, J. et al. An efficient error correction and accurate assembly tool for noisy long reads. 2023.03.09.531669 Preprint at https://doi.org/10.1101/2023.03.09.531669 (2023).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics. 0, 1–3 (2013).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020).
Jebb, D. et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature 583, 578–584 (2020).
Godfrey, A. K. et al. Quantitative analysis of Y-Chromosome gene expression across 36 human tissues. Genome Res. 30, 860–873 (2020).
Scheben, A. et al. Long-read sequencing reveals rapid evolution of immunity- and cancer-related genes in bats. Genome Biol. Evol. 15, evad148 (2023).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10.1–4.10.14 (2004).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP plus and AUGUSTUS supported by a protein database. Nar Genom. Bioinform. 3, lqaa108 (2020).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, 89–89 (2016).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. BMC Bioinformatics 19, 189 (2018).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Haas, B. J. et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Kiełbasa, S., Wan, R., Sato, K., Horton, P. & Frith, M. Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–93 (2011).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25459631 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25476260 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25470059 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25470058 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25490035 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25461847 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25461853 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR25461918 (2023).
Lan, L. Genome assembly and annotation of Aselliscus stoliczkanus. Figshare https://doi.org/10.6084/m9.figshare.23902812.v2 (2023).
Genome assembly of Aselliscus stoliczkanus. GenBank https://identifiers.org/ncbi/insdc:JAWWOG000000000 (2023).
Acknowledgements
We thank Sun Haijian and Wang jiaying for their help in filed work. This work was supported by Program for Oriental Scholars of Shanghai Universities (to Li L.J.), Shanghai Frontiers Science Center of Cellular Homeostasis and Human Diseases (to Li L.J.).
Author information
Authors and Affiliations
Contributions
Mao X.G. and Li L.J. conceived and supervised the project. Lan L.J., Zhang X. and Yang S.X. analyzed data. Mao X.G. wrote the manuscript with the input of Lan L.J. All authors edited the manuscript and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lan, L., Zhang, X., Yang, S. et al. Chromosome-level genome assembly of the Stoliczka’s Asian trident bat (Aselliscus stoliczkanus). Sci Data 10, 902 (2023). https://doi.org/10.1038/s41597-023-02838-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02838-0
- Springer Nature Limited