
Abstract
Chinese sea bass (Lateolabrax maculatus) is a highly sought-after commercial seafood species in Asian regions due to its excellent nutritional value. With the rapid advancement of bioinformatics, higher standards for genome analysis compared to previously published reference genomes are now necessary. This study presents a gapless assembly of the Chinese sea bass genome, which has a length of 632.75 Mb. The sequences were assembled onto 24 chromosomes with a coverage of over 99% (626.61 Mb), and telomeres were detected on 34 chromosome ends. Analysis using Merqury indicated a high level of accuracy, with an average consensus quality value of 54.25. The ONT ultralong and PacBio HiFi data were aligned with the assembly using minimap2, resulting in a mapping rate of 99.9%. The study also identified repeating elements in 20.90% (132.25 Mb) of the genome and inferred 22,014 protein-coding genes. These results establish meaningful groundwork for exploring the evolution of the Chinese sea bass genome and advancing molecular breeding techniques.
Similar content being viewed by others


Background & Summary
The Chinese sea bass (Lateolabrax maculatus) (Fig. 1), a member of the Moronidae family in the Perciformes order, displays a distinctive feature of multiple prominent black dots on its lateral body region1. Recently, it has been distinguished as a new species with obvious morphological and genetic differences from the Japanese sea bass (Lateolabrax japonicus)2. Compared to L. japonicus, L. maculatus has a wider ecological range and is found along the coast and estuaries of China, Japan, and the Korean Peninsula1. The Chinese sea bass shows excellent adaptability to a wide range of temperatures and salinity environments and possesses a delicate taste and high nutritional value1,3,4. Therefore, it has been extensively cultivated in freshwater ponds and seawater net cages in China5. In 2021, the yearly production of Chinese sea bass in China reached 199,106 tons, which accounted for 10.79% of the aggregate aquafarming output of marine fish. Consequently, the Chinese sea bass is regarded as a much sought-after marine economic fish in China6.

Genomic landscape of the Chinese sea bass. The rings, from the outermost to the innermost layer, represent the chromosomes of the L. maculatus genome (a), gene density (b), GC density (c), DNA transposons (d), LTRs (e), LINEs (f), and SINEs (g). The identified telomere ends are represented by black dots in (a). The analysis of (a) was conducted using 500-kb genomic windows, while (b–g) were analysed using 50-kb sliding windows.
Recently, extensive molecular genetics research has been conducted on the Chinese sea bass, and the genomes of Chinese sea bass from both the Bohai Gulf and subtropical regions have been assembled7,8. Besides, numerous transcriptomic databases have been generated, and extensive research on functional genes has been conducted by researchers9. However, with advancements in genome sequencing procedures and DNA assembly methodologies, seamless telomere-to-telomere (T2T) genome assembly has now become a reality, enabling the identification of almost the entire genome. Recently, there has been a surge in deciphering seamless genomes for several species, such as Arabidopsis thaliana, Homo sapiens, Citrullus lanatus, Clarias gariepinus, Musa acuminata, Oryza sativa, and Fragaria vesca10,11,12,13,14,15,16. However, assembly of the L. maculatus genome at an equivalent level has not yet been reported.
To this end, we integrated Pacific Biosciences (PacBio) HiFi sequencing, Oxford Nanopore Technologies (ONT) ultralong sequencing, and Hi-C technology to assemble a high-quality T2T genome of L. maculatus. Our assembly significantly improves upon the two previously published genome assemblies, as it is nearly complete without any gaps (Fig. 1). This not only facilitates population genetic research and evolutionary analysis of the Chinese sea bass but also provides important resources for optimizing genetic breeding.
Methods
Sample collection and sequencing
Mature male Chinese sea bass were captured from the Yantai Jinghai Marine Fisheries Co., Ltd, Yantai Shandong, China. High molecular weight genomic DNA (gDNA) was isolated from muscle tissue using a standard sodium dodecyl sulfate (SDS) extraction method for ONT ultralong sequencing. For PacBio HiFi sequencing, a Blood & Cell Culture DNA Kit (Qiagen 13323) was utilized to extract the gDNA. Three methods were used for DNA quality and quantification testing, including (i) a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA), (ii) gel electrophoresis, and (iii) a Qubit fluorometer (Invitrogen, USA). Total DNA was purified by AMPure PB beads (PacBio 100-265-900, USA). High-quality gDNA was prepared for the next step of library construction.
The PacBio HiFi sequencing technique included the construction of a standard SMRTbell library using the SMRTbell Express Template Prep Kit 2.0, following the prescribed guidelines from the manufacturer. Subsequently, the SMRTbell libraries underwent sequencing using a PacBio Sequel II system (Pacific Biosciences, CA, USA). For ONT ultralong sequencing, a library was produced with the Oxford Nanopore SQK-ULK001 kit following the instructions provided by the manufacturer and then sequenced on a PromethION flow cell. As a result, 73.83 Gb (117×) of PacBio HiFi read data and 62.58 Gb (99×) of ONT ultralong read data were obtained (Table 1).
The Hi-C library was produced using a blood sample from the same Chinese sea bass used for gDNA sequencing. Library construction involved the following steps17,18: initial crosslinking of cells using formaldehyde, DNA digestion, end filling and biotin labelling, ligation of the generated blunt-end fragments, purification, and random shearing of DNA into 300–500 bp fragments. After a quality control test of the libraries using Qubit 2.0 (Invitrogen, USA), an Agilent 2100 instrument (Agilent Technologies, CA, USA), and q-PCR, 150 bp PE sequencing of the Hi-C library was implemented on the Illumina NovaSeq. 6000 platform. In total, 92.61 Gb (146×) of Hi-C read data was obtained (Table 1).
Genome assembly and telomere identification
With the ultralong ONT, PacBio HiFi, and Hi-C sequencing data described above, the contigs were assembled utilizing the initial values of Hifiasm19 (v0.19.5). We obtained a gapless-level genome assembly of L. maculatus (YSFRI_Lmacu_1.1), where the genome length was approximately 632.75 Mb and N50 was 27.95 Mb (Table 2). The 3D-DNA pipeline and Juicer-box20 (v1.91) were utilized to examine and visualize the interaction frequencies among different chromosomes (Fig. 2a). Both karyotype analysis and the published genome assembly of ASM402354v1 indicate that the species has a total of 24 chromosomes8,21. Subsequently, we employed minimap222 (v2.17) to compare the L. maculatus genome with the two published genomes. Our assembly appears to be significantly more complete than the current reference genome (ASM402354v1), and it exhibits a distinct mount order in comparison to the other assembly (ASM402866v1) (Fig. 2b). To assess the assembled telomere sequences in the Chinese sea bass genome, we utilized the Telomere Identification toolkit (v0.2.31) (https://github.com/tolkit/telomeric-identifier) to identify occurrences of a 6 bp motif (TTAGGG) within the genome sequence. A total of 34 telomeres were identified, and telomeres were detected on both ends of 11 chromosomes (Fig. 1 and Table 3).

Overview of the genome-wide Hi-C heatmap and collinearity diagram comparing the old and new versions of the genome assembly. (a) The Hi-C heatmap illustrates the interaction frequencies among various chromosomes in Chinese sea bass. Chromosomes are represented by blue squares. (b) Dot plots illustrate the collinear relationship between the L. maculatus assembly and its two previously published assemblies.
Repetitive sequence annotation
We utilized a combined approach involving de novo explorations and homologous alignments for the annotation of repeat elements. Homologue prediction was performed using RepeatMasker23 (v4.0.6) and RepeatProteinMask24 (v4.0.6) based on the Repbase library25 (v202101). Tandem Repeats Finder26 (v4.07) was utilized specifically for the detection of tandem repeats. RepeatModeler24 (v1.0.8) and LTR-Finder27 (v1.06) were employed for de novo prediction of repeat elements. The resultant predictions were merged to create a library utilized by RepeatMasker for the identification of repeat elements. The assembly results indicated that repeat sequences constituted approximately 20.90% of the genome. Among these repeats, long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), and long terminal repeats (LTRs) accounted for 4.60%, 0.27%, and 3.93% of the genome, respectively (Fig. 1 and Table 4).
Gene prediction and functional annotation
We performed gene annotation on the assembled genome, encompassing both structural and functional annotation. Before annotating gene sequences, we masked the observed repetitive sequences. We employed de novo, homologue-based, and transcriptomic approaches to predict the location and structure of genes. Subsequently, functional annotation was conducted to unveil the biological roles of these coding genes within the Chinese sea bass genome.
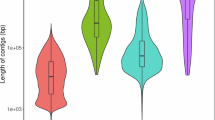
We obtained RNA sequencing data from 14 samples of muscle, testis, liver, gill, stomach, spleen, and brain tissues from the NCBI database. These datasets were subsequently aligned to the genome assembly using HISAT228 (v2.1.0) and assembled using StringTie29 (v2.1.4). De novo prediction of the gene structure within the genome was performed by employing two established fish models, namely, zebrafish and sea lamprey, with the use of the AUGUSTUS26 (v3.3.0) gene prediction tool. For homology-based prediction, we utilized Miniport30 (v0.11) to conduct a comparative analysis of the protein sequences from 12 closely related species, including Dicentrarchus labrax, Branchiostoma belcheri, Gasterosteus aculeatus, Cynoglossus semilaevis, Lates calcarifer, Oreochromis niloticus, Danio rerio, Oryzias latipes, Oryzias melastigma, Salmo salar, Tetraodon nigroviridis, and Takifugu rubripes. The protein sequences were downloaded from the NCBI database and compared to the genome to infer gene structure according to homology-based evidence. To synthesize the findings obtained from the three methods, we employed EvidenceModeler31 (v1.1.1). This powerful tool facilitated the amalgamation and integration of the gene predictions, resulting in the definitive identification of 22,014 protein-coding genes. Gene sets were downloaded from the NCBI database for three species closely related to L. maculatus, namely, D. labrax, L. calcarifer, and G. aculeatus. mRNA length distribution and the number of exons in each mRNA was compared among the different gene sets using various length windows (Fig. 3a). The analysis revealed that the statistical characteristics of the gene elements of closely related species exhibited a similar distribution.

Comparison map of gene sets among closely related species and an UpSet diagram of functional annotation of the Chinese sea bass genome. (a) The distribution of mRNA length and the number of exons in each mRNA were compared between gene sets of closely related species using 1 kbp mRNA length as a window. (b) Gene function annotation was used to generate a statistical UpSet diagram using 5 public databases: Kofam, Pfam, KEGG, SwissProt, EggNOG, and NR.
After gene prediction, the finalized gene sets derived from the preceding methods underwent functional annotation through matching with a variety of databases. In particular, functional annotation of the inferred genes for L. maculatus was performed using diamond32 (v2.1.6) against the SwissProt33, KEGG34, EggNOG35, Pfam36, NR37, and Kofam38 databases with an e-value cut-off of 1e-5. Finally, 21,522 genes were annotated, which accounted for 97.77% of all inferred genes of L. maculatus (Fig. 3b and Table 5).
Data Records
The genome assembly data can be accessed at GenBank using the accession number JAUTWU00000000039.
The raw sequencing data have been deposited into the CNGB Sequence Archive (CNSA) with the accession number CNP000461040 and Genome Sequence Archive (GSA) in NGDC under the accession number CRA01444341.
The genome annotation files, gene CDS, and protein data have been submitted to Figshare42.
Technical Validation
To assess the completeness of the L. maculatus genome assembly, we utilized BUSCO43 (v5.4.7) with the Actinopterygii database (actinopterygii_odb10) to identify conserved single-copy genes in the assembly. Of the 3,640 conserved genes searched, an impressive 97.9% were identified as complete, indicating a high level of gene content preservation. Among these, 97.2% were both complete and present as single-copy genes, further emphasizing the quality of the assembly. Additionally, only 0.2% were fragmented, and 1.9% were missing from the assembly (Table 6). To ensure the quality and accuracy of the Chinese sea bass assembly, we employed a two-step validation process. First, the assembly quality value (QV) was quantified using Merqury44 (v1.4), resulting in a QV score of 54.25, reflecting a high-quality assembly. Subsequently, we aligned the raw sequencing data to the assembly using minimap222 (v2.15). For PacBio HiFi and ONT ultralong sequencing, this alignment approach achieved mapping rates of 99.93% and 99.99%, respectively.
Code availability
No custom software code was written for this research. All bioinformatics tools and pipelines were executed as per the manual and protocols provided by their respective software developers. The software versions used, along with their corresponding parameters, have been thoroughly described in the Methods section.
References
Liu, J., Gao, T., Yokogawa, K. & Zhang, Y. Differential population structuring and demographic history of two closely related fish species, Japanese sea bass (Lateolabrax japonicus) and spotted sea bass (Lateolabrax maculatus) in Northwestern Pacific. Molecular Phylogenetics and Evolution 39, 799–811 (2006).
Yokogawa, K. Genetic Divergence of Fishes in Genus Lateolabrax (Perciformes: Percichthyidae). Preprint at https://doi.org/10.11233/aquaculturesci1953.46.315 (1998).
An, H. S. et al. Genetic differences between wild and hatchery populations of Korean spotted sea bass (Lateolabrax maculatus) inferred from microsatellite markers. Genes Genom 35, 671–680 (2013).
Chen, J., Jayachandran, M., Xu, B. & Yu, Z. Sea bass (Lateolabrax maculatus) accelerates wound healing: A transition from inflammation to proliferation. Journal of Ethnopharmacology 236, 263–276 (2019).
Zhu, Q. et al. Transcriptomic Modulation Reveals the Specific Cellular Response in Chinese Sea Bass (Lateolabrax maculatus) Gills under Salinity Change and Alkalinity Stress. IJMS 24, 5877 (2023).
Wang, D., Wu, F. X., Song, D. D. & Gao, H. Q. China Fishery Statistics Yearbook. (China Agricultural Press, 2022).
Chen, B. et al. Chromosome-Level Assembly of the Chinese Seabass (Lateolabrax maculatus) Genome. Front. Genet. 10, 275 (2019).
Shao, C. et al. Chromosome-level genome assembly of the spotted sea bass, Lateolabrax maculatus. GigaScience 7, giy114 (2018).
Liu, Y. et al. First High-Density Linkage Map and QTL Fine Mapping for Growth-Related Traits of Spotted Sea bass (Lateolabrax maculatus). Mar Biotechnol 22, 526–538 (2020).
Nguinkal, J. A., Zoclanclounon, Y. A. B., Brunner, R. M. & Goldammer, T. Haplotype-resolved assembly of the African catfish (Clarias gariepinus) provides insights for semi-terrestrial adaptation of airbreathing catfishes. Preprint at https://www.biorxiv.org/content/10.1101/2023.03.23.533919v2 (2023).
Zhang, Y. et al. The telomere‐to‐telomere gap‐free genome of four rice parents reveals SV and PAV patterns in hybrid rice breeding. Plant Biotechnology Journal 20, 1642–1644 (2022).
Deng, Y. et al. A telomere-to-telomere gap-free reference genome of watermelon and its mutation library provide important resources for gene discovery and breeding. Molecular Plant 15, 1268–1284 (2022).
Nurk, S. et al. The complete sequence of a human genome. Science 376, 44–53 (2022).
Hou, X., Wang, D., Cheng, Z., Wang, Y. & Jiao, Y. A near-complete assembly of an Arabidopsis thaliana genome. Molecular Plant 15, 1247–1250 (2022).
Belser, C. et al. Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun Biol 4, 1047 (2021).
Ma, F. et al. Gap-free genome assembly of anadromous Coilia nasus. Sci Data 10, 360 (2023).
Rao, S. S. P. et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680 (2014).
Xie, T. et al. De Novo Plant Genome Assembly Based on Chromatin Interactions: A Case Study of Arabidopsis thaliana. Molecular Plant 8, 489–492 (2015).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18, 170–175 (2021).
Durand, N. C. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Sola, L. et al. A karyotype analysis of the genus Dicentrarchus by different staining techniques. Journal of Fish Biology 43, 329–337 (1993).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Tarailo‐Graovac, M. & Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. CP in Bioinformatics 25, (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33, 290–295 (2015).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014 (2023).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Buchfink, B., Reuter, K. & Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18, 366–368 (2021).
Bairoch, A. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 28, 45–48 (2000).
Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 28, 27–30 (2000).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research 47, D309–D314 (2019).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Research 49, D412–D419 (2021).
Pruitt, K. D. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Research 33, D501–D504 (2004).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252 (2020).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_031216445.1 (2023).
CNGB Sequence Archive https://db.cngb.org/search/project/CNP0004610/ (2023).
NGDC Genome Sequence Archive https://bigd.big.ac.cn/gsa/browse/CRA014443 (2023).
Sun, Z. L. Genome assembly of the spotted sea bass. Figshare https://doi.org/10.6084/m9.figshare.23807889.v2 (2023).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution 38, 4647–4654 (2021).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol 21, 245 (2020).
Acknowledgements
This research was funded by Marine S&T Fund of Shandong Province for Qingdao Marine Science and Technology Center (2022QNLM030001), the Key Research and Development Project of Shandong Province (2022LZGC016), the National Key R&D Program of China (grant number 2022YFD2400100), the National Science and Technology Innovation 2030-Major Program(2022ZD0401803), the AoShan Talents Cultivation Program Supported by Qingdao National Laboratory for Marine Science and Technology (grant number 2017ASTCP-ES06), the Taishan Scholars Program (NO. tstp20221149) to C.S, the National Ten-Thousands Talents Special Support Program to C.S., the Central Public-interest Scientific Institution Basal Research Fund, CAFS (grant number 2020TD19), the Key R&D Program of Hebei Province, China (21326307D) and the project by the National Marine Genetic Resource Center.
Author information
Authors and Affiliations
Contributions
C.S. conceived the project. X.C., J.L. and Y.L. collected the samples. Z.S. analysed the data and drafted the manuscript. S.L. supervised the data analysis and revised the manuscript. W.L. contributed to the analysis. K.L., H.W. and Q.W. revised the manuscript. All authors have reviewed and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, Z., Li, S., Liu, Y. et al. Telomere-to-telomere gapless genome assembly of the Chinese sea bass (Lateolabrax maculatus). Sci Data 11, 175 (2024). https://doi.org/10.1038/s41597-024-02988-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-02988-9
- Springer Nature Limited