
Abstract
Photoaging is the premature aging of the skin caused by prolonged exposure to solar radiation. The visual alterations manifest as wrinkles, reduced skin elasticity, uneven skin tone, as well as other signs that surpass the expected outcomes of natural aging. Beyond these surface changes, there is a complex interplay of molecular alterations, encompassing shifts in cellular function, DNA damage, and protein composition disruptions. This data descriptor introduces a unique dataset derived from ten individuals, each with a minimum of 18 years of professional experience as a driver, who are asymmetrically and chronically exposed to solar radiation due to their driving orientation. Skin samples were independently collected from each side of the face using a microdermabrasion-like procedure and analyzed on an Exploris 240 mass spectrometer. Our adapted proteomic statistical framework leverages the sample pairing to provide robust insights. This dataset delves into the molecular differences in exposed skin and serves as a foundational resource for interdisciplinary research in photodermatology, targeted skincare treatments, and computational modelling of skin health.
Similar content being viewed by others


Background & Summary
The skin is the body’s primary line of defense, forming a protective barrier against environmental factors. It also serves as a dynamic indicator of our body’s overall health. This complex organ plays a pivotal role in fluid conservation, temperature moderation, and sensory communication. Moreover, it houses a unique immunological zone crucial for tissue stability, protection, and repair1.
As the skin ages, a series of modifications become evident. The structural integrity begins to decline, leading to decreased elasticity and suppleness. The skin’s capacity to retain moisture diminishes, making it more susceptible to dehydration2. Furthermore, cellular regeneration decelerates, hindering the skin’s innate repair mechanisms. These shifts go beyond mere surface changes; they indicate foundational alterations in the skin’s functions. Its defense capabilities, thermal regulation, and sensory communication all begin to falter3.
The mechanisms of skin aging are primarily divided into chronological and premature pathways. Chronological or intrinsic aging is an inevitable process driven by genetics and time, resulting in reduced collagen production, diminished cellular turnover, and elastin degradation. On the other hand, premature or extrinsic aging is largely influenced by environmental stressors4. Key contributors include exposure to pollutants, lifestyle habits like smoking, and notably, solar radiation. Among these, solar radiation stands out as a paramount factor, inducing oxidative stress, and the breakdown of collagen and elastin structures, thereby accelerating the visible signs of skin aging5.
Solar radiation encompasses ultraviolet (UV), visible light (VL), and infrared radiation (IRA). UV is subdivided into UVC (100–280 nm), UVB (280–320 nm), and UVA (320–400 nm), both UVA and UVB have known effects on skin health6. UV exposure leads to photoaging, characterized by skin roughness and age spots, while VL, especially blue-violet light (around 415 nm), can induce prolonged pigmentation. At the cellular level, both UV and VL produce reactive oxygen species, damaging DNA, proteins, and lipids7. IRA also affects dermal structure and skin lipid composition5. Given these varied effects, understanding the skin’s molecular changes due to different solar components is crucial for maintaining skin health8.
In response of the intricate nature of these molecular dynamics, proteomic advancements over the past decade have revolutionized our understanding of skin conditions and the underlying molecular mechanisms9. Utilizing mass spectrometry, proteomics not only pinpoints and measures individual proteins but also offers insights on post-translational modifications, protein interactions, and pathways. This technique has been particularly effective in deciphering the complex molecular mechanisms behind photoaging and associated skin disorders; however, despite its groundbreaking potential, the incorporation of proteomics into dermatological research hasn’t kept pace with its technological evolution10.
Proteomic studies in humans represent significant challenges, primarily due to the inherent complexity and diversity of the human proteome10. Obtaining consistent and representative human samples is a considerable hurdle, compounded by inter-individual variability and the dynamic nature of protein expression in response to various internal and external factors. The importance of employing paired studies to achieve robust scientific outcomes has been demonstrated in diverse areas of proteomic research, including a notable study on breast cancer11. In the latter, nipple aspirate fluid was collected from breasts both with and without cancer across different participants. The application of paired statistical approaches was instrumental in identifying differentially abundant proteins, thereby underscoring the nature of paired datasets12.
With the motivation of pairing samples to minimize inter-individual variability and maximize result reliability, this dataset focuses on photoaging among professional drivers. All drivers belonging to this dataset have a minimum of 18 years of professional experience, are non-smokers, and where particularly susceptible to asymmetric solar exposure due to the orientation of their driver-side windows. This chronic, side-specific exposure to solar radiation makes them an ideal cohort for photoaging studies, allowing for a nuanced understanding of the effects of the exposure. In our research, we used a microdermabrasion-like, non-invasive, and painless skin collection technique, that, through mass spectrometry, can offer a proteomic overview of changes associated with photoaging.
The quality control of our dataset was performed using RawVegetable, a tool designed for the nuanced evaluation of mass spectrometry data. Among its features are the charge state chromatogram and TopN density estimation modules, which aided in honing our chromatography processes13. Furthering our analysis, we utilize PatternLab for Proteomics V (PLV) to gauge the quantity of peptides and estimate the number of proteins in the sample. PLV’s suite of integrated tools offers an intricate view of the sample’s proteomic landscape. Leveraging its peptide spectrum matching and data filtering capabilities, we’re poised to both identify and quantify peptides, resulting in precise protein identification14. Finally, we also employed DiagnoMass15; this tool relies on spectral clustering to perform sample comparison without the bias of the search engine (in this case, PLV). As such, DiagnoMass allows us to probe how much of the proteome is discriminative between the two conditions and what proportion of it was missed by PLV, thus revealing how much is yet to be explored from our contribution16.
We believe this dataset will not only shed light on molecular differences in exposed skin, especially between the right (lower exposure) and left (higher exposure) sides of the face, but also provide a robust foundation for future interdisciplinary investigations in photodermatology and beyond.
Methods
Sample collection
This study was approved by the Fiocruz Research Ethics Committee (CAAE 38352020.8.0000.5248. Male Caucasian participants, aged between 35 and 70 years and with a phototype ranging from II to IV, met our anamnesis criteria, which included specific inclusion and exclusion standards. Exclusion criteria encompassed conditions such as skin diseases, smokers, or having diabetes. All participants provided written informed consent for data collection and sharing. Prior to sample collection, the skin was thoroughly cleaned with a cotton pad soaked in micellar water to remove surface contaminants and excess oils. We then employed a microdermabrasion technique using the Dermotonus Slim Vacuum Therapy equipment produced by Ibramed to gently exfoliate the skin without causing harm or discomfort; the exfoliated skin is trapped in a 3D printed device adapted by us (under patent) for attachment to the equipment and to minimize sample manipulation. Each participant provided one sample from each side of their face, resulting in a total of 20 samples across 10 participants.
Sample preparation
The skin samples were subjected to lysis for 10 vortex cycles using the equipment (FlexVortex 2 – Loccus) at maximum intensity. The first five cycles incorporated 0.1 mm zirconium beads (Loccus), and the remaining cycles also incorporated RapiGest detergent at a concentration of 0.1%, following the manufacturer’s recommendations. Each cycle involved one minute of vortexing followed by one minute of cooling on ice.
The skin proteins extraction was performed with RapiGest detergent at a concentration of 0.1% according to the manufacturer’s recommendations. One hundred micrograms of proteins from each sample were reduced with dithiothreitol (DTT) (final concentration of 10 mM) for 30 min, at 60 °C. After being cooled to room temperature, the samples were alkylated with iodoacetamide (final concentration of 30 mM) for 25 min at room temperature, in the dark, and finally digested with high sequence grade modified trypsin in the proportion of 1/50 (E/S) for 20 h, at 37 °C.
Desalting and sample quantification
In due course, the enzymatic reaction was stopped by adding trifluoroacetic (0.4% v/v final) and the peptides were incubated for additional 40 min to degrade the RapiGest. Afterward, the samples were centrifuged at 18,000 g for 10 min to remove any insoluble materials. Subsequently, the peptides were quantified using the fluorometric assay—Qubit 2.0 (Invitrogen) according to the manufacturer’s recommendations. Each sample was desalted and concentrated using Stage-Tips (STop and Go-Extraction TIPs)17.
Mass spectrometry analysis
Each peptide mixture was twice subjected to reversed-phase liquid chromatography followed by tandem mass spectrometry (LC–MS/MS) analysis with an UltiMate 3000 nanoHPLC (Thermo Scientific®) coupled online with an Exploris 240 Orbitrap mass spectrometer (Thermo Scientific®). The peptide mixture was chromatographically separated on a column (15 cm in length with a 75 μm I.D., C18-AQ 3.0 μm resin, SNC20442712 - Thermo Scientific) with a flow of 250 nL/min from 1% to 40% ACN (acetonitrile) in 0.1% formic acid, in a 120 min gradient. The Exploris 240 Orbitrap was set to the data-dependent acquisition (DDA) mode to automatically switch between full scan MS and MS/MS acquisition with 30 s dynamic exclusion. Survey scans (200–2000 m/z) were acquired in the Orbitrap system with a resolution of 60,000 at m/z 200. The most intense ions captured in a 2 s cycle time were selected, excluding those unassigned and in a 1 + charge state, sequentially isolated and HCD (Higher-energy collisional dissociation) fragmented using a stepped normalized collision energy of 25, 30, and 35. The fragment ions were analyzed with a resolution of 15,000 at 200 m/z. The general mass spectrometric conditions were as follows: 2.5 kV spray voltage, no sheath or auxiliary gas flow, heated capillary temperature of 40 °C, predictive automatic gain control (AGC) enabled, and an S-lens RF level of 40%. Mass spectrometer scan functions and nLC solvent gradients were controlled by the Xcalibur 4.1 data system (Thermo Scientific®).
Peptide spectrum matching (PSM)
The data analysis was performed with the PatternLab for proteomics V (PLV) software that is freely available at https://www.patternlabforproteomics.org 14. Homo sapiens’ sequences were downloaded on July 7th, 2023, from the Swiss-Prot and then a target-decoy database was generated to include a reversed version of each sequence plus those from 104 common mass spectrometry contaminants. The data was preprocessed with the Y.A.D.A. 3.0 deconvolution algorithm to enable multiplexed spectra identification18. Comet 2021 search engine19, which is embedded into PLV, was used for identifying the mass spectra. The search parameters considered: fully and semi-tryptic peptide candidates with masses between 500 and 6000 Da, up to two missed cleavages, 35 ppm for precursor mass, and bins of 0.02 m/z for MS/MS. The modifications were carbamidomethylation of cysteine and oxidation of methionine as fixed and variable, respectively.
Validation PSM
The validity of the PSMs was assessed using Search Engine Processor (SEPro)20. The identifications were grouped by charge state (2 + and ≥ 3 + ), and then by tryptic status, resulting in four distinct subgroups. For each group, the XCorr, DeltaCN, DeltaPPM, and Peaks Matches values were used to generate a Bayesian discriminator. The identifications were sorted in nondecreasing order according to the discriminator score. A cutoff score accepted a false-discovery rate (FDR) of 2% at the peptide level based on the number of decoys21. This procedure was independently performed on each data subset, resulting in an FDR independent of charge state or tryptic status. Additionally, a minimum sequence length of five amino-acid residues and a protein score greater than 3 were imposed. Finally, identifications deviating by more than 10 ppm from the theoretical mass were discarded. These last filters led to FDRs, now at the protein level, to be lower than 1% for all search results.
Technical Validation
Dataset quality control with RawVegetable 2.0
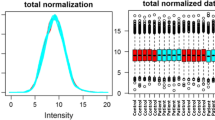
To assess the quality of our mass spectrometry data, especially when comparing experimental technical replicates, we employed RawVegetable, a specialized software tool designed for mass spectrometry data assessment13. We employed several functionalities of this tool that are now described. The charge state chromatogram module and the TopN density estimation module allowed us to optimize our chromatography before generating the final dataset. TopN is notably significant as it governs the number of MS/MS scans generated per cycle. This feature aided in pinpointing retention time intervals where under-sampling or over-sampling occurred, making gradient adjustments more straightforward. The chromatography reproducibility module enabled direct comparisons across experiments, ensuring data consistency as shown in Fig. 1. A detailed quality control analysis with images for each replicate pair can be found in ‘Supplementary_QC.docx’ file. Finally, we assessed the quality of MS/MS spectra by examining their Xrea scores24 throughout the run and using the precursor signal ratio distribution to gauge fragmentation efficiency.

Ion chromatogram comparison for technical replicates: face skin Left and Right sides of the same driver.
We provide Table 1, that summarizes the number of identifications provided by PLV on our samples. Details regarding the identification can be found in the ‘Supplementary_ID.rar’ file.
Our mass spectrometry analysis generated a total of 40 raw files, comprising 2,560,226 mass spectra. These spectra were clustered into 240,369 unique spectral clusters using DiagnoMass software. It is important to note that peptides with identical sequences, but different charge states were classified into separate clusters, suggesting that the actual number of unique biological molecules could be approximately half of the total clusters. The hierarchical clustering algorithm employed by DiagnoMass required a minimum spectral angle of 0.75 for spectra to be grouped together. Spectra that did not meet this criterion were excluded from the analysis. We considered only those clusters containing three or more spectra with a spectral angle greater than or equal to 0.75. The results of this analysis are summarized in Table 2, which also outlines the number of these spectral clusters subsequently identified by PLV.
This table provides a comprehensive breakdown of the spectral clusters identified in the skin samples, focusing on the differences between the solar-exposed (left side) and non-exposed (right side) areas of the face. It enumerates the number of unique spectral clusters detected using DiagnoMass and further delineates the number of these clusters identified by PatternLab. The columns are labeled as follows: ‘Number of Replicates’ indicates the number of biological replicates that exhibited the corresponding count of unique spectral clusters; for instance, 3,709 unique spectral clusters were identified exclusively on the solar-exposed left side in three or more biological replicates. ‘Spectral Clusters (Solar-Exposed)’ and ‘Spectral Clusters (Non-Exposed)’ represent the number of unique spectral clusters identified on the solar-exposed left and non-exposed right sides of the face, respectively. ‘Identified Clusters (Solar-Exposed)’ and ‘Identified Clusters (Non-Exposed)’ specify the number of spectral clusters from each side that were identified by PatternLab. The table serves as a critical resource for understanding the proteomic alterations induced by solar exposure, thereby providing a foundation for future research in photodermatology and targeted skincare treatments.
Usage Notes
Our updated and comprehensive dataset on proteomic changes resulting from solar exposure in facial skin provides not only a profound insight into direct protein alterations and broader implications of the exposome but also lays the foundation for understanding photoaging, investigating the links between solar exposure and skin diseases, developing personalized treatments, researching the combined effects of environmental factors on skin health, and assisting cosmetic and pharmaceutical industries in refining products to address specific protein changes. We now suggest 3 usage cases for this dataset.
Advancements in photodermatology research
The dataset is particularly useful for researchers in the field of photodermatology. It can serve as a foundational resource for understanding the molecular mechanisms that underlie solar-induced skin aging. Researchers can employ this dataset to validate existing theories or generate new hypotheses on how prolonged solar exposure leads to specific proteomic alterations.
Development of targeted skincare treatments
Personalized skincare has become an area of burgeoning interest in both the cosmetic and pharmaceutical industries. The proteomic data can be used to identify key proteins or pathways that are differentially regulated due to solar exposure. This information ultimately aids in the development of targeted treatments that can either upregulate or downregulate specific proteins to mitigate the effects of photoaging. For example, if a certain protein is less abundant in solar-exposed skin, a treatment could be formulated to boost its expression, thereby potentially slowing the aging process at the molecular level. However, we acknowledge the limitations posed by the dataset’s size and suggest that these findings serve as an exploratory step toward more comprehensive studies. Further research with expanded cohorts is essential to develop reliable, personalized skincare treatments based on proteomics.
Computational modeling and algorithm development
The dataset’s high complexity and dimensionality make it an invaluable resource for scholars in the fields of computational biology and bioinformatics. Its unique structure provides fertile ground for the development of innovative algorithms tailored for the analysis of paired mass spectrometry data, a critical aspect for ensuring robust statistical outcomes. Moreover, the dataset can be amalgamated with other “omics” data types, such as genomics or transcriptomics, to formulate multi-layered computational models that deepen our understanding of skin health and aging processes.
A noteworthy aspect is the role of DiagnoMass in this dataset’s utility. The tool reveals that only a minor fraction of spectral clusters unique to solar exposed versus non-exposed conditions are currently identified. This suggests the existence of potentially undiscovered alterations not yet cataloged in public databases, or post-translational modifications that present detection challenges for existing tools. DiagnoMass thus serves as a critical benchmark, setting an upper limit on what is currently known and highlighting the expansive scope for future discoveries in skin proteomics.
Code availability
In this study, no custom code was utilized. All software used in this study is open access. A comprehensive list of software used in this study is provided in this section as well.
• PatternLab for Proteomics V (http://patternlabforproteomics.org/).
• DiagnoMass (https://www.diagnomass.com/).
• RawVegetable (http://patternlabforproteomics.org/rawvegetable/).
References
Nguyen, A. V. & Soulika, A. M. The Dynamics of the Skin’s Immune System. IJMS 20, 1811 (2019).
Csekes, E. & Račková, L. Skin Aging, Cellular Senescence and Natural Polyphenols. IJMS 22, 12641 (2021).
Liu, Y. et al. Targeting the stem cell niche: role of collagen XVII in skin aging and wound repair. Theranostics 12, 6446–6454 (2022).
Wong, Q. Y. A. & Chew, F. T. Defining skin aging and its risk factors: a systematic review and meta-analysis. Sci Rep 11, 22075 (2021).
Krutmann, J., Bouloc, A., Sore, G., Bernard, B. A. & Passeron, T. The skin aging exposome. Journal of Dermatological Science 85, 152–161 (2017).
Gromkowska‐Kępka, K. J., Puścion‐Jakubik, A., Markiewicz‐Żukowska, R. & Socha, K. The impact of ultraviolet radiation on skin photoaging — review of in vitro studies. J of Cosmetic Dermatology 20, 3427–3431 (2021).
Pandel, R., Poljšak, B., Godic, A. & Dahmane, R. Skin Photoaging and the Role of Antioxidants in Its Prevention. ISRN Dermatology 2013, 1–11 (2013).
Lee, H., Hong, Y. & Kim, M. Structural and Functional Changes and Possible Molecular Mechanisms in Aged Skin. IJMS 22, 12489 (2021).
Camillo-Andrade, A. C. et al. Proteomics reveals that quinoa bioester promotes replenishing effects in epidermal tissue. Sci Rep 10, 19392 (2020).
Zheng, S. et al. Proteomics as a tool to improve novel insights into skin diseases: what we know and where we should be going. Front. Surg. 9, 1025557 (2022).
Brunoro, G. V. F. et al. Differential proteomic comparison of breast cancer secretome using a quantitative paired analysis workflow. BMC Cancer 19, 365 (2019).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol 18, 83 (2017).
Kurt, L. U. et al. RawVegetable - A data assessment tool for proteomics and cross-linking mass spectrometry experiments. J Proteomics 225, 103864 (2020).
Santos, M. D. M. et al. Simple, efficient and thorough shotgun proteomic analysis with PatternLab V. Nat Protoc 17, 1553–1578 (2022).
Santos, M. D. M. et al. DiagnoMass: A proteomics hub for pinpointing discriminative spectral clusters. Journal of Proteomics 277, 104853 (2023).
Lin, A. D. et al. Beyond the identifiable proteome: Delving into the proteomics of polymyxin-resistant and non-resistant Acinetobacter baumannii from Brazilian hospitals. Journal of Proteomics 105012 https://doi.org/10.1016/j.jprot.2023.105012 (2023).
Rappsilber, J., Ishihama, Y. & Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 (2003).
Clasen, M. A. et al. Increasing confidence in proteomic spectral deconvolution through mass defect. Bioinformatics 38, 5119–5120 (2022).
Eng, J. K. et al. A deeper look into Comet–implementation and features. J. Am. Soc. Mass Spectrom. 26, 1865–1874 (2015).
Carvalho, P. C. et al. Search engine processor: Filtering and organizing peptide spectrum matches. Proteomics 12, 944–949 (2012).
Barboza, R. et al. Can the false-discovery rate be misleading? Proteomics 11, 4105–4108 (2011).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 50, D543–D552 (2022).
Camillo-Andrade, A. et al. PRIDE. https://identifiers.org/pride.project:PXD045887 (2024).
Na, S. & Paek, E. Quality Assessment of Tandem Mass Spectra Based on Cumulative Intensity Normalization. Journal of Proteome Research 5, 3241–3248 (2006).
Acknowledgements
The authors thank Vichy Exposome Award, CNPq (442655/2023-1), CAPES, Fiocruz - INOVA Fundação Araucária, and FOCEM-COF 03/11 for financial support.
Author information
Authors and Affiliations
Contributions
A.C.C.A. was responsible for participant selection, sample collection and preparation, data generation and analysis, and manuscript writing. M.D.M.S. contributed to sample preparation, data generation, and data analysis. A.L. was involved in data generation. P.S.N. and A.B.L.L. handled sample collection. L.S. contributes to data analysis. J.S.F.G. participated in sample preparation. P.C.C. and R.D. acted as advisors, overseeing the entire project, and contributing to discussions at all stages of the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that part of this research was funded by the Vichy Exposome Award. Vichy had no role in the study design, data collection, data analysis, data interpretation, or writing of the manuscript. The authors declare no conflicting interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Camillo-Andrade, A.C., Santos, M.D.M., Nuevo, P.S. et al. Intra-Individual Paired Mass Spectrometry Dataset for Decoding Solar-Induced Proteomic Changes in Facial Skin. Sci Data 11, 441 (2024). https://doi.org/10.1038/s41597-024-03231-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03231-1
- Springer Nature Limited