
Abstract
Broomcorn millet (Panicum miliaceum L.), known for its traits of drought resistance, adaptability to poor soil, short growth period, and high photosynthetic efficiency as a C4 plant, represents one of the earliest domesticated crops globally. This study reports the telomere-to-telomere (T2T) gap-free reference genome for broomcorn millet (AJ8) using PacBio high-fidelity (HiFi) long reads, Oxford Nanopore long-read technologies and high-throughput chromosome conformation capture (Hi-C) sequencing data. The size of AJ8 genome was approximately 834.7 Mb, anchored onto 18 pseudo-chromosomes. Notably, 18 centromeres and 36 telomeres were obtained. The assembled genome showed high quality in terms of completeness (BUSCO score: 99.6%, QV: 61.7, LAI value: 20.4). In addition, 63,678 protein-coding genes and 433.8 Mb (~52.0%) repetitive sequences were identified. The complete reference genome for broomcorn millet provides a valuable resource for genetic studies and breeding of this important cereal crop.
Similar content being viewed by others

Background & Summary
Broomcorn millet (Panicum miliaceum L.), a member of the Paniceae tribe in the Gramineae family, exhibits remarkable adaptability to marginal regions due to its short growing season (60–90 days), low water requirements, high salt tolerance, and efficient nutrient resource utilization1,2. Being a C4 plant, broomcorn millet demonstrates enhanced carbon fixation and efficient utilization of water and nitrogen resources. Additionally, its grains are characterized by their gluten-free nature and exceptional nutritional value, containing higher protein content, mineral composition, and antioxidant levels compared to most other cereals3. Consequently, broomcorn millet has been extensively cultivated in semiarid regions across Asia, Europe, and other continents and is considered one of the oldest crops worldwide4. The cultivation of broomcorn millet holds promise for enhancing food security, diversifying agriculture, and promoting a healthier diet5. Broomcorn millet has an allotetraploid genome consisting of 36 (2n = 4x = 36) chromosomes6. Although four chromosome-level of broomcorn millet, Jinshu77, LM_v18, LM_v29, and Pm_03908, have been made available, there are still missing segments within the genome due to the presence of highly repetitive sequences clustered across the genome, particularly in the telomere and centromere regions. In recent years, T2T and gap-free genomes have been successfully obtained in various important crops, including rice10, barley11, and maize12.
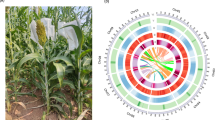
In the present study, we assembled the first T2T gap-free genome of broomcorn millet (AJ8) (Fig. 1a), achieved through PacBio HiFi long reads, Nanopore technologies and Hi-C sequencing data. The resulting complete genome assembly has a final size of 834.7 Mb and is organized into 18 pseudo-chromosomes (Table 1; Fig. 1b). Gene annotation identified 52.0% repetitive sequences and 63,678 protein-coding genes (Fig. 1b). This complete reference genome provides a robust foundation for future studies on population and conservation genetics of broomcorn millet.

An overview of the AJ8 genome. (a) The photograph of the AJ8 plant. (b) Circos plot illustrating the genome of the AJ8 genome. The plot includes the following components, arranged from inside to outside: (I) Collinear regions within the AJ8 assembly; (II) GC content in non-overlapping 1 Mb windows; (III) Percentage of repeats in 1-Mb sliding windows; (IV) Gene density in 1-Mb sliding windows; (V) Length of pseudo-chromosome in megabases (Mb).
Methods
Plant materials and growth conditions
The broomcorn millet landrace sequenced in this study was originally collected from Center for Agricultural Genetic Resources Research, Shanxi Agricultural University, Taiyuan, Shanxi Province (coordinates: E 112° 34′ 26.66″, N 37° 46′ 37.16″). The plants were planted under controlled conditions with a temperature of 25 °C, humidity of 60%, and a light intensity following a 14-hour day and 10-hour night cycle. Twenty seedlings with consistent growth at the fourth leaf stage were carefully chosen and sampled from various organs, including roots, stems, and leaves. A weight of 2 g was measured for each tissue organ, which was immediately placed in a freezing chamber with liquid nitrogen and subsequently stored at −80 °C.
Long insert libraries preparation and sequencing
Genomic DNA was extracted from leaf tissue using DNeasy Plant Maxi kit (Qiagen). The PacBio long insert libraries were prepared according to manufacturers’ instructions with an insert size of approximately 20 kb (Pacific Biosciences, USA). Subsequently, the libraries were subjected to sequencing using PacBio Sequel II platforms in circular consensus sequencing mode. The subreads were processed using SMRTLink (v11.1.0)13 with parameters “–minPasses 3 –minPredictedAccuracy 0.99 –minLength 500”, yielding approximately 77.0 Gb high-fidelity (HiFi) reads with a N50 size of about 18.0 kb (Table 2).
The ONT ultra-long insert libraries were generated using the Oxford Nanopore SQK-LSK109 kit, and then sequenced on a PromethION flow cell (Oxford Nanopore Technologies, Oxford, UK). A total of 165.6 Gb of ONT data with 196x coverage was generated, and the N50 value was 55,765 bp (Table 2). After error correction and length filtering of the data, 34.8 Gb ultra long ONT reads with the N50 value 92, 975 bp were obtained (Table 2).
Short insert libraries preparation and sequencing
For chromosomal conformational capture (Hi-C) sequencing, Hi-C libraries based on DpnII restriction enzymes were generated as previously described14, and sequenced on the MGISEQ-2000 platform. A total of 253.0 Gb of clean data were obtained from 255.1 Gb of sequencing data using software SOAPnuke (v2.0)15 with parameters “-n 0.01 -l 20 -q 0.1 -i -Q 2 -G 2 -M 2 -A 0.5” (Table 2).
RNA-seq libraries from leaf tissues were constructed using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA) following the manufacturer’s protocol. Then the RNA libraries were sequenced on a MGISEQ-2000 instrument and generated 150 bp paired-end reads. After quality control by fastp (0.19.5)16 with parameters of “–adapter_sequence AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA–adapter_sequence_r2 AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG–average_qual 15 -l 150”, each library contained more than 7.8 Gb of clean data. More than 98.1% of the clean data had scores greater than Q20 in each library (Table 3).
Genome assembly
Using HiFi reads, ultra-long ONT reads, and Hi-C clean data, the primary contigs were assembled by Hifiasm (v 0.19.5)17 with default parameters. To anchor contigs onto chromosomes, we used BWA (v 0.7.12)18 to align the Hi-C clean data to the assembled contigs. Low-quality reads were filtered out using the HiC-Pro pipeline19 with default parameters. The remaining valid reads were employed to anchor chromosomes with Juicer20 and 3d-dna pipeline21. Excitingly, our results showed that the hifiasm assembly consists of contiguous sequences covering the entire length of all chromosomes. This achievement can be attributed to the remarkable accuracy of HiFi data, the utilization of ultra-long ONT data, and the ongoing enhancements in assembly algorithms. Analogous to the T2T genome of rapeseed22, the hifiasm assembly comprises continuous sequences spanning the entirety of nine chromosomes. For further refinement of the genome, the T2T assembly was polished using a similar method described by Mc Cartney et al.23. In brief, the HiFi reads were aligned to the T2T assembly using Winnowmap2 (v 2.03)24. The resulting alignments were filtered to exclude secondary alignments and alignments with excessive clipping by using ‘falconc bam-filter-clipped’ tool. Finally, racon (v 1.5.0)25 was performed with the filtered alignments. The final assembled genome had a length of 834,678,208 bp and a contig N50 of 48.3 Mb (Table 1). The assembled sequences were successfully anchored to 18 pseudo-chromosomes (Table 1). The completeness of the assembly was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO) (v 5.5.0)26 with the embryophyta_odb10 (parameters: -m genome -l embryophyta_odb10). We appylied Merqury (v1.3)27 using PacBio HiFi long reads with a K-mer value of 17-bp to estimate the quality value.
Annotation of repetitive sequences
Tandem repeats and interspersed repeats were identified using the method described in Qu et al.28. In brief, RepeatModeler (v1.0.4)29 and LTR-FINDER (v1.0.7)30 were employed to construct of a de novo repeat sequence library. This compiled library was subsequently deployed for the detection of interspersed repeats and low-complexity sequences utilizing RepeatMasker (v4.0.7)31. DNA and protein transposable elements (TEs) were identified through RepeatMasker (v4.0.7) and RepeatProteinMasker (v4.0.7), respectively. Tandem repeats were discerned utilizing Tandem Repeat Finder (v4.10.0)32. A total of 433.8 Mb (~52.0%) of repetitive sequences were obtained. Among the interspersed repeats, three types of repetitive elements, namely class I (retrotransposons), class II (DNA transposons), and unclassified elements, accounted for 49.6% of the genome assembly (Table 4). The telomeric sequences and centromere region in the AJ8 genome assembly were identified using quartet (v1.1.36)33 with “-c plant”.
Protein-coding genes prediction and functional annotation
Gene prediction in this study was conducted using a combination of transcriptome-based prediction, homology-based prediction, and ab initio prediction methods. For transcriptome-based prediction, 194.2 Gb Illumina clean reads from 21 samples were assembled by Trinity (v 2.15.1)34 with parameters of ‘–max_memory 200 G–CPU 40–min_contig_length 200–genome_guided_bam merged_sorted.bam–full_cleanup–min_kmer_cov 4–min_glue 4–bfly_opts ‘-V 5–edge-thr = 0.1–stderr’–genome_guided_max_intron 10000’, which generated 227,501 transcripts with a N50 of 2,698. These assembled transcripts were aligned against the AJ8 T2T assembly using Program to Assemble Spliced Alignment (PASA) (v 2.4.1)35. Gene structures were generated from valid transcript alignments (PASA-set). RNA-seq clean reads were also mapped to the AJ8 T2T assembly using Hisat2 (v 2.0.1)36. Stringtie (v 1.2.2)37 and TransDecoder (v 5.7.1) (https://github.com/TransDecoder/TransDecoder) were employed to assemble the transcripts and identify candidate coding regions into gene models (Stringtie-set). Homologous genomes from seven plants, including rice (T2T-NIP)10, maize (T2T Mo17)12, A. thaliana (Col-PEK)38, sorghum (GCA_000003195.3)39, pearl millet (Tift23D2B1-P1-P5)40, foxtail millet (GCA_000263155.2)41, and two previous versions of broomcorn millet (Jinshu7: GCA_026771285.1; Longmi4: GCA_002895445.3)7,9 were downloaded and used as queries to search against the AJ8 T2T assembly using GeMoMa (v 1.9)42. These homology predictions were referred to as “Homology-set”. For ab initio prediction methods, AUGUSTUS (v 3.2.3)43 was used to predict coding regions in the repeat-masked genome. All gene models were combined using EvidenceModeler (v 2.1.0)44 with different weight parameters assigned to evidence from different sources (10 for PASA-set, 5 for Stringtie-set, 5 for Homology-set, and 1 for AUGUSTUS gene prediction). The resulting protein-coding genes that were only derived from ab initio prediction were filtered out. Finally, the produced gene models were further refined with PASA (v 2.4.1)37 to generate untranslated regions and alternative splicing variation information. The final comprehensive gene set comprised 63,678 genes.
The integrated gene set was translated into amino-acid sequences and annotated using the method described in Zhou et al.45. As a result, 98.8% of the predicted protein-coding genes were functionally annotated (Table 5). In addition, we utilized the Diamond (v 0.9.30)46 software (E value <= 1e-05) to perform homologous comparison analyses between AJ8 and Arabidopsis (TAIR10)47, as well as between AJ8 and rice (Osativa_v7_0)48. Among the AJ8 genes, 48,284 (75.83%) genes exhibited homology with Arabidopsis, while 56,777 (89.16%) genes showed homology with rice (Table S1).
Gene expression analysis
The gene expression analysis was used the same method as previously reported49. The expression heatmap was constructed using heatmap R package. The expression matrix of genes in different transcriptome samples was displayed in Table S2.
Synteny analysis
The identification of syntenic regions was based on conducting homology searches using MCScan (Python version)50, with a minimum requirement of 30 genes per block.
Subgenome phasing
By employing repetitive k-mers as “differential signatures” and utilizing the SubPhaser software, we successfully phased the subgenomes of AJ8. The results obtained from SubPhaser51 were found to be consistent with the Jinshu7 genome, as indicated in Table 6 and Fig. 4.
Data Records
The sequencing data and assembled genome sequence have been deposited in the Sequence Read Archive with accession numbers SRP48256652 under project number PRJNA1059665. The genome assembly has been deposited at GenBank under the WGS accession
GCA_038442765.153. Files of the gene structure annotation, repeat predictions and gene functional annotation were deposited at Figshare54.
Technical Validation
Genome assembly and gene prediction quality assessment
The accuracy and integrity of AJ8 T2T assembly were assessed through several analyses. Firstly, the Hi-C heatmap displayed consistent results across all chromosomes, indicating the correct ordering and orientation of contigs in the assembly (Fig. 2a). Secondly, the assembly successfully captured 18 centromeres and all 36 telomeres, providing strong evidence for their integrity (Fig. 2b; Tables 7, 8). Thirdly, the assembly showed high collinearity with Jinshu7 (GCA_026771285.1)7 and Panicum hallii (GCA_002211085.2)55 (Fig. 2c). Fourthly, alignment results from minimap2 (v 2.24-r1122)56 revealed that 100.0% of ONT reads and 99.98% of HiFi reads could be aligned to the AJ8 T2T assembly. Additionally, the average genome alignment rate of the transcriptome was 98.7% (Table 3). Lastly, AJ8 T2T demonstrated an LTR assembly index of 20.4, a quality value of 61.7, and a BUSCO score of 99.6%, indicating its high completeness (Table 1).

The high-quality of the AJ8 genome. (a) Heatmap displaying Hi-C interactions of AJ8 pseudomolecules. (b) Telomere detection map. Triangles and circles represent telomeres and centromere within the AJ8 assembled chromosomes. The orange color represents regions with high gene density, while the dark sky blue color represents regions with low gene density. (c) Synteny analysis of Panicum hallii, AJ8 and Jinshu7.
We compared the length distribution of genes among the AJ8, maize12, sorghum (GCF_000003195.3)39, LM_18, and Jinshu77 and found similar patterns (Fig. 3a). The BUSCO analysis showed that 99.5% (single-copy gene: 19.5%, duplicated gene: 80.0%) of 1,614 embryophyta single-copy orthologs were successfully identified as complete, while 0.2% were fragmented and 0.3% were missing in the assembly (Fig. 3b). 62,885 (98.8%) gene models were successfully annotated in diverse databases and 51,958 gene models (81.6%) exhibited detectable transcriptional activity (FPKM value ≥ 1) across 21 RNA-seq samples (Fig. 3c; Table 5; Fig. 3d). Taken together, these results provide strong evidence that a high-quality AJ8 genome has been obtained. The high-quality genome provides a solid foundation for uncovering the drought resistance and adaptive mechanisms of AJ8, and also serves as an important reference for the rapid breeding of AJ8 and other crops.

Gene prediction quality assessment. (a) The composition of gene length in the AJ8 genome compared to the genomes of other species. (b) BUSCO assessments of the AJ8 gene. (c) Venn diagram showing the number of genes with homology or functional classification by each method. (d) The expression heatmap illustrates the expression levels among 21 RNA-seq samples. The color bar in the lower right corner represents log2-transformed FPKM values. Blue and red boxes indicate genes with lower and higher expression levels, respectively.

Phased subgenomes of the AJ8 genome. (a) The histogram of differential k-mers among homoeologous chromosome sets. (b) Heatmap and clustering of differential k-mers. The x-axis, differential k-mers; y-axis, chromosomes. The vertical color bar, each chromosome is assigned to which subgenome; the horizontal color bar, each k-mer is specific to which subgenome (blank for non-specific kmers). (c) Principal component analysis of differential k-mers. Points indicate chromosomes.
Code availability
No specific code or script was used in this work. Commands used for data processing were all executed according to the manuals and protocols of the corresponding software.
References
Yue, H. et al. Transcriptome-wide identification and expression profiles of the WRKY transcription factor family in Broomcorn millet (Panicum miliaceum L.). BMC genomics 17, 343 (2016).
Rajput, S. G., Santra, D. K. & Schnable, J. Mapping QTLs for morpho-agronomic traits in proso millet (Panicum miliaceum L.). Molecular breeding: new strategies in plant improvement 36, 37 (2016).
Saleh, A. S. M., Zhang, Q., Chen, J. & Shen, Q. Millet Grains: Nutritional Quality, Processing, and Potential Health Benefits. Comprehensive Reviews in Food Science and Food Safety 12, 281–295 (2013).
Lu, H. et al. Earliest domestication of common millet (Panicum miliaceum) in East Asia extended to 10,000 years ago. Proceedings of the National Academy of Sciences 106, 7367–7372 (2009).
Habiyaremye, C. et al. Proso Millet (Panicum miliaceum L.) and Its Potential for Cultivation in the Pacific Northwest, U.S.: A Review. Frontiers in Plant Science 7 (2017).
Hunt, H. V. et al. Reticulate evolution in Panicum (Poaceae): the origin of tetraploid broomcorn millet, P. miliaceum. Journal of Experimental Botany 65, 3165–3175 (2014).
Sun, Y. et al. Biased mutations and gene losses underlying diploidization of the tetraploid broomcorn millet genome. The Plant journal 113, 787–801 (2022).
Shi, J. et al. Chromosome conformation capture resolved near complete genome assembly of broomcorn millet. Nature communications 10, 464 (2019).
Wang, Z. et al. A high-quality, phased genome assembly of broomcorn millet reveals the features of its subgenome evolution and 3D chromatin organization. Plant Communications 4 (2023).
Shang, L. et al. A complete assembly of the rice Nipponbare reference genome. Molecular plant 16, 1232–1236 (2023).
Navrátilová, P. et al. Prospects of telomere‐to‐telomere assembly in barley: Analysis of sequence gaps in the MorexV3 reference genome. Plant Biotechnology Journal 20, 1373–1386 (2021).
Chen, J. et al. A complete telomere-to-telomere assembly of the maize genome. Nature Genetics 55, 1221–1231 (2023).
Chin, C. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nature Methods 10, 563–569 (2013).
Belton, J.-M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58(3), 268–76 (2012).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 7 (2017).
Chen, S., Zhou, Y., Chen, Y. & Jia, G. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Servant, N. et al. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biology 16 (2015).
Durand, N. et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Systems 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, eaal3327 (2017).
Li, B. et al. A gap-free reference genome reveals structural variations associated with flowering time in rapeseed (Brassica napus). in Horticulture research Vol. 10 uhad171 (2023).
Mc Cartney, A. M. et al. Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. Nature methods 19, 687–695 (2022).
Jain, C., Rhie, A., Hansen, N. F., Koren, S. & Phillippy, A. M. Long-read mapping to repetitive reference sequences using Winnowmap2. Nature methods 19, 705–710 (2022).
Vaser, R., Sovic, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome research 27(5), 737–746 (2017).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods in molecular biology (Clifton, N.J.) 1962, 227–245 (2019).
Rhie, A., Walenz, B. P., Koren, S. & Phillippy, A. M. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21 (2020).
Qu, C. et al. Comparative genomic analyses reveal the genetic basis of the yellow-seed trait in Brassica napus. Nature Communications 14, 5194 (2023).
Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Current Protocols in Bioinformatics 5 (2004).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic acids research 35, W265–8 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics (Oxford, England) 21(Suppl 1), i351–8 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Lin, Y. et al. quarTeT: a telomere-to-telomere toolkit for gap-free genome assembly and centromeric repeat identification. Horticulture Research (2023).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology 29(7), 644–52 (2011).
Haas, B. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Research 31, 5654–5666 (2003).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology 20, 278 (2019).
Hou, X., Wang, D., Cheng, Z., Wang, Y. & Jiao, Y. A near-complete assembly of an Arabidopsis thaliana genome. Molecular plant 15, 1247–1250 (2022).
Paterson, A. H. et al. The Sorghum bicolor genome and the diversification of grasses[J]. Nature 457(7229), 551–556 (2009).
Ramu, P. et al. Improved pearl millet genomes representing the global heterotic pool offer a framework for molecular breeding applications. Communications Biology 6 (2023).
Bennetzen, J. L. et al. Reference genome sequence of the model plant Setaria[J]. Nature biotechnology 30(6), 555–561 (2012).
Jens et al. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods in Molecular Biology (2019).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic acids research 33, W465–7 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biology 9, R7 (2008).
Zhou, Y. et al. Gap-free genome assembly of Salangid icefish Neosalanx taihuensis. Scientific data 10, 768 (2023).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nature Methods 12, 59–60 (2015).
Phytozome database, https://phytozome-next.jgi.doe.gov/info/Athaliana_TAIR10 (2022).
Phytozome database, https://phytozome-next.jgi.doe.gov/info/Osativa_v7_0 (2022).
Sun, C. et al. Chromosome-level genome assembly for the largemouth bass Micropterus salmoides provides insights into adaptation to fresh and brackish water. Molecular ecology resources 21, 301–315 (2021).
Tang, H. et al. Synteny and Collinearity in Plant Genomes. Science 320, 486–488 (2008).
Zhang, R.-G. et al. Subgenome-aware analyses suggest a reticulate allopolyploidization origin in three Papaver genomes. Nature Communications 14 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP482566 (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_038442765.1 (2024).
Wang, Haigang. Genome assembly and annotation of Panicum miliaceum AJ8. figshare https://doi.org/10.6084/m9.figshare.24947565.v1 (2024).
Lovell, J. T. et al. The genomic landscape of molecular responses to natural drought stress in Panicum hallii. Nature Communications 9 (2018).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2017).
Zou, C. et al. The genome of broomcorn millet. Nature communications 10, 436 (2019).
Acknowledgements
This work was funded by Key Research and Development Program of Shanxi Province (2022ZDYF110), Project of Conservation and Utilization of Agricultural Germplasm Resources in Shanxi Province (sxzyk202201), the Central Guiding Local Science and Technology Development Fund Project (YDZJSX20231A035), China Agriculture Research System of MOF and MARA (CARS-06-14.5-A16).
Author information
Authors and Affiliations
Contributions
Z.X.M., Z.J.Q., and L.W. conceived the study. H.G.W., J.J.W., H.B.Q., and M.L. collected and prepared the samples. C.H.C., H.G.W., X.K.Y., and J.B.J. performed bioinformatics analysis. H.G.W., C.H.C. and L.C. wrote the manuscript with significant contributions. X. T., S.Hou., X.K.Y., P.G. and J.B.J. revised the manuscript. All authors read, edited, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, H., Wang, J., Chen, C. et al. A complete reference genome of broomcorn millet. Sci Data 11, 657 (2024). https://doi.org/10.1038/s41597-024-03489-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03489-5
- Springer Nature Limited