
Abstract
The Eastern Pamir, distinguished with high altitude, extremely arid and cold climate, limited nutrients and sparse vegetation, is a unique ecological reservoir. Microbial communities play a central role in maintaining Eastern Pamir’s ecosystem functioning. Despite the ecological significance, due to the difficulty of sample collection and microbial isolation, the microbial diversity and its functionality at the Pamir Plateau have been rarely documented. To fill this gap, 80 soil samples from 17 sites across different elevations were collected, performed the rDNA amplicon sequencing to present the first large-scale overview of bacterial, archaeal, and fungal communities in the Eastern Pamir. Microbiome analysis revealed that the bacteria Actinobacteria, Alphaproteobacteria and Bacteroidia, alongside such as archaea Nitrososphaeria and Halobacteria, and fungi including Dothideomycetes, Sordariomycetes and Eurotiomycetes were dominant lineages at class level in soil microbial communities. The community structure and biodiversity of soil microorganisms provided by this dataset would be pivotal for future studies aimed at understanding the biogeographical distribution, ecological functions and environmental responses of microbial communities of the Pamir Plateau.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background & Summary
The plateau ecosystems, characterized by their cold climates and extensive expanses of tundra, lakes, and glaciers, are integral to the terrestrial ecosystems. These high-latitude and high-elevation ecosystems are particularly susceptible to global climate change, as evidenced by Johnson et al.1. Microorganisms play a pivotal role in the biogeochemical cycling of carbon and nitrogen in such extreme environments, marked by such as high altitude, low temperatures, aridity, and low oxygen levels (only about 70% of that in the plains). Furthermore, microorganisms are essential in shaping and maintaining ecosystems under these harsh conditions2.
The Pamir Plateau is the second largest plateau in the world, extending across southwestern Xinjiang in China, southeastern Tajikistan, and northeastern Afghanistan, covering an area of approximately 100,000 square kilometers with an average elevation of 4,500 meters above sea level3,4. The vast mountains, elevated terrain, and varying topography in the region foster an extraordinary arid and cold climate with severe temperature fluctuations5,6. The region also encompasses diverse natural habitats like lakes, water systems, and glaciers, which collectively shape a distinctive heterogeneous zone7,8. The region is known for its abundant and unique microbial resources, making it a significant constitution to the high-altitude bioresource repository and global genetic diversity and becoming a key zone for biodiversity conservation9,10. However, the region confronts challenges due to anthropogenic disturbances, natural disasters, and climate change, which have accelerated the loss and extinction of microbial species in fragile ecosystems11. Soil microbial diversity and community functionality are particularly vulnerable to these extreme environmental changes.
Soil microorganisms constitute one of the most diverse groups of organisms on Earth. They play a crucial role in soil biogeochemical processes, participate in almost all material transformations in the soil, and have a significant impact on ecosystems such as forests, grasslands, wetlands, and arable lands12,13. The extreme aridity, nutrient scarcity, diurnal temperature fluctuations, and high ultraviolet radiation in the Pamir Plateau present a challenging environment. These conditions disrupt soil productivity and ecosystem functioning14,15. To date, prior studies on microorganisms in plateau regions have focused on the Tibetan, Loess, and Mongolian Plateaus. These studies have involved variations in soil enzyme activities and microbial communities along elevation gradients16, reflections of functional microorganisms in soil to natural factors such as nitrogen deposition and precipitation17,18, or different responses of soil microorganisms from different plant communities to changes in environmental stresses19.
There were sporadic reports on the microorganisms of the Pamir Plateau. As early as the 1960s, researchers studied soil actinomycetes and their antagonistic properties on the Pamir Plateau20,21. Aksenov et al.22 studied the adaptive mechanisms of Cryptococcus in the Pamir region under very low humidity conditions. In the 1970s, Szymon et al.23 isolated 16 species of fungi from 12 species of endemic plants of the Pamir Aly Mountains, which were found to be predominantly cotyledonous mycorrhizal fungi. Nataliia et al.24,25 carried out studies on microbial diversity and colonization strategies in rock of cold desert ecosystems in the Eastern Pamir Mountains of Tajikistan, and Bu et al.26 isolated numerous cold-adapted bacteria as well as archaeal resources from the Pamir Plateau. However, a systematic investigation of the microbial resources of the Pamir Plateau, comprehensive data on the microbial resources of the Pamir region, including the distribution, composition, and function of the microbial communities is still an uncharted area. Therefore, research into soil microorganisms and the data collection on microbial resources in the Pamir Plateau are of great importance for understanding regional and global climatic and environmental changes, biodiversity, and carbon cycling.
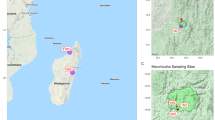
In this study, we collected 85 soil samples from 17 sites along the G314 highway, stretching approximately 300 kilometers from Kashgar City to Tashkurgan County, with altitudes ranging from 1715 meters to 4069 meters (Table 1 and Fig. 1). Since five of the soil samples were unsuccessful in DNA extraction, resulting in a final dataset of 80 soil samples. Amplicon sequencing using Illumina NovaSeq sequencing platform yielded 7,644,450, 6,821,012, and 6,627,693 amplicon reads for soil archaeal, bacterial, and fungal datasets, respectively. Subsequent analysis yielded a count of 480 taxa in the archaeal dataset, 9,829 taxa in the bacterial dataset, and 1,778 taxa in the fungal dataset across all soil samples (Fig. 2 and Table 2). In Fig. 2, the rarefaction curves of all samples exhibit a plateauing trend, suggesting that the current sampling strategy is adequate to capture all the microbial taxa in soil communities. However, it is noteworthy that the species richness levels for archaea and fungi in this study were relatively low.

Contour map of sampling site distribution in the Eastern Pamir.

Microbial diversity of the soil microbiota profile in Eastern Pamir. (a) Rarefaction curves of the bacterial (purple), archaeal (blue) and fungal (red) communities. (b–d) The richness of the archaeal (b), bacterial (c), and fungal (d) diversity index for each sampling site in Pamir, respectively.
In addition, 224 taxa could not be taxonomically classified within the fungal community at the phylum level, representing 12.60% of the overall detected fungal taxa with the cumulative relative abundance. Similarly, 422 bacterial taxa could not be classified at the phylum level, comprising 4.29% of the total detected bacterial taxa. Moreover, all archaeal taxa were annotated into 14 families, but 191 archaeal taxa remained unclassified at the genus level, accounting for a substantial 39.79% of the total detected archaeal taxa. This suggests that there may be a considerable volume of unrecognized microbial resources in this region waiting for further exploration.
The analysis of the bacterial communities revealed that the Actinobacteria (15.61%) and Alphaproteobacteria (11.99%) emerged as the dominant groups, alongside notable prevalence of Bacteroidia (8.72%) and Gammaproteobacteria (7.01%) (Fig. 3a). Fungal communities were dominantly represented by Dothideomycetes (32.06%), Sordariomycetes (23.66%), and Eurotiomycetes (9.35%) (Fig. 3b). In archaeal communities, Nitrososphaeria class was predominant, representing 70.46% of the relative abundance, followed by Halobacteria at 27.90% (Fig. 4a). Further analysis of the composition of archaeal community at the genus level showed that Candidatus Nitrososphaera (26.48%) was the predominant genus, followed by Candidatus Nitrocosmicus (25.9%), Nitrososphaeraceae (18.08%), Haloferacaceae (8.58%), and Halalkalicoccus (3.74%) (Fig. 4b). Notably, the highest species richness for archaea is observed at Subashi Bridge 8th (SbsBrdg8), whereas for bacteria and fungi, it was at Gez River site 2 (Gez2) (Table 3). The results of the amplicon data provided insights into the composition of the microbial community and its spatial distribution patterns in the region.

Soil microbial community structures in Pamir. The bar plots show the taxonomic distribution of the bacterial community (a) and fungal community (b) at the class level.

Average relative abundance of the predominant archaea at the class level (a) and genus level (b) in Pamir.
Multiple Regression on Distance Matrices (MRM) analysis explored the influence of environmental factors on the composition of the three types of microbial communities (Table 4). It also reveals that site geographical distances significantly influenced the compositions of all three microbial communities. Archaeal communities are best explained by the environmental factors (R² = 0.4121), followed by fungal (R² = 0.2517) and bacterial communities (R² = 0.1446). Soluble salt emerges as the most influential factor for archaeal communities, followed by total nitrogen, pH, precipitation seasonality, and available nitrogen. For bacterial communities, precipitation seasonality and soil moisture were significantly influential, while total nitrogen, moisture, total organic matter, soluble salt, and available nitrogen were key factors shaping fungal communities. Annual average solar radiation, annual mean temperature, and temperature seasonality strongly correlate with altitude, suggesting elevation is a key determinant of temperature and solar radiation in this region (Fig. 5).

Kendall correlation indicated self-correlated environmental factors in the present study.
Methods
Sampling
In July 2020, 85 soil samples were collected from 17 sites along the eastern slope of the Eastern Pamir Plateau in Xinjiang, China (Fig. 1 and Table 1). At each site, five soil samples were taken as replicates. These replicates were collected from 5 m × 5 m plots using a five-point sampling strategy. The 2 cm of topsoil was removed to discard litters, and approximately 1 kg of soil was collected from depths of 2–20 cm and sieved to remove rocks and debris. The soil samples were immediately transported to the laboratory in an ice box. For subsequent DNA extraction, 50 g of soil from each sample was stored at −80 °C, and the remainder was stored under cool, dry conditions for geochemical analysis. The contour map of sampling sites was drawn using ArcGIS mapping software (Arc Geographic Information System, Environmental Systems Research Institute, Inc. USA) and downloading 30 m resolution DEM data from the Geospatial Data Cloud website (https://www.gscloud.cn/), contour data is generated through the splicing, cropping, and contour tools in the ArcGIS toolbox. Smooth the generated contour data and add a grid, legend, compass, scale bar, and drawing name.
DNA extraction and PCR amplification
Total genomic DNA was extracted from 5 g of each soil sample using CTAB/SDS method. DNA concentration and quality was assessed on 1% agarose gels. Due to the failure to extract DNA from 5 soil samples, data from 80 soil samples were finally obtained. DNA was then diluted to 1 ng/µL using sterile deionized water. The specific amplification process employed barcode-attached universal primers targeting the 16S rRNA V4-V5 region for archaea (Arch519F/Arch915R)27, the 16S rRNA V4 region for bacteria (515 F/806 R)28, and the ITS1 region for fungi (ITS1-1F-F/ITS1-1F-R)29. All PCR reactions were carried out with Phusion® High-Fidelity PCR Master Mix (New England Biolabs). All PCR reactions were performed using 15 µL systems, composing of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 µM of both forward and reverse primers, and about 10 ng DNA templates. The thermal cycling consisted of an initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, and elongation at 72 °C for 30 s, with a final elongation 72 °C for 5 min. PCR products were mixed with 1X loading buffer (contained SYB green) and subjected to electrophoresis on a 2% agarose gel for quality assessment and purified with GeneJETTM Gel Extraction Kit (Thermo Scientific).
Library preparation and sequencing
The sequencing libraries were generated using TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, USA) following the manufacturer’s recommendations. The library quality was assessed on the Qubit@ 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. The qualified libraries were mixed in equal amounts and then sequenced on an Illumina NovaSeq platform with a 250 bp paired-end model. The DNA extraction, Amplicon library preparation, and sequencing services were provided by Novogene Co., Ltd. (Beijing, China).
Bioinformatics analysis
The forward and reverse amplicon reads acquired through the Illumina NovaSeq sequencer were demultiplexed utilizing the QIIME2 plugins (version 2018.11). Primer trimming was performed with Cutadapt tool30. The subsequent quality filtering, denoising, paired-end merging, and dereplication were conducted via the DADA2 workflow31. Chimeric sequences were identified and removed using the removeBimeradenovo function (Fig. 6).

The Schematic overview of sampling and details of the workflow and tools used for data processing and analysis performed in this study.
Taxonomic assignment for archaeal and bacterial ASVs was conducted using the Naïve Bayes approach (with a minimum of 75 bootstrap cutoff value) following the DADA2 workflow31, in reference to the SILVA training set (version 138.1)32. Fungal ASVs were classified against the UNITE Fungi database (version 10.05.2021_dev)33. The taxonomically annotated ASVs were subsequently agglomerated at the species level using the tax_glom function in the “phyloseq” package31,34. ASVs not assignable at the species level were clustered into operational taxonomic units (OTUs) based on 97% similarity using the OTU function in the “kmer” package35. Representative sequences for these OTUs were classified with the Naïve Bayes approach. In each microbial dataset, singletons, doubletons, and samples with fewer than 1,000 sequences were excluded. Taxonomic assignment reads among the samples were Hellinger transformed using the decostand function in the R “vegan” package36, and these transformed values were subsequently considered as abundance measures for statistical analyses. Figure 6 depicts the overall course of the production of all datasets.
Environmental factors analysis
A standard soil test series (NY/T 1121) was conducted. Organic matter (OM) was determined using the K2Cr2O7 oxidation method. The total nitrogen (TN) was measured using the Kjeldahl method. Available nitrogen (AN) was determined using the sulfate extraction method. Available phosphorus (AP) was detected using the hydrochloric acid–ammonium fluoride extraction–molybdenum antimony colorimetric method. Available potassium (AK) was detected using the ammonium acetate extraction–flame photometric method. Soluble salt (Salt) was detected using the mass method. The pH was determined using a potentiometric method37. Kendall’s τ statistic was employed to estimate a rank-based measure of associations between environmental factors, utilizing pairs function within the “graphics” package and panel-related functions in the “MESS” package38. Multiple Regression on Distance Matrices (MRM) analysis was conducted to test the principal environmental factors shaping microbial communities, using the MRM function in the “ecodist” package39. MRM analysis employed the 1-distanceBray-Curtis similarity measure to represent the microbial community composition.
Data Records
The processed data along with metadata have been deposited in the Sequence Read Archive (SRA) database of the National Center for Biotechnology Information (NCBI) under the BioProject IDs PRJNA1032247 (https://identifiers.org/ncbi/insdc.sra:SRP468803)40. The ASV tables, fasta sequences, and taxonomy data for archaea, bacteria, and fungi were respectively uploaded to Figshare repository, resulting in distinct links for each dataset (https://doi.org/10.6084/m9.figshare.2608753341; https://doi.org/10.6084/m9.figshare.2608755442; https://doi.org/10.6084/m9.figshare.2608756643).
Technical Validation
Sampling procedure
Strict aseptic procedures were implemented during soil sample collection to prevent contamination from the human body or between samples. The shoves and sieves were sprayed and wiped with 75% ethanol before and after every sampling, and latex gloves worn by collectors were changed for every sampling. Plastic bags and containers were newly opened or sterilized with ethanol before taking soil samples.
Qualification strategy
Eighty soil samples for each microbial dataset were successfully sequenced, yielding 98,765.09, 89,198.56, and 95,629.65 raw reads per sample in average in archaeal, bacterial, and fungal communities, respectively. The observed and estimated error rates were evaluated after error learning, to determine the optimal quality control strategy. In our research, sequencing quality thresholds for archaeal and bacterial raw reads were expected errors lower than 2 for both pair ends, trimming forward reads to 180 bp and reverse reads to 160 bp, truncating at the first instance of a quality score less than or equal to 2, and removing the reads of final lengths shorter than 100 bp; quality thresholds for fungal raw reads were expected errors at 2 and lengths trimmed to 210 bp for both pair ends, with the same quality truncating and final lengths criteria. The quality filtration, error denoising, pair-ends merging, and chimera removal yielded 56,988.48, 65,828.96, and 71,913.94 clean reads per sample on average in archaeal, bacterial, and fungal communities, respectively, which were subjected to subsequent analysis.
Taxonomy annotation
In order to take into account both ASV-based high taxonomy resolution and diversity evaluation at species level, this study employed a two-step approach for taxonomic annotation. ASVs assigned with species names were agglomerated and thus were not analogous to remaining ASVs at the taxonomic hierarchy. Consequently, the remaining ASVs that were unable to be annotated at species level, were clustered into OTUs based on 97% similarity, and annotated at genus level or above. The microbial diversity and community composition were assessed at species level with species agglomerated from ASVs and OTUs clustered from ASVs. The threshold of bootstrap value was set to 75 rather than the default 50.
Code availability
R codes applied in the present study are available at: https://github.com/xnus/PamirSoilMicrobes.
References
Johnson, D., Campbell, C. D., Lee, J. A., Callaghan, T. V. & Gwynn-Jones, D. Arctic microorganisms respond more to elevated UV-B radiation than CO2. Nature. 416, 82–83 (2002).
Kong, W. D. A review of microbial diversity in polar terrestrial environments. Biodiversity Science. 21, 456–467 (2013).
Yang, S. P., Jiang, J. & Yan, P. Eco-geographic distribution of rare and endangered plants and endemism in the Pamirs Region of China. Journal of Arid Land Resources and Environment. 32, 115–120 (2018).
Mętrak, M., Sulwiński, M., Chachulski, Ł., Wilk, M.B., Suskamalawska, M. Creeping environmental problems in the pamir mountains: Landscape conditions, climate change, wise use and threats. In Climate Change Impacts on High-Altitude Ecosystems. Springer Science and Business Media LLC: Berlin, Germany. 665–694 (2015).
Yao, T. et al. Different glacier status with atmospheric circulations in Tibetan Plateau and surroundings. Nat. Clim. Change. 2(9), 663–667 (2012).
Mętrak, M. et al. Nature’s Patchwork: How water sources and soil salinity determine the distribution and structure of halophytic plant communities in arid environments of the Eastern Pamir. PLoS ONE 12, e0174496 (2017).
Xiang, S. R., Yao, T. D., An, L. Z., Xu, B. L. & Wang, J. X. 16S rRNA Sequences and Differences in Bacteria Isolated from the Muztag Ata Glacier at Increasing Depths. Applied and Environmental Microbiology. 71, 4619–4627 (2005).
Singh, V.P., Singh, P., Haritashya, U.K. (Eds.). Pamirs. Encyclopedia of Snow, Ice and Glaciers. 13–815 Springer, Netherlands (2011).
Mölg, N., Bolch, T., Rastner, P., Strozzi, T. & Paul, F. A consistent glacier inventory for Karakoram and Pamir derived from Landsat data: distribution of debris cover and mapping challenges. Earth Syst. Sci. Data 10, 1807–1827 (2018).
Barandun, M. et al. Hot Spots of glacier mass balance variability in central Asia. Geophysical Research Letters 48, e2020GL092084 (2021).
Monika, M. et al. Morphology and distribution of biological soil crusts and their potential role in soil-forming processes under dry high-altitude periglacial conditions (Eastern Pamir, Tajikistan). Geoderma Regional. 33, e00636 (2023).
Zhang, Q. & Duan, K. Q. Characteristics of precipitation in the Pamirs in 2017 based on WRF simulation. Arid Land Geography 44, 1707–1716 (2021).
Veresoglou, S. D., Halley, J. M. & Rillig, M. C. Extinction risk of soil biota. Nature Communications. 6, 8862–8871 (2015).
Ma, B., Wang, H., Dsouza, M. & Xu, J. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME Journal. 10, 1891–1901 (2016).
Li, M. Y., Wang, J. L. & Gulbahar, S. Culturable bacterial diversity in snow, ice and melt water of theYangbark Glacier, Muztag Ata. Jourmal of Glaciology and Geocryology. 37, 1634–1641 (2015).
Fan, S. Y. et al. Variations in Soil Enzyme Activities and Microbial Communities along an Altitudinal Gradient on the Eastern Qinghai–Tibetan Plateau. Forests 12, 681–681 (2021).
Li, Y. M. et al. Function and distribution of nitrogen-cycling microbial communities in the Napahai plateau wetland. Archives of Microbiology 205, 357–357 (2023).
Xu, M. J. et al. Sensitivity of soil nitrifying and denitrifying microorganisms to nitrogen deposition on the Qinghai-Tibetan plateau. Annals of Microbiology volume 71, 6 (2021).
Xu, Y. T., Sun, R., Yan, W. M. & Zhong, Y. Q. W. Divergent response of soil microbes to environmental stress change under different plant communities in the Loess Plateau. Catena 230, 107240 (2023).
Agre, N. S. & Orleanskii, V. K. Thermophil actinomycetes in Pamir and their antagonistic properties. Mon Bull Minist Health Public Health Lab Serv. 31, 95–102 (1962).
KuznetsovM, V. D. Actinomycetes of various Pamir soils and their antagonistic properties. Mikrobiologiia 29, 563–570 (1969).
Aksenov, S. I., Babyeva, I. P. & Golubev, V. I. On the mechanism of adaptation of microorganisms to conditions of extreme low humidity. Life Sci Space Res. 11, 55–61 (1973).
Szymon, Z., Marcin, N., Janusz, B., Piotr, M. & Arkadiusz, N. Fungal root endophyte associations of plants endemic to the Pamir Alay Mountains of Central Asia. Symbiosis 54, 139–149 (2011).
Khomutovska, N. et al. Life in extreme habitats: diversity of endolithic microorganisms from cold desert ecosystems of eastern Pamir. Polish Journal of Ecology 65, 303–319 (2017).
Khomutovska, N., de los Ríos, A. & Jasser, I. Diversity and Colonization Strategies of Endolithic Cyanobacteria in the Cold Mountain Desert of Pamir. Microorganisms 9, 6 (2021).
Bu, X. Y., Ren, M., Wan, C. X. & Zhang, L. L. Diversity of aerobic cold-adapted bacteria and archaea isolated from the Pamir Plateau. Acta Microbiologica Sinica. 62, 2568–2581 (2022).
Wei, S. et al. Comparative evaluation of three archaeal primer pairs for exploring archaeal communities in deep-sea sediments and permafrost soils. Extremophiles 23, 747–757 (2019).
Walters, W. et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 22, e00009–15 (2015).
Usyk, M., Zolnik, C. P., Patel, H., Levi, M. H. & Burk, R. D. Novel ITS1 Fungal Primers for Characterization of the Mycobiome. mSphere 13, e00488–17 (2017).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 17, 10–2 (2011).
Callahan, B. J., Sankaran, K., Fukuyama, J. A., McMurdie, P. J. & Holmes, S. P. Bioconductor workflow for microbiome data analysis: From raw reads to community analyses. F1000Research. 5, 1492 (2016).
Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Research. 42, D643–D8 (2014).
Nilsson, R. H. et al. K. The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D64 (2018).
McMurdie, P. J. & Holmes, S. phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE. 8, e61217 (2013).
Wilkinson, S. kmer: an R package for fast alignment-free clustering of biological sequences. https://cran.r-project.org/package=kmer Accessed (2018).
Oksanen, J. et al vegan: Community Ecology Package. R package version 2.5-6. (2019).
Zhu, J., Wang, H. N., Tang, Q. Y., Gu, M. Y. & Zhang, Z. D. Composition and distribution of bacterial communities and potential radiation-resistant bacteria at different elevations in the eastern Pamirs. Front. Microbiol. 15, 1427806 (2024).
Ekstrøm, C. MESS: Miscellaneous Esoteric Statistical Scripts. CRAN.R-project.org/package=MESS. Accessed (2022).
Goslee, S. C. & Urban, D. L. The ecodist package for dissimilarity-based analysis of ecological data. Journal of Statistical Software 22, 1–19 (2007).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP468803 (2023).
Zhu, J. Amplicon data of 80 soil archaea from the Pamir Plateau. figshare https://doi.org/10.6084/m9.figshare.26087533 (2024).
Zhu, J. Amplicon data of 80 soil bacteria from the Pamir Plateau. figshare https://doi.org/10.6084/m9.figshare.26087554 (2024).
Zhu, J. Amplicon data of 80 soil fungi from the Pamir Plateau. figshare https://doi.org/10.6084/m9.figshare.26087566 (2024).
Acknowledgements
This work was supported by the Project of Fund for Stable Support to Agricultural Sci-Tech Renovation (xjnkywdzc-2023005).
Author information
Authors and Affiliations
Contributions
J.Z., X.S., Z.D.Z. and J.W.C. designed the study. J.Z., Q.Y.T., M.Y.G. and Z.D.Z. collected the samples. X.S. and J.Z. performed the analysis. J.Z. and X.S. wrote the paper and prepared the figure and tables. All co-authors commented on the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, J., Sun, X., Tang, QY. et al. The first large scale rDNA amplicon database of soil microbiomes from Pamir Plateau, China. Sci Data 11, 856 (2024). https://doi.org/10.1038/s41597-024-03711-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03711-4
- Springer Nature Limited