
Abstract
MADS-box genes encode transcription factors that are involved in plant development control (particularly in floral organogenesis) and signal transduction pathways, though a comprehensive analysis of MADS-box family proteins in Chinese jujube (Ziziphus jujuba Mill.) is still missing. Here, we report a genome-wide analysis of the MADS-box gene family in Chinese jujube. Based on phylogenetic analyses, 52 jujube MADS-box genes were classified into 25 MIKCC-type, 3 MIKC*-type, 16 Mα, 5 Mβ and 3 Mγ genes. 37 genes were randomly distributed across all 12 putative chromosomes. We found that the type II genes are more complex than the type I genes and that tandem duplications have occurred in three groups of MADS-box genes. Meanwhile, some gene pairs in the same clade displayed similar or distinct expression profiles, suggesting possible functional redundancy or divergence. MIKCC-type genes exhibited typical temporal and spatial expression patterns in the four whorls of floral tissues. The expressions of B, C/D and E-type genes were significantly suppressed in phyllody as compared to flower, providing valuable evidence for their involvement in flower development. This study is the first comprehensive analysis of the MADS-box family in jujube, and provides valuable information for elucidating molecular regulation mechanism of jujube flower development.
Similar content being viewed by others

Introduction
MADS-box genes are transcription factors that play a significant role in plant development, especially in determining floral organ identities. Floral organ identity genes have traditionally been subdivided into five different classes (class A, B, C, D, and E genes) that provide five different “homeotic functions”, with A-class genes specifying sepals, A + B + E specifying petals, B + C + E specifying stamens, C + E specifying carpels, and D specifying ovules1,2,3.
Phylogenetically, MADS-box genes have been divided into two types, type I and type II. The type I genes have, in turn, been divided into three phylogenetic groups (Mα, Mβ and Mγ), whereas the type II genes have been further divided into MIKCC and MIKC*-type genes based on both the different lengths of their encoded K-domains and on phylogenetic standards4,5,6. In angiosperms, MIKCC-type genes were further subdivided into 12 clades7.
Structurally, MADS-box genes possess a conserved DNA-binding domain, which is defined by a highly conserved 60-amino-acid sequence that is involved in binding to DNA based on a consensus CC(A/T)6GG (also known as the CArG box) sequence8,9,10. In addition to the highly conserved MADS domain, members of the type II lineage contain three additional domains (from N- to C-terminus): the Intervening (I) domain, the Keratin-like (K) domain and the C-terminal (C) region11,12,13,14. The K domain is the second most-conserved region and is responsible for dimerization via a coiled-coil structure, while the less conserved I domain may contribute to DNA-binding specificity and dimerization. The C-terminal region is highly variable, and it has been shown to function in protein complex formation and transcriptional activation15,16,17.
Chinese jujube (Ziziphus jujuba Mill.) is one of the most economically important fruit trees in China. Compared to other fruit trees, this tree has some unique reproductive features such as a fast flowering time (of approximately 7 days), a short juvenile phase and first-year fruiting ability. Flower bud differentiation and fruiting occur in the same year, and there has been no flower bud dormancy reported for the Chinese jujube. Hence, a genome-wide analysis of the jujube MADS-box gene family may be useful for revealing the unique flowering mechanism of this species at the molecular level.
In this study, we identified 52 MADS-box genes in the jujube genome, analyzed their phylogenetic relationships and gene structures, and predicted their chromosomal localization. Additionally, to study the role of these genes in jujube flower development, we used semi-quantitative RT-PCR and real-time quantitative RT-PCR (qRT-PCR) to determine expression profiles.
Results
Identification and classification of MADS-box genes in the Chinese jujube
After the screening process, a total of 52 non-redundant MADS-box proteins were identified and serially named as ZjMADS1 through ZjMADS52 (Table 1). The CDS length of the jujube MADS-box genes ranged from 387 bp to 1053 bp; the encoded proteins ranged from 128 to 350 amino acids (aa) in length (with an average of 235.92 aa), had a predicted molecular mass of 14.60–40.24 KDa, and protein pIs ranged from 4.34 to 9.91.
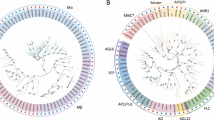
Based on the phylogenetic analysis, 24 type I and 28 type II jujube genes were further divided into more detailed subgroups. Twenty-four type I genes were divided into Mα, Mβ, Mγ subgroups (Fig. 1) and, similar to P. mume, Mα was the group with the most genes. Sixteen out of 24 type I genes were classified into the Mα subgroup, while 5 were classified into the Mβ subgroup and 3 into the Mγ subgroup. Twenty-eight type II genes were further classified into 25 MIKCC-type and 3 MIKC*-type genes (Fig. 2). The 25 MIKCC-type genes were further divided into 13 clades: FLOWERING LOCUS C (FLC), SHORT VEGETATIVE PHASE (SVP), AGL6, TOMATO MADS-box 8 (TM8), AGL17, AGL15, AGL12, AG, AP3/PI, SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), B-sister (BS/TT16), SEPALLATA (SEP), and APETALA1/FRUITFULL (AP1/FUL), each clade containing 1 to 4 MADS-box genes. In contrast to the P. mume genome, where an extreme expansion of the SVP clade was observed, phylogenetic analysis of the jujube type II genes showed that these genes were evenly distributed among different clades (Fig. 2).

Phylogenetic tree of Z. jujuba and P. mume type I MADS-box genes. The phylogenetic tree was constructed based on the alignable region of protein sequence alignment of Chinese jujube MADS-box genes using the neighbor-joining method and no of differences model with bootstrapping analysis (1000 replicates). The subgroups are marked by different colors.

Phylogenetic tree of type II MADS-box genes in Z. jujuba, P. mume and Arabidopsis. The tree was generated using the neighbor-joining method implemented in MEGA 5.1. The FLC subfamily (absent in P. mume) and the TM8 subfamily (absent in Arabidopsis) are shown in frame sets.
Gene structure and conserved motif distribution analysis
To better understand the structural diversity of MADS-box genes, we compared various intron/exon arrangements and conserved motifs based on their phylogenetic relationships. We obtained each gene structure by comparing their ORFs with their genomic sequences. As shown in Fig. 3, closely related genes were generally more structurally similar, differing only in intron and exon lengths. However, some close gene pairs exhibited distinct intron/exon arrangements. For example, ZjMADS31 consisted of 7 exons, whereas its close paralog ZjMADS24 had only 4 exons. In addition, we found that jujube MADS-box genes contained between 1 to 11 exons. The average number of exons in type II genes (7) was considerably greater than the average number of exons of the type I genes (1.12), which suggests that the type II MADS-box genes may be more complex.

Phylogenetic relationships and structural analysis of MADS-box genes in the Chinese jujube. The unrooted neighbor-joining tree was constructed from the alignment of full-length amino acid sequences. Lengths of exons and introns of each MADS-box gene are displayed proportionally. Green solid boxes represent exons; black lines represent introns.
We then used the MEME program to analyze conserved motifs in the MADS-box proteins, which had previously been subjected to SMART annotation. A total of 20 conserved motifs were identified (Supplementary Fig. S1). The commonly shared motifs tended to be in the same group: for instance, the SVP group included the ZjMADS36, ZjMADS37 and ZjMADS38 genes, all of which contain motifs 1, 2, 6, 7 and 9. Motif 1 was comprised of approximately 60 amino acids and was the most typical MADS-box domain. Motif 2 represented the K domain, which has been described to be the most conserved domain and essential for protein-protein interactions among MADS-box transcriptional factors. The K domain was identified in the majority of type II proteins, with one exception (ZjMADS40). This result is consistent with previous studies that showed that the K-box domain was only found in type II MADS-box genes18. However, in our study, we found that one type I gene, ZjMADS51 also contained a K-box domain.
Genomic distribution and duplication of jujube MADS-box genes
We found that 37 of the 52 ZjMADS-box genes were randomly distributed across all 12 putative chromosomes, while 12 genes were assigned to unanchored scaffolds (Table 1 and Fig. 4). Three genes obtained by cloning were not anchored on chromosomes or scaffolds. Fourteen type I genes were mapped to 7 chromosomes, though most of the genes were located on chromosome 1. Twenty-three of the 28 type II genes were mapped to 9 chromosomes, and most of these were distributed on chromosomes 6, 8, 10 and 12. We also found clusters of some genes on chromosome 1, scaffold 1156 and chromosome 10 (ZjMADS14, ZjMADS7 and ZjMADS8; ZjMADS9, ZjMADS10 and ZjMADS11; ZjMADS30 and ZjMADS31, respectively), and then we confirmed that tandem duplications have occurred in these genes. Further analyses should be performed to study the role of these duplications in the expansion of this family.

Positions of ZjMADS genes on the jujube pseudo-chromosomes or scaffolds.
Expression pattern of the jujube MADS-box genes in different tissues/organs
We investigated the expression patterns of 28 MADS-box genes in seven different tissues/organs (Fig. 5). Twenty-one MADS-box genes were expressed in at least one organ (especially in the flower bud), whereas the other 7 type I genes (ZjMADS1, ZjMADS7, ZjMADS10, ZjMADS11, ZjMADS14, ZjMADS20 and ZjMADS21) were not detected in any of the organs tested. While most type I genes were only expressed in the flower bud, most type II genes were highly expressed in all reproductive tissues. Almost all of the MIKCC genes exhibited the highest expressions in the flower bud and flowers, which further confirms their roles in jujube flower development.

Expression patterns of jujube MADS-box genes in vegetative and reproductive organs by RT-PCR. Sources of the samples are as follow: 1-Root (R), 2-Young branch (YB), 3-Old branch (OB), 4-Leaf (L), 5-Flower Bud (B), 6-Flower (F) and 7-Young fruit (YF).
Previous studies had shown that SOC1 and its homologs in Arabidopsis were pivotal genes for flower transition and that SOC1 overexpression could cause an early flowering phenotype19, 20. In our study, we found that ZjMADS29 (the SOC1 homolog) was highly expressed in the vegetative tissues. A high expression of SOC1 in leaves was also found in Arabidopsis and tree peonies21, 22. In addition, we found that ZjMADS38, the SVP homolog, was widely expressed in various vegetative and propagation tissues/organs, which suggests that this gene may play multiple roles in jujube development.
The critical MADS-box genes involved in jujube floral organ development
To further investigate the role of MADS-box genes in floral organ development, the expression patterns of 8 MIKCC type genes were determined in four whorls (the sepal, petal, stamen and pistil) using a qRT-PCR assay. The expression of these 8 genes exhibited typical temporal and spatial expression patterns consistent with floral development (Fig. 6), suggesting a possible role in flower differentiation. The expression of ZjMADS31 (an A-type gene) was notably high in the sepal; the expression of ZjMADS39 and ZjMADS40 (two B-type genes), was high in the petal but low in the pistil; transcripts of two C/D-type genes, ZjMADS32 and ZjMADS46, were highly detected in the pistil and stamen; the expression of ZjMADS30, ZjMADS47 and ZjMADS48 (three E-type genes of the SEP subfamily), was high in the sepal, petal and pistil, indicating their overlapping expression profiles.

Expression patterns of 8 MIKC genes in four whorls of floral organs (sepal, petal, stamen and pistil) by RT-qPCR. ZjACT primers were used as the internal standard for each gene. The mean expression value was calculated from 3 independent replicates. The vertical bars indicate the standard deviation.
Phyllody caused by phytoplasma is one of typical symptoms in jujube trees infected by Jujube Witches’ Broom (JWB). The petal, stamen and pistil of phyllody in diseased jujube trees are the abnormal development of floral parts into sepal/leaf structures. The expression of ZjMADS31 (A type gene) in flower and phyllody was higher than that in leaf (Supplementary Fig. S2), suggesting it played normal function in phyllody; however the increased expressions of B and C/D genes were observed in flower but not elevated in leaf and phyllody (Supplementary Fig. S2), indicating their function was nearly completely suppressed in phyllody and causing the abnormal development of petal, stamen and pistil; the expressions of three E-type genes in phyllody were higher than in leaf and lower than in flower (Supplementary Fig. S2), showing their function was partially inhibited in phyllody as compared to flower. The expressions of those genes were in agreement with above expression patterns in four whorls (Fig. 6). ZjMADS47 is the homolog of Arabidopsis SEP3 (NP_850953.1) in Chinese jujube and they shares 78% identity at the amino acid sequence level. Over-expression of ZjMADS47 in Arabidopsis showed early flowering phenotype when compared with wild-type plants (Supplementary Fig. S3). RT-PCR analysis demonstrated that AtSEP3 expression was up-regulated in transgenic lines. The results further highlighted that these genes were involved in jujube flower development.
Discussions
MADS-box genes have been identified in several species, including Arabidopsis 5, poplar (Populus trichocarpa)23, grape (Vitis vinifera L.)24, apple (Malus domestica)25 and P. mume 26. In this study, we identified 52 non-redundant ZjMADS-box genes; this number may increase in the future when assembling or annotation problems in the jujube genome are addressed. Except for the tandem duplication, the segmental duplication should also contribute to the expansion of jujube MADS box genes. The difference in the number of MADS-box genes among the above-mentioned species (Supplementary Table S1) might be caused by a recent whole-genome duplication event. During the process of plant genome evolution, one or two whole-genome duplication events have previously been described in different species. According to previous studies, there has been one α and one β duplication event but no recent whole-genome duplication in Chinese jujube and P. mume, whereas two duplication events have occurred in apple. Thus, the number of apple MADS-box genes is greater than that of jujube and P. mume.
The average number of exons in the jujuba type II MADS-box genes was greater than that of the type I genes. Among the type II genes, 27 out of 28 (96.4%) contained more than 4 exons, while most type I genes had only one or two exons. The same phenomenon has also been observed in other species, such as Arabidopsis 5, apple25 and P. mume 26. The exon length distribution analysis also produced a similar pattern (Supplementary Table S2). These observations further highlight the conserved evolution among plants.
In contrast to type II MADS-box genes, information about type I genes is limited. Recent studies in Arabidopsis suggest that type I genes are important for plant reproduction and are required for proper development, especially for determining female gametophyte, and for proper embryo27 and endosperm development28,29,30. As in Arabidopsis, most type I genes in jujube were mainly expressed in flower buds or flowers, though the expression of some genes was too weak to detect. Compared to type I genes, most jujube type II genes were expressed in at least three organs, which suggests that these genes may have multiple functions in different organs. We found that gene pairs in the same clade can display similar or distinct expression profiles, which suggests possible functional redundancy or divergence, respectively. For instance, two AP3/PI genes (ZjMADS39 and ZjMADS40) were highly expressed in the petal, implying that both genes may be able to perform B-related functions. In contrast, ZjMADS40 was uniquely expressed in young fruits, whereas ZjMADS39 was not, which indicates that these genes may have functionally diverged.
We believe that the expression profiles of MIKCC type genes that we obtained in our study will be an important tool for elucidating flower development mechanisms in the Chinese jujube. Based on the bioinformatics analyses, the expression of the ABCDE genes in four whorls of jujube flower was very consistent with the predicted expression patterns, which suggests that they may indeed be involved in flower differentiation. As one of perennial woody plants, the genetic transformation system of Chinese jujube was still not established, therefore it is very difficult to perform functional study in jujube. Phyllody as the suitable tissue for studying jujube flower development was applied in this study (Supplementary Fig. S2). The expression pattern of A type gene in phyllody was similar to flower, suggesting A type gene should play the normal function in phyllody. However the low level expression of B, C/D and E type genes in phyllody indicated that their function were remarkable inhibited, causing the abnormal development of petal, stamen and pistil. We demonstrated that over-expressing of ZjMADS47 (homolog with SEP3 in Arabidopsis) caused early flowering in Arabidopsis plants, which is in accordence with the SEP3 function in Arabidopsis and lily31, 32. This result was supported the functional conservation of MADS-box genes between jujube and other plants.
In addition, ZjMADS46 is a SHATTERPROOF-like gene that showed high similarity to Prunus persica (85%), Prunus triloba (85%) and Fragaria x ananassa (81%) genes. SHATTERPROOF-like genes have been shown to regulate fruit ripening in Arabidopsis33, peach34, strawberry35, and oil palm36. In our study, ZjMADS46 exhibited the highest expression in the pistil (Fig. 6); more studies are needed to further investigate the role of ZjMADS46 in jujube fruit development.
In summary, our results have laid the foundation for a thorough functional characterization of the MADS-box gene family in jujube and have hopefully enabled a better understanding of the structure-function relationship between MADS-box gene family members. Additionally, our study provides comprehensive information and novel insights into the evolution and divergence of the MADS-box genes in plants. Finally, studies similar to this may potentially aid in the understanding of the molecular basis of many agriculturally important jujube traits such as flower and fruit development and other physiological processes.
Materials and Methods
Plant material
Roots (R), young branches (YB), old branches (OB), leaves (L), flower buds (B), flowers (F) and young fruits (YF) were collected from three jujube trees. Four different floral organs (the sepal, petal, stamen and pistil) were harvested at florescence time (May 29, 2016). The Chinese jujube flowers are very small (approximately 5 mm). Four whorls were quickly separated and frozen in liquid nitrogen in the field. Phyllody is one of typical symptoms in jujube trees infected by JWB disease. Three tissues, i.e. leaf, phyllody and flower, were applied to study the expression of genes related to floral development. Three biological replicates were collected in each treatment. The samples were stored at −80 °C for RNA extraction and expression analysis.
Identification of MADS-box genes
To identify MADS-box genes in the jujube genome, previously identified Arabidopsis MADS-box sequences were submitted to the Pfam database (http://pfam.sanger.ac.uk)37 to obtain the domain architecture of this family. The hidden Markov model (HMM) profiles of the SFR (type I) domain (PF00319) and the Myocyte Enhancer Factor-2 MEF2 (type II) domain (PF09047) were retrieved from Pfam38. MADS-box genes were then identified in the jujube genome database39 using the hidden Markov model (HMM) profile corresponding to the Pfam MADS-box family PF00319 and PF09047 domains, using the HMMER version 3.0 software40. Finally, we further verified these sequences using the SMART tool (http://smart.embl-heidelberg.de/)41 combined with the Pfam (http://Pfam.sanger.ac.uk/) and the NCBI databases (http://www.ncbi.nlm.nih.gov/). The sequences lacking MADS domains were rejected in this analysis. A total of 49 MADS-box proteins were obtained and used for further analysis. 3 MADS-box proteins obtained from homologous cloning were also included in this study. The online tool ProtParam (http://web.expasy.org/protparam/) was employed to predict the molecular weight and isoelectric point (pI) of each protein.
Phylogenetic analysis
The Arabidopsis thaliana MADS proteins were retrieved from the TAIR database (http://www.arabidopsis.org/) based on a previous report5. The Prunus mume genome sequences were downloaded from the P. mume genome project website (http://prunusmumegenome.bjfu.edu.cn/), and the dataset of the predicted P. mume MADS proteins was retrieved from previous analyses26. Following the convention established for P. mume and Arabidopsis MADS proteins, the jujube MADS proteins were classified into different groups. Multiple sequence alignments were performed using Clustal X2.0 with default parameters42. A phylogenetic tree was then constructed using the neighbor-joining method, and bootstrap values were calculated with 1,000 replications using MEGA5.143.
Conserved motif and gene structure analysis
To identify the conserved motifs in the Chinese jujube full-length MADS proteins, the Multiple Expectation-maximization for Motif Elicitation (MEME) program version 4.9.044 was used with mostly default parameters, except for the following: (1) the optimum motif width was set to ≥6 and ≤60 and (2) the maximum number of motifs was set to 20. The MEME motifs were then annotated using the SMART program (http://smart.embl-heidelberg.de) and the Pfam database.
The coding domain sequences (CDS) and DNA sequences of the MADS-box genes were used to predict gene structure using the online tool GSDS45 (http://gsds.cbi.pku.edu.cn), which allowed us to infer both exon position and gene length. Then, exon lengths of the jujube MIKCC genes were estimated and compared with those of Arabidopsis and apple genes.
Chromosomal location and gene duplication
To determine the chromosomal location of the MADS-box genes, the MADS-box gene sequences were further used as query sequences in BLASTN searches against the jujube genome sequence. Each MADS-box gene was thus mapped to the jujube genome according to their coordinates on the genome. Tandem duplications were identified according to previously described methods46.
RNA isolation and expression analysis
Total RNA was isolated from 100 mg of frozen tissue using an RNA kit (RNAprep Pure Plant Kit, Tiangen, Beijing, China) according to the manufacturer’s instructions. First-strand cDNA was synthesized from 2 μg of RNA using the TIANScript First Strand cDNA Synthesis Kit (Tiangen, Beijing, China) according to manufacturer’s instructions. The resulting cDNA was then diluted nine-fold and stored at −20 °C for the subsequent semi-quantitative RT-PCR and qRT-PCR assays.
For gene expression quantification, specific primers were designed for each MADS-box gene using the Primer Premier 5.0 software and expression patterns were assayed by semi-quantitative RT-PCR. PCR reactions were performed using the following program: initial denaturation at 95 °C for 3 min; 30 cycles at 95 °C for 15 s, 55 °C for 15 s, and 72 °C for 30 s, and a final extension cycle at 72 °C for 10 min. ZjACT was used as reference gene45. RT-PCR products were then sequenced to ensure that they were derived from the desired target genes. Primer details primers are listed in the Supplementary Table S3.
The expression of 8 MADS-box genes in different floral organs was examined using qRT-PCR. Total RNA was extracted from the sepal, petal, stamen and pistil. qRT-PCR was performed using the Bio-Rad iQ5 detection system. Reactions were performed in a 20 μL volume containing 1 μL of cDNA, 400 nM of each primer and 10 μL of SYBR Green mix, according to the TransStart Top Green qPCR SuperMix instructions. The reactions were performed under the following conditions: 94 °C for 30 s, and 40 cycles of 94 °C for 5 s, 55 °C for 15 s and 72 °C for 15 s. The specificity of the amplicon for each primer pair was verified by melting curve analysis. All the experiments were performed in three biological replicates, and each replicate was measured in triplicate. The relative expression levels were calculated using the 2−ΔCt method and with ZjACT as the reference gene47.
References
Angenent, G. C. & Colombo, L. Molecular control of ovule development. Trends Plant Sci. 1, 228–232, doi:10.1016/S1360-1385(96)86900-0 (1996).
Theiben, G. & Saedler, H. Plant biology: floral quartets. Nature 409, 469–471, doi:10.1038/35054172 (2001).
Weigel, D. & Meyerowitz, E. M. The ABCs of floral homeotic genes. Cell 78, 203–209, doi:10.1016/0092-8674(94)90291-7 (1994).
Henschel, K. et al. Two ancient classes of MIKC-type MADS-box genesare present in the moss Physcomitrella patens. Mol Biol Evol. 19, 801–814, doi:10.1093/oxfordjournals.molbev.a004137 (2002).
Parenicová, L. et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis new openings to the MADS world. Plant Cell 15, 1538–1551, doi:10.1105/tpc.011544 (2003).
Kwantes, M., Liebsch, D. & Verelst, W. How MIKC* MADS-box genes originated and evidence for their conserved function throughout the evolution of vascular plant gametophytes. Mol. Biol. Evol. 29, 293–302, doi:10.1093/molbev/msr200 (2012).
Becker, A. & Theibn, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol. 29, 464–489, doi:10.1016/S1055-7903(03)00207-0 (2003).
Pellegrini, L., Tan, S. & Richmond, T. J. Structure of serum response factor core bound to DNA. Nature 376, 490–498, doi:10.1038/376490a0 (1995).
Sasaki, K. et al. Functional divergence within class B MADS-box genes TfGLO and TfDEF in Torenia fournieri Lind. Mol Genet Genomics 284, 399–414, doi:10.1007/s00438-010-0574-z (2010).
Shore, P. & Sharrocks, A. D. The MADS-box family of transcription factors. Eur J Biochem. 229, 1–13, doi:10.1111/ejb.1995.229.issue-1 (1995).
Cho, S. et al. Analysis of the C-terminal region of Arabidopsis thaliana APETALA1 as a transcription activation domain. Plant Mol Biol. 40, 419–429, doi:10.1023/A:1006273127067 (1999).
De Bodt, S., Raes, J., Van de Peer, Y. & Theibn, G. And then there were many: MADS goes genomic. Trends Plant Sci. 8, 475–483, doi:10.1016/j.tplants.2003.09.006 (2003).
Norman, C., Runswick, M., Pollock, R. & Treisman, R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 55(6), 989–1003, doi:10.1016/0092-8674(88)90244-9 (1988).
Smaczniak, C., Immink, R. G., Angenent, G. C. & Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139, 3081–3098, doi:10.1242/dev.074674 (2012).
Honma, T. & Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529, doi:10.1038/35054083 (2001).
Kaufmann, K., Melzer, R. & Theißen, G. MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 347, 183–198, doi:10.1016/j.gene.2004.12.014 (2005).
Riechmann, J. L., Krizek, B. A. & Meyerowitz, E. M. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc Natl Acad Sci USA 93, 4793–4798, doi:10.1073/pnas.93.10.4793 (1996).
Lyons, E. et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148, 1772–1781, doi:10.1104/pp.108.124867 (2008).
Borner, R. et al. A MADS domain gene involved in the transition to flowering in Arabidopsis. Plant J. 24, 591–599, doi:10.1046/j.1365-313x.2000.00906.x (2000).
Dorca-Fornell, C. et al. The Arabidopsis SOC1-like genes AGL42, AGL71 and AGL72 promote flowering in the shoot apical and axillary meristems. Plant J. 67, 1006–1017, doi:10.1111/tpj.2011.67.issue-6 (2011).
Samach, A. et al. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 288, 1613–1616, doi:10.1126/science.288.5471.1613 (2000).
Zhang, Y. et al. Isolation and characterization of a SOC1-Like gene from tree peony (Paeonia suffruticosa). Plant Mol Biol Rep. 33(4), 855–866, doi:10.1007/s11105-014-0800-7 (2015).
Leseberg, C. H., Li, A. L., Kang, H., Duvall, M. & Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 378, 84–94, doi:10.1016/j.gene.2006.05.022 (2006).
Diaz-Riquelme, J., Lijavetzky, D., Martinez-Zapater, J. M. & Carmona, M. J. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant Physiology 149, 354–369, doi:10.1104/pp.108.131052 (2009).
Velasco, R. et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat Genet. 42(10), 833–839, doi:10.1038/ng.654 (2010).
Xu, Z. D. et al. Genome-wide identification, characterisation and expression analysis of the MADS- box gene family in Prunus mume. Mol Genet Genomics. 289, 903–920, doi:10.1007/s00438-014-0863-z (2014).
Bouyer, D. et al. Polycomb repressive complex 2 controls the embryo-to-seedling phase transition. PLoS Genet. 7, e1002014, doi:10.1371/journal.pgen.1002014 (2011).
Kang, I. H., Steffen, J. G., Portereiko, M. F., Lloyd, A. & Drews, G. N. The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 20, 635–647, doi:10.1105/tpc.107.055137 (2008).
Bemer, M., Wolters-Arts, M., Grossniklaus, U. & Angenent, G. C. The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 20, 2088–2101, doi:10.1105/tpc.108.058958 (2008).
Portereiko, M. F. et al. AGL80 is required for central cell and endosperm development in Arabidopsis. Plant Cell 18, 1862–1872, doi:10.1105/tpc.106.040824 (2006).
Pelaz, S. et al. APETALA1 and SEPALLATA3 interact to promote flower development. Plant J. 26, 385–394, doi:10.1046/j.1365-313X.2001.2641042.x (2001).
Tzeng, T. Y. et al. Two lily SEPALLATA-like genes cause different effects on floral formation and floral transition in Arabidopsis. Plant Physiol. 133, 1091–1101, doi:10.1104/pp.103.026997 (2003).
Liljegren, S. J. et al. SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404, 766–770, doi:10.1038/35008089 (2000).
Tadiello, A. et al. A PLENA-like gene of peach is involved in carpel formation and subsequent transformation into a fleshy fruit. J Exp Bot. 60, 651–661, doi:10.1093/jxb/ern313 (2009).
Daminato, M., Guzzo, F. & Casadoro, G. A SHATTERPROOF-like gene controls ripening in non- climacteric strawberries, and auxin and abscisic acid antagonistically affect its expression. J Exp Bot. 64(12), 3775–3786, doi:10.1093/jxb/ert214 (2013).
Tranbarger, T. J. et al. Regulatory mechanisms underlying oil palm fruit mesocarp maturation, ripening and functional specialization in lipid and carotenoid metabolism. Plant Physiol. 156, 564–584, doi:10.1104/pp.111.175141 (2011).
Punta, M. et al. The Pfam protein families database. Nucleic Acids Res. 40, D290–D301, doi:10.1093/nar/gkr1065 (2012).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42 (Database issue), D222–230, 10.1093/nar/gkt1223 (2014).
Liu, M. J. et al. The complex jujube genome provides insights into fruit tree biology. Nat Commun. 5, 5315, doi:10.1038/ncomms6315 (2014).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39 (Web Server issue), W29–37, 10.1093/nar/gkr367 (2011).
Letunic, I., Doerks, T. & Bork, P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–305, doi:10.1093/nar/gkr931 (2012).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics. 23, 2947–2948, doi:10.1093/bioinformatics/btm404 (2007).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28, 2731–2739, doi:10.1093/molbev/msr121 (2011).
Bailey, T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–208, doi:10.1093/nar/gkp335 (2009).
Guo, A., Zhu, Q., Chen, X. & Luo, J. GSDS: a gene structure display server. Yi Chuan. 29, 1023–1026, doi:10.1360/yc-007-1023 (2007).
Kong, A. Y. Y., Fonte, S. J., van Kessel, C. & Six, J. Soil aggregates control N cycling efficiency in long-term conventional and alternative cropping systems. Nutr. Cycl. Agroecosys. 79, 45–58, doi:10.1007/s10705-007-9094-6 (2007).
Bu, J., Zhao, J. & Liu, M. Expression stabilities of candidate reference genes for RT-qPCR in Chinese jujube (Ziziphus jujuba Mill.) under a variety of conditions. PLoS One. 11(4), 1–11 (2016).
Acknowledgements
This study was supported by grants from Funds for Hebei Distinguished Young Scholar (C2016204145), National science and technology support plan of China (2013BAD14B03) and Agricultural University of Hebei Foundation for Leaders of Disciplines in Science Technology. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J.Z. and M.L. conceived and designed the study. J.Z., L.Z. and Y.H. contributed to data collection and bioinformatics analysis. L.Z., C.F., J.Z. and J.W. were responsible for sample collection and RT-qPCR analysis. J.Z. and L.Z. wrote the manuscript and prepared the figures. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, L., Zhao, J., Feng, C. et al. Genome-wide identification, characterization of the MADS-box gene family in Chinese jujube and their involvement in flower development. Sci Rep 7, 1025 (2017). https://doi.org/10.1038/s41598-017-01159-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-01159-8
- Springer Nature Limited
This article is cited by
-
Genome-wide identification and expression pattern analysis of MIKC-Type MADS-box genes in Chionanthus retusus, an androdioecy plant
BMC Genomics (2024)
-
Overexpression of Polypogon fugax Type I–Like MADS-Box Gene PfAGL28 Affects Flowering Time and Pod Formation in Transgenic Arabidopsis
Plant Molecular Biology Reporter (2022)
-
ZjSEP3 modulates flowering time by regulating the LHY promoter
BMC Plant Biology (2021)
-
Analysis of MADS-box genes revealed modified flowering gene network and diurnal expression in pineapple
BMC Genomics (2020)
-
Genome-wide analysis of the bZIP gene family in Chinese jujube (Ziziphus jujuba Mill.)
BMC Genomics (2020)