
Abstract
Maternal folate deficiency is linked to restricted fetal growth, however the underlying mechanisms remain to be established. Here we tested the hypothesis that mTOR functions as a folate sensor in vivo in mice and that maternal folate deficiency inhibits placental mTOR signaling and amino acid transporter activity and causes fetal growth restriction. Folate deficient mice had lower serum folate (−60%). In late pregnancy, fetal weight in the folate deficient group was decreased (−17%, p < 0.05), whereas placental weight, litter size and crown rump length were unaltered. Maternal folate deficiency inhibited placental mTORC1 and mTORC2 signaling and decreased trophoblast plasma membrane System A and L amino acid transporter activities and transporter isoform expression. Folate deficiency also caused a decrease in phosphorylation of specific functional readouts of mTORC1 and mTORC2 signaling in multiple maternal and fetal tissues. We have identified a novel specific molecular link between maternal folate availability and fetal growth, involving regulation of placental mTOR signaling by folate, resulting in changes in placental nutrient transport. mTOR folate sensing may have broad biological significance because of the critical role of folate in normal cell function and the wide range of disorders, including cancer, that have been linked to folate availability.
Similar content being viewed by others


Introduction
Folate is critical for normal fetal development and growth and maternal folate deficiency is associated with poor pregnancy outcomes1, 2. Periconceptional folate deficiency is associated with neural tube defects (NTDs)3 and animal experiments, epidemiological studies and interventional trials have demonstrated that folate supplementation decreases the incidence of these structural fetal malformations4,5,6. Low maternal folate levels are also linked to restricted fetal growth2, 7,8,9 and preterm birth10, and two cohort studies showed that supplementation with high-dose folic acid (3.7–5 mg/d) reduced the risk of low birth weight and preterm delivery11, 12.
It is now well-established that exposures during early life modulate the risk of developing non communicable diseases in childhood and in adult life, a concept known as the developmental origins of health and disease (DOHaD) or fetal programming13, 14. For example, there is substantial evidence for an association between low birth weight and risk to develop type-2 diabetes, coronary heart disease, and hypertension later in life, which has been attributed to poor nutrition in utero 15, 16. Reduced folate availability has been implicated in fetal programming17, which may in part be mediated by link between folate deficiency and fetal growth restriction7, 8 and in part due altered methylation of critical genes. Low birth weight is a predictor for metabolic disease in later life18. Recently, folic acid supplementation during pregnancy in a generally undernourished population was shown to reduce the prevalence of metabolic syndrome (MS) in children at 6 to 8 years of age19. Furthermore, folate supplementation in pregnant rats modified growth patterns and the metabolic response to fasting in adult offspring20, providing further evidence that maternal folate intake during pregnancy affects metabolic health in offspring.
Folate is a water-soluble B vitamin that is essential for the synthesis of purine and thymidine nucleotides, which are needed for DNA replication and repair21. In addition, folate is a critical methyl donor for DNA methylation, which is a key mechanism of epigenetic regulation. Although impaired DNA synthesis/repair and gene methylation are generally believed to explain the association between folate deficiency and poor pregnancy outcomes, the mechanistic links remain elusive.
The mechanistic target of rapamycin (mTOR) is master regulator of cell growth, aging, ribosome biogenesis, protein synthesis, actin-cytoskeletal organization, autophagy and metabolism22. mTOR forms two distinct heteromeric complexes, mTOR Complex 1 (mTORC1) and mTORC2. mTORC1 contains mTOR, raptor (regulatory associated protein of mTOR), mLST8 and PRAS4023, while mTOR in mTOR Complex 2 associates with rictor (rapamycin-insensitive companion of mTOR), mLST8, mSin1 and protor24. Thus, raptor and rictor constitute specific components of mTORC1 and mTORC2, respectively. We have previously demonstrated that both mTORC1 and mTORC2 are powerful positive regulators of placental amino acid transporters25,26,27,28,29. mTORC1 modulate System A and System L amino acid transport in primary human trophoblast cells at the posttranslational level by regulating Nedd4-2 mediated ubiquitination, which influences the trafficking of specific amino acid transporter isoforms to the plasma membrane28. Placental mTORC1 activity is inhibited in human IUGR25, 29, 30 and activated in placentas of large babies born to obese mothers31. Furthermore, placental mTORC1 activity has been reported to be decreased in hyperthermia-induced IUGR in the sheep32, in response to a maternal low protein diet in the rat33 and following maternal calorie restriction in the baboon34 and activated in a mouse model of maternal obesity associated with fetal overgrowth35, 36. Importantly, placental amino acid transport has been reported to be changed in the same direction as mTOR signaling in these clinical conditions and animal models of altered fetal growth29, 31, 33,34,35, 37,38,39. Collectively, this data suggest that trophoblast mTOR signaling regulates fetal growth by modulating placental nutrient transport. mTORC1 signaling is regulated by a multitude of upstream signals, including amino acids, ATP, glucose, oxygen and growth factors40,41,42. We recently reported that mTOR signaling functions as a folate sensor in primary human trophoblast cells, representing a previously unknown molecular mechanism by which folate regulates trophoblast cell function43. Folate sensing by mTOR in PHT cells involves both mTOR Complex 1 and 2 and requires the proton-coupled folate transporter (PCFT)43. Herein, we tested the hypothesis that mTOR functions as a folate sensor in vivo in mice and that maternal folate deficiency inhibits placental mTOR signaling and placental amino acid transporter activity and causes fetal growth restriction.
Materials and Methods
Experimental Design (Mice)
Animals
Experiments were carried out in ICR mice in accordance with the ‘Principles of Laboratory Animal Care’ (1996) and with the approval of the Institutional Animal Care and Use Committee at the University of Texas Health Science Center, San Antonio. Weanling female ICR mice (n = 20) were housed individually in polypropylene cages with wire mesh bottom and maintained at 22 °C ± 2 °C under standard lighting conditions (12-h light/dark cycle). They were divided into two groups (n = 10 each) and fed a control diet (TD110609, Harlan, WI) with 2 mg folate/kg diet for 6 weeks ad libitum or the same diet deficient in folate (TD 00434, Harlan, WI). Detailed information on the composition of the control and folate deficient diets is shown in Table 1. All the animals had free access to deionized water. Daily food intake and weekly body weights were determined. At the end of 6 weeks of feeding, blood was collected from the supraorbital sinus to determine serum folate concentrations and blood glucose. Subsequently female mice were mated with control males (2 females to 1 male) and the day a vaginal plug was detected was defined as embryonic day (E) 0.5. Animals were maintained on their respective diets throughout gestation. At E18.5, dams were euthanized for collection of blood and tissue samples.
Biochemical measurements
Folate was measured in maternal serum by a microbiological assay using a commercially available kit (ALPCO Diagnostic Products, NH, USA), according to the manufacturer’s instructions. Blood glucose was measured in triplicate using a One Touch Ultra-2 (Life Scan Inc, Milpitas, CA, USA).
Tissue collection
Dams were fasted (4 hr) and then euthanized at E18.5 by carbon dioxide inhalation. After laparotomy, fetuses and placentas were collected and quickly dried on blotting paper, any remaining fetal membranes were removed. Fetal weight, crown–rump length (CRL), measured with digital calipers, and placental weight were recorded for each pup. Pups were rapidly submerged in ice and decapitated by cervical dislocation. Maternal and fetal liver, heart, kidneys, and spleen were dissected out, weighed and immediately snap frozen in liquid nitrogen, and stored at −80 °C until further analysis. All placentas in each litter were pooled and washed in phosphate buffer saline and transferred to buffer D [250 mM sucrose, 10 mM Hepes-Tris, and 1 mM EDTA (pH 7.4) at 4 °C], protease and phosphatase inhibitor cocktail (Sigma-Aldrich Corp., St. Louis, MO, USA) was added at a dilution of 1:1000, and the mixture was homogenized using a Polytron (Kinematica, Bohemia, NY, USA).
Isolation of trophoblast plasma membranes (TPM)
TPM were isolated from frozen placental homogenates using differential centrifugation and Mg2+ precipitation as described35, 44. This protocol results in the isolation of the maternal-facing plasma membrane of trophoblast layer II of mouse placenta, and accumulating evidence suggests that this membrane is functionally analogous to the syncytiotrophoblast microvillus plasma membrane of the human placenta45. Protein concentration was determined using the Lowry assay (Bio Rad, CA). Using electron microscopy, it has been previously shown that alkaline phosphatase is localized to the apical (maternal facing) plasma membrane of trophoblast layer II (TPM) in the mouse45. Thus, the ratio of the alkaline phosphatase activity in the TPM fraction over the activity in the homogenate ais used to determine the enrichment of TPM. The mean alkaline phosphatase enrichment for TPM vesicles isolated from placentas of FD animals was not significantly different from the enrichment in TPM vesicles obtained from control placentas (Supplementary Figure 1).
TPM amino acid transporter activity measurements
The activity of System A and L amino acid transporters in TPM was determined using radiolabelled amino acids and rapid filtration techniques slightly modified from procedures previously described for human syncytiotrophoblast plasma membranes and mouse TPM44. TPM vesicles were preloaded by incubation in 300 mM mannitol and 10 mM Hepes-Tris, pH 7.4 overnight at 4 °C. Subsequently, TPM vesicles were pelleted and resuspended in a small volume of the same buffer (final protein concentration 5–10 mg ml−1). Membrane vesicles were kept on ice until immediately prior to transport measurements when samples were warmed to 37 °C. At time zero 30 μl vesicles were rapidly mixed (1:2) with the appropriate incubation buffer containing [14C] methyl-aminoisobutyric acid (MeAIB, 150 μM) with or without Na+ or [3 H] L-leucine (0.375 μM). Based on initial time course studies (0–30 s), uptake at 15 s was used in all subsequent experiments. Uptake of radiolabelled substrate was terminated by addition of 2 ml of ice cold PBS. Subsequently, vesicles were rapidly separated from the substrate medium by filtration on mixed ester filters (0.45 μm pore size, Millipore Corporation, Bedford, MA, USA) and washed with 3 × 2 ml of PBS. In all uptake experiments, each condition was studied in duplicate for each membrane vesicle preparation. Filters were dissolved in 2 ml liquid scintillation fluid (Filter Count, PerkinElmer, Waltham, MA, USA) and counted. Appropriate blanks were subtracted from counts and uptakes expressed as pmol x (mg protein)−1. Na+-dependent uptake of MeAIB (corresponding to system A activity) was calculated by subtracting Na+-independent from total uptakes. For leucine, mediated uptake was calculated by subtracting non-mediated transport, as determined in the presence of 20 mM unlabelled leucine, from total uptake.
Western blot analysis
Placental homogenates
Protein expression and phosphorylation of well-established functional readouts in the mTORC1 and mTORC2 signaling pathways was determined in placental homogenates using commercial antibodies (Cell Signaling Technology, Boston, MA). In addition, we determined TPM protein expression of the System A amino acid transporter isoforms (SNAT) 1, 2 and 4, the System L amino acid transporter isoforms LAT1 and LAT2. A polyclonal SNAT2 antibody generated in rabbits was received as a generous gift from Dr V. Ganapathy and Dr P. Prasad at the University of Georgia, Augusta. Western blotting was carried out as previously described33. Analysis of the blots was performed by densitometry.
Maternal and fetal tissues
Snap frozen maternal and fetal organs were homogenized in ice-cold radioimuno precipitation assay buffer (0.5 mol/L Tris-Cl, pH 7.4, 1.5 mol/L NaCl, 10 mmol/L ethylenediaminetetraacetic acid [EDTA], 2.5% deoxycholic acid, 10% NP-40, protease inhibitor, and phosphatase inhibitor), by rapid agitation in the presence of beads and spun at 13 000 g at 4 °C for 10 minutes. mTORC1 and mTORC2 signaling activity in maternal and fetal tissue homogenates was assessed by determining total protein expression and phosphorylation of key downstream targets using Western blot and commercial antibodies (Cell Signaling Technology, Boston, MA).
Proximity ligation assay (PLA) and confocal microscopy
Placentas were serially cryosectioned at 3 μm and sections were stored at −80 °C, until ready for use. The sections were blocked using 5% new born calf (NCS) serum in PBS for 1 hour followed by incubation in anti-mTOR (anti-rabbit) and anti-LAMP-2 (anti-mouse) antibodies for 2 hours. PLA probes anti-rabbit PLUS and anti-mouse MINUS were diluted in Duolink dilution buffer and incubated in a pre-heated humidity chamber for 1 hour. This was followed by ligation, amplification and detection according to the Duolink In Situ Orange kit (Sigma-Aldrich) manufacturer’s protocol. Confocal microscopy was performed using Zeiss LSM 780 microscope at 63x magnification using oil immersion. Images were captured in the same laser settings with four Z-step of 0.4 um. In each section, at least ten randomly selected microscopic fields were used to calculate the number of mTOR-LAMP2 interaction positive sites (yellow dots) per mm2 and data were averaged to represent a single placenta.
Experimental design (baboons)
Animals and diets
All procedures were approved by the Texas Biomedical Research Institute Institutional Animal Care and Use Committee and conducted in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care. Baboons (Papio species) were housed in outdoor metal and concrete gang cages, each containing 10–16 females and 1 male. Details of housing, environmental enrichment and system for controlling and recording individual feeding have been described elsewhere46. Animals were fed Purina Monkey Diet 5038 (Supplementary Table 1, Purina, St. Louis, MO, USA). Females were mated, pregnancy confirmed by ultrasound at gestational day (GD) 30, subsequently animals subjected to maternal nutrient restriction (MNR) were fed 70% of the total food intake of contemporaneous controls on a per-kilogram basis as previously described in detail47.
Collection of tissue and blood samples
Cesarean sections were performed under isoflurane anesthesia at GD 165 (term 184). Briefly, animals were tranquilized with ketamine hydrochloride (10 mg/kg), intubated, and anesthetized using isoflurane (starting rate 2% with oxygen: 2.0 L/min). Conventional cesarean sections using standard sterile technique were performed as described previously46. At cesarean section, fetuses and placentas were towel dried and weighed. Postoperative analgesia was provided using buprenorphine (0.015 mg/kg/d as 2 doses) for 3 d46.
Maternal serum folate analysis
Serum was prepared and frozen at −80 °C until analysis of folate concentrations using an Immulite 1000 (Siemens Healthcare Diagnostics, Deerfield IL), according to the manufacturer’s instructions. Intra- and inter-assay coefficients of variation were 6.7 and 7.9% at 700 pg/ml.
Western blot analysis
Placental homogenates
Placental mTORC1 and mTORC2 signaling functional readouts were measured as described for mice placental homogenates and reported elsewhere34.
Placental microvillus membrane system A and L amino acid transport activity and expression
Syncytiotrophoblast plasma microvillous membrane vesicles (MVM) were isolated as described in detail previously48, with minor modifications34. MVM system A and L amino acid transporter activity and isoform expression were measured as described previously33, 49 and reported elsewhere34. In the current study we determined the correlation between placental mTORC1 and mTORC2 signaling/amino acid transporter activity or expression34 and maternal serum folate and fetal weight34.
Experimental design (humans)
This study was approved by the Institutional Review Board at the University of Texas Health Science Center at San Antonio (approval no. HSC20070723H). Women with uncomplicated singleton term (37–40 weeks’ gestation) pregnancies were recruited with written consent before undergoing scheduled cesarean deliveries, which routinely were scheduled at 39 weeks gestation. Informed consent was obtained from all subjects. These women had had one or more previous cesarean delivery and were either not a candidate for, or declined, a trial of labor. Gestational age was estimated from the date of the last menstrual period and confirmed by ultrasound dating.
Maternal venous blood samples were obtained prior to c-section. Serum was prepared and frozen at −80 °C until analysis of folate concentrations using an Immulite 1000 (Siemens Healthcare Diagnostics, Deerfield IL), according to the manufacturer’s instructions. Intra- and inter-assay coefficients of variation were 6.7 and 7.9% at 700 pg/ml. The placenta was collected within 15 min of delivery, the decidua basalis and chorionic plate were removed and villous tissue was dissected and rinsed in cold physiological saline. The villous tissue was transferred to cold buffer D (250 mM sucrose, 10 mM HEPES, pH 7.4) containing 1:100 dilution of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO) and homogenized on ice with a Polytron (Kinematica, Luzern, Switzerland). The placental homogenates were frozen in liquid nitrogen and stored at −80 °C until further processing. MVM system A and L amino acid transporter activity was measured as described previously48.
Data presentation and statistics
In mice, placental and fetal data were averaged within each litter and the litter mean was used as the observation for that dam. Thus, n represents the number of litters. Data are presented as means ± S.E.M or + S.E.M. Statistical significance of differences between control and experimental groups was assessed using Two-way repeated-measures (RM) ANOVA or Student’s t-test. A P value < 0.05 was considered significant.
Results
Dietary folate restriction before pregnancy results in decreased serum folate levels
Dietary folate restriction between 3 and 9 weeks of age in female mice (Fig. 1a) did not influence body weight gain (Fig. 1b), food intake (Fig. 1c) or fasting blood glucose (Fig. 1d). The serum folate concentration was decreased by 60% after 6 weeks of dietary folate restriction (p = 0.0004, n = 8/each group, mean ± SEM, Fig. 1e).

Effect of dietary folate restriction before pregnancy on body weight gain, food consumption and biochemical parameters in mice. (a) Study design illustrating the period of folate deficiency in mice. (b) #Preconceptional body weight gain and (c) food intake were largely unaffected. (d) Fasting blood glucose was not influenced by folate deficiency but (e) serum folate levels were decreased by feeding a folate deficient diet for 6 weeks. Values are given as mean + SEM; *P < 0.05 vs. control; unpaired Student’s t-test; n = 6–10/each group; #Two-way repeated-measure ANOVA and post-hoc pairwise comparisons with Bonferroni correction (diet × weight) did not reveal any significant interaction between folate diet and weight gain at different time points (F = 0.958, p = 0.45, n = 10 each group).
Effect of maternal folate deficiency on placental and fetal growth in mice
Low maternal serum folate is associated with restricted fetal growth in pregnant women1, 2, 50, providing the rationale to examine the effects of maternal folate deficiency in mice on fetal-placental growth characteristics. Food intake during pregnancy was comparable between control and folate deficient groups (Fig. 2a). At E18.5, fetal weight (−17%, p < 0.05, Fig. 2b) and fetal/placental weight ratio (−14%, p < 0.05, Fig. 2c) were decreased in the folate deficient group, whereas placental weight, crown rump length and litter size were unaltered (Fig. 2d–f).

Effect of folate deficiency on food consumption, fetal and placental weights in mice. Feeding a folate deficient diet prior to and during pregnancy did not influence daily food intake (a) but did decreased fetal weight (b) and fetal/placental weight ratio (c). In addition, folate deficiency did not affect placental weight (d) crown rump length (e), or litter size (f). Placental and fetal data were averaged within each litter and the litter mean was used as the observation for that dam. Thus, n represents the number of litters. Values are given as mean + SEM; *P < 0.05 vs. control; unpaired Student’s t-test; n = 6–7.
Maternal folate deficiency inhibits placental mTORC1 signaling in mice
Raptor interacts with mTOR to form a nutrient-sensitive complex that signals to the cell growth machinery51. We studied the raptor protein expression in placental homogenates of the control and folate deficient mice. Raptor protein expression in the placenta was not significantly different in control and folate deficient mice (Fig. 3a,b). Upon activation, mTOR is phosphorylated on several residues, including T2446, S2448 and S2481. Recent reports have shown that p70S6 kinase is a major effector of mTOR phosphorylation at Ser-2448 in response to both mitogen- and nutrient-derived stimuli52. Whereas total placental expression of mTOR was unaffected, phosphorylation of mTOR at Ser-2448 was inhibited by folate deficiency in mice (Fig. 3a,b). Next, we examined the phosphorylation of p70S6K1 and S6 downstream kinases regulated by the mTORC1 complex. Maternal folate deficiency significantly decreased the phosphorylation of S6K (Thr-389) and S6 (Ser-235/236) but did not affect the total expression of any of these kinases (Fig. 3a,b).

Maternal folate deficiency in mice inhibits placental mTORC1 signaling. (a) Placental mTORC1 signaling was inhibited in response to maternal folate deficiency. Histogram (b) summarizes the immunoblot data of placental mTORC1. Values are given as mean + SEM; *P < 0.05 vs. control; unpaired Student’s t-test; n = 6–7. Full-length gels and blots are included in the Supplementary Information File.
In most instances, translation is regulated at the initiation phase, when a ribosome is recruited to the 5′ end of an mRNA. The eIF4E-binding proteins (4E-BPs) inhibit translation initiation by binding to the translation factor eIF4E, and preventing recruitment of the translation machinery to mRNA. However, phosphorylation of the binding protein decreases its affinity for eIF4E, thereby activating protein translation. In mammals, the 4EBP family consists of 3 proteins, 4EBP1, 4EBP2, and 4EBP3 and the best characterized 4EBP is 4EBP1. There are six phosphorylation sites have been identified in 4EBP1 (Thr 37, Thr 46, Ser 65, Thr 70, Ser 83, and Ser 112)53 and we studied four of them. Folate deficiency in mice significantly inhibited the phosphorylation of placental 4E-BP1 at Thr-37 and 46 as compared to control. Similarly, phosphorylation of 4E-BP1 at Thr-70 was decreased in placenta of folate deficient mice as compared to control. However, there was no significant differences between the control and folate deficient group placenta, when analyzing 4E-BP1 phosphorylation at Ser-65. Compared to control, total expression of 4E-BP1 was higher in placentas of mice fed the folate deficient diet. Thus, both the increased total expression and decreased phosphorylation of 4E-BP is expected to inhibit protein translation in placentas of folate deficient mice.
Maternal folate deficiency inhibits placental mTORC2 signaling
Rapamycin-insensitive companion of mammalian target of rapamycin (rictor) is a cytosolic protein that was originally recognized as a specific component of mTORC2. An increasing body of evidence however shows that rictor may also function independently of mTORC2 through association with other proteins and complexes54, 55. We found that the protein expression of rictor was significantly decreased in placentas of folate deficient dams (Fig. 4a,b). Protein kinase C (PKC) is one of the most extensively studied kinase family and has been implicated in cell proliferation and differentiation56. Recent study demonstrates that mTORC2 is involved in post‐translational processing of PKC by facilitating phosphorylation and revealed a novel function of mTORC2 in cellular regulation57. The phosphorylation of placental PKCα at Ser-657 was decreased in folate deficiency. There was no significant difference in the total PKCα expression level between control and FD group (Fig. 4a,b).

Maternal folate deficiency in mice inhibits placental mTORC2 signaling. (a) Placental mTORC2 signaling was inhibited in response to maternal folate deficiency. Histogram (b) summarizes the immunoblot data of placental mTORC2. Values are given as mean + SEM; *P < 0.05 vs. control; unpaired Student’s t-test; n = 6–7. Full-length gels and blots are included in the Supplementary Information File.
SGK is a downstream target of mTORC255, serving as a critical node in a highly conserved signaling pathway that integrates multiple inputs from the environment58. Activation of SGK1 is triggered by phosphorylation of a threonine residue within the T-loop of the kinase domain and a serine residue lying within the C-terminal hydrophobic motif (Ser422 in SGK1)58. Phosphorylation of SGK1 at Ser-422 was significantly decreased in the placenta of folate deficient mice as compared to control. However, total SGK1 expression was comparable between control and folate deficient group mice (Fig. 4a,b).
The activation of Akt involves the phosphorylation of two residues. Threonine 308 (Thr308) in the activation loop of Akt is phosphorylated by PDK1, and therefore represents a functional readout of the insulin/IGF-I growth factor signaling pathway. In contrast, serine 473 (Ser473) in the hydrophobic motif is phosphorylated by mTORC259. The phosphorylation of placental Akt at Ser-473 was markedly decreased in folate deficient mice. Importantly, folate deficiency did not influence Akt phosphorylation at Thr-308 (Fig. 4a,b), suggesting that the impact of low folate on cellular signaling is specific. There was no significant difference in the total Akt expression level between folate deficient and control placentas (Fig. 4a,b).
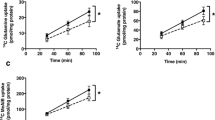
Maternal folate deficiency decreases placental amino acid transport activity in mice
We have previously demonstrated that mTOR signaling regulates placental amino acid transport and that mTOR inhibition decreases amino acid transport by preventing the trafficking of specific transporter isoforms to the plasma membrane27. To examine the effect of maternal folate deficiency on placental amino acid transport we isolated trophoblast plasma membranes (TPM)44, 45 from folate-deficient and control pregnancies. Importantly, the activity of System A and L amino acid transporters was significantly decreased in TPM isolated from folate deficient dams (Supplementary Figure 2a,b). To explore if the marked reduction in TPM amino acid transport activity was associated with a decrease in the transporter abundance, we determined the isoform protein expression by Western blot in isolated TPM. The TPM protein expression of SLC38A2 (Sodium-coupled Neutral Amino acid Transporter 2, SNAT2, an isoform of the System A amino acid transporter) and SLC7A5 (Large neutral Amino acid Transporter, LAT1, a System L amino acid transporter isoform), but not SLC38A1 (SNAT1) or SLC7A8 (LAT2), was markedly decreased in response to maternal folate deficiency (Supplementary Figure 2c,d).
Folate deficiency in pregnant mice decreases the interaction between mTOR and LAMP2 in the placenta
Activation of the mTORC1 pathway by amino acids has been shown to promote the translocation of mTORC1 from an unidentified vesicular compartment to a membrane-bound compartment containing Rab7, a marker of both late endosomes and lysosomes60. More recent data show that the precise location of mTORC1 is at the lysosomal surface, characterized by LAMP2-positive staining61. To test the possibility that folate also regulate the mTOR subcellular localization, we used confocal microscopy and proximity ligation assay to determine the extent of co-localization between mTOR and LAMP2 in placentas of control and folate deficient mice. Whereas mTOR co-localized with LAMP2 in placentas of control mice, the proximity ligation assay signal was markedly attenuated in placentas of folate deficient mice, suggesting a decrease in mTOR/LAMP2 interaction (Fig. 5a,b,c). These data are consistent with the model proposed for mTORC1 amino acid sensing whereby mTORC1, in response to amino acids, is shuttled to the lysosome where it interacts with the nutrisome, resulting in mTORC1 activation62.

Folate deficiency in pregnant mice decreases the interaction between mTOR and LAMP2 in the placenta. (a,b) Proximity ligation assay and confocal microscopy was used to determine interactions between mTOR and LAMP2, a lysosomal marker, in the placenta of control and folate deficient pregnant mice. In control placentas (top) mTOR and LAMP2 were in close proximity of each other as evidenced by a large number of yellow dots, suggesting localization of mTOR at the surface of the lysosomes. In contrast, folate deficiency markedly decreased the interaction between mTOR and LAMP2, consistent with localization of mTOR away from the lysosomes. Blue = nuclei; Scale bar-50µm. (c) Histogram summarizes the proximity ligation assay data of placental mTOR-LAMP2 interactions. In each section, at least ten randomly selected microscopic fields were used to calculate the number of mTOR-LAMP2 interaction positive sites (yellow dots) per mm2 and data were averaged to represent a single placenta. Values are given as mean + SEM; *P < 0.05 vs. control; unpaired Student’s t-test; n = 3 placenta/each group.
Effect of maternal folate deficiency on maternal tissue growth and mTOR signaling
Maternal folate deficiency significantly decreased the weights of maternal liver, heart, spleen and kidney (Fig. 6a,b).

Effect of maternal folate deficiency on maternal tissue growth and mTOR signaling. (a) Maternal liver and (b) heart, spleen, kidney weights were decreased in folate deficient dams. Values are given as mean + SEM; *P < 0.05 vs. control; unpaired Student’s t-test; n = 6–7. (c) mTORC1 (S6-S-235/236, S6, 4E-BP1-T-37/46) and mTORC2 (Rictor, Akt-S-473) downstream signaling in maternal skeletal muscle was inhibited in folate deficient dams. Using Western blot analysis, two distinct bands were observed for total 4E-BP1 in maternal muscle and analyzed both bands together. Histogram (d,e) summarizes the immunoblot data of maternal skeletal muscle mTORC1and mTORC2 signaling. Values are given as mean + SEM; *P < 0.05 vs. control; unpaired Student’s t-test; n = 6–7. Full-length gels and blots are included in the Supplementary Information File.
Maternal muscle
The phosphorylation of ribosomal protein S6 at S-235/236 and 4E-BP1 at T-37/46 were significantly decreased in maternal muscle of FD group as compared to control (Fig. 6c,d,e). However, the phosphorylation of 4E-BP1 at S-65 was unaffected by FD. No significant differences were found in the expression of maternal muscle raptor, total 4E-BP1 expression between control and FD group. In contrast, total S6 expression in maternal muscle was significantly decreased by FD. Furthermore, folate deficiency significantly decreased the expression of rictor and phosphorylation of Akt at S-473. No significant differences were found in the expression of maternal muscle total Akt and phosphorylated Akt-T-308 between control and FD group (Fig. 6c,d,e).
Maternal heart
As shown in Supplementary Figure 3(a,b,c), FD significantly decreased the phosphorylation of S6 at S-235/236 and 4E-BP1 at T-37/46 in heart homogenates. No significant differences were found in the expression of maternal heart raptor, total mTOR, S6, 4E-BP1 and phosphorylated mTOR- S-2448 and 4E-BP1-T-70 between control and FD group. In addition, maternal folate deficiency caused a marked decrease in Akt phosphorylation at S-473 in the heart. However, total Akt expression was comparable between FD and control group.
Maternal liver
Maternal folate deficiency significantly decreased the phosphorylation of mTOR-S-2448, S6-S-235/236, 4E-BP1-T-37/46 (mTORC1 signaling) and SGK-S-422, Akt-S-473 (mTORC2 signaling) in the liver. In contrast, total mTOR and 4E-BP1 expression were increased in liver homogenates of FD as compared to control. However, total S6 and Akt expression were comparable between FD and control group Supplementary Figure 3(d,e,f).
Effect of maternal folate deficiency on fetal tissue growth and mTOR signaling
At E18.5, the weights of fetal liver and heart were decreased (Supplementary Figure 4a,b) in response to maternal folate deficiency.
Fetal liver
Maternal folate deficiency decreased the expression/phosphorylation of the downstream targets of mTORC1 signaling such as raptor, mTOR-S-2448, S6K-T-389, 4E-BP1-T-70 in the fetal liver (Supplementary Figure 4c). Total mTOR and 4E-BP1 expression was increased in fetal liver of FD group as compared to control. Moreover, maternal folate deficiency significantly decreased the expression/phosphorylation of rictor, SGK-S-422, Akt-S-473 in fetal liver. However, fetal liver S6-S-235/236, total S6K, S6 (mTORC1 signaling) and total SGK, Akt (mTORC2 signaling) expression was comparable between control and FD mice.
Fetal heart
FD caused a marked decrease in phosphorylation of mTORC1 downstream targets such as S6-S-235/236, 4E-BP1- T-70, 37/46, 4E-BP1-S-65 and mTORC2 functional downstream targets such as PKC-α-S-657, SGK-S-422, Akt-S-473 in fetal heart (Supplementary Figure 4d). Total 4E-BP1 expression was significantly increased in the heart homogenates of FD group as compared to control. However, fetal heart mTOR-S-2448, total mTOR, S6, rictor, PKC α, SGK, Akt-T-308 expression/phosphorylation was unaffected by FD.
Fetal kidney
In contrast to fetal liver and heart, mTORC1 (raptor, mTOR-S-2448, mTOR, S6, 4E-BP1 T-70, 37/46, and total 4E-BP1) and mTORC2 (Akt-S-473) signaling were comparable in fetal kidney homogenates between FD and control group mice (Supplementary Figure 4e).
Maternal serum folate is associated with fetal weight and placental mTORC1 signaling in the baboon
Next we examined the relationship between maternal serum folate and fetal weight and placental mTORC1 and mTORC2 signaling in baboon pregnancy. Maternal nutrient restriction in the baboon significantly reduced fetal weights (Supplementary Figure 5a). Furthermore, maternal serum folate levels were positively correlated with fetal weights in both the control and MNR group (Supplementary Figure 5b). However, maternal serum folate levels were not significantly different between the control and MNR group (Supplementary Figure 5c). Placental mTORC1 and mTORC2 signaling in the same placentas has been reported elsewhere34. Placental mTORC1 signaling functional readouts phosphorylated S6 (Serine-235/236), 4E-BP1 (Thr-37/46) and S6 kinase (Thr-389) were positively correlated with maternal serum folate and fetal weights (Fig. 7a–f).

(a,c,e) Correlation between maternal serum folate and placental mTORC1 functional readouts in baboon at gestational day 165. (a) Relationship between maternal serum folate and placental S6 (Serine-235/236); Control, r = 0.97, p = 0.0001; MNR, r = 0.88, p = 0.001. (c) Relationship between maternal serum folate and placental 4E-BP1 (Threonine-37/46); Control, r = 0.86, p = 0.006; MNR, r = 0.81, p = 0.008. (c) Relationship between maternal serum folate and placental 4E-BP1 (Threonine-70); Control, r = 0.87p = 0.005; MNR, r = 0.89, p = 0.001. (b,d,f) Correlation between placental mTORC1 functional readouts and fetal weight in baboon at gestational day 165. (b) Relationship between placental S6 (Serine-235/236) and fetal weight; Control, r = 0.86, p = 0.005; MNR, r = 0.96, p = 0.0001. (d) Relationship between placental 4E-BP1 (Threonine-37/46) and fetal weight; Control, r = 0.83, p = 0.01; MNR, r = 0.91, p = 0.0006. (e) Relationship between placental 4E-BP1 (Threonine-70) and fetal weight; Control, r = 0.83, p = 0.01; MNR, r = 0.92, p = 0.0005. Control, n = 8; MNR, n = 9; r = Pearson’s correlation coefficient.
Placental microvillus membrane amino acid transport is associated with maternal serum folate and fetal weight in the baboon
Syncytiotrophoblast microvillus plasma membrane SNAT2 and LAT1 expression were positively correlated with maternal serum folate and fetal weights. (Supplementary Figure 6a–d). Similarly, microvillus membrane system A and system L activity were positively correlated with maternal serum folate and fetal weights. (Supplementary Figure 7a–d).
Maternal serum folate is associated with placental microvillus membrane system L activity in human pregnancy
To explore the clinical relevance of our findings, we examined the relationship between maternal serum folate and placental microvillus membrane system L activity in term placentas from a cohort of healthy women (n = 13) undergoing cesarean section at term. Even in this small cohort, maternal serum folate was positively correlated with birth weight (Supplementary Figure 8a). Furthermore, we observed a significant positive correlation between maternal serum folate levels and placental MVM system L activity (Supplementary Figure 8b). In addition, MVM system L activity positively correlated with birth weight (Supplementary Figure 8c). Phosphorylation of S6 (Serine-235/236), a functional readout of mTORC1 activity, was positively correlated with maternal serum folate (r = 0.7290, p = 0.0047, n = 13). Similarly, phosphorylation of Akt (Serine-473), representing mTORC2 activity, was positively correlated with maternal serum folate (r = 0.7497, p = 0.0032, n = 13).
Discussion
We show that mTOR functions as a folate sensor in vivo in mice extending our recent report that folate is a positive regulator of mTORC1 and mTORC2 signaling in cultured primary human trophoblast cells43. Collectively these studies identify a novel mechanism by which folate regulates cell growth and function, which may have broad biological implications. This is also the first study exploring changes in placental signaling and nutrient transporter function and fetal growth in response to maternal folate deficiency in mice. Our findings suggest that placental mTOR folate sensing constitutes a mechanistic link between folate availability and fetal growth, a model supported by the observation that maternal serum folate levels, placental mTOR signaling, nutrient transport and fetal growth are strongly correlated in baboon and human pregnancy.
Although the literature is not entirely consistent1, several studies demonstrate a strong positive relationship between maternal red blood cell folate and birth weight1, 2, 9, 50. Furthermore, a meta analysis of 8 randomized controlled trials investigating the effect of folate supplementation on fetal growth clearly shows a dose-response relationship between folate intake and birth weight2. However, the mechanisms linking folate availability to fetal growth are poorly understood. Fetal growth is largely determined by nutrient availability and trophoblast mTOR signaling is a positive regulator of placental amino acid26, 27, 63 and folate transport64 and mitochondrial function65. mTOR signaling is influenced by an array of upstream regulators, including growth factor signaling, amino acid availability, cellular energy and oxygen levels40, 41, 66,67,68. We recently reported that both mTORC1 and mTORC2 signaling pathways in primary human trophoblast cells are influenced by folate availability with folate deficiency causing a profound inhibition of mTOR signaling activity43. The present study provides compelling evidence that mTOR functions as a folate sensor in vivo and identifies down-regulation of placental nutrient transporters following inhibition of mTOR signaling as a possible underlying mechanism linking low maternal folate levels to fetal growth restriction. First, maternal folate deficiency in mice resulted in inhibition of placental mTORC1 and mTORC2 signaling pathways, decreased expression of key amino acid transporter isoforms, and lowered placental in vitro system A and L amino acid transport activity. Second, maternal folate levels in baboons positively correlated with functional read outs of placental mTORC1 signaling and fetal weights. Third, maternal serum folate correlated positively with birth weight, placental mTORC1 and mTORC2 signaling and placental MVM system L amino acid transport activity in small cohort of pregnant women, suggesting that our finding have clinical relevance. Because trophoblast growth and function plays a critical role in determining fetal growth69,70,71, folate sensing by trophoblast mTOR may represent a mechanism by which maternal folate status modulates fetal growth and development.
It is possible that the effect of maternal folate deficiency on placental function is mediated by indirect effects rather by placental mTOR folate sensing. For example, it cannot be excluded that folate deficiency resulted in changes in maternal circulating levels of metabolic hormones, adipokines and nutrient levels, which are well established to regulate placental function72. However, the striking similarity in trophoblast responses to folate deficiency in vivo and in vitro 43 suggest that this is unlikely. Specifically, the signature of mTORC1 and mTORC2 inhibition, including down-regulation of rictor expression and markedly decreased Ser-473 Akt phosphorylation, leaving Ser-308 Akt phosphorylation unaffected was remarkably similar the mouse placenta in vivo (present study) and in primary trophoblast cells in vitro 43. Furthermore, given that we previously have reported that mTORC1 and 2 regulates the plasma membrane trafficking of specific amino acid transporter isoforms (SNAT 2 and LAT1)27, the finding that folate deficiency in vivo in mice specifically decreased the protein expression of these two isoforms in the trophoblast plasma membrane, suggest that that the down regulation of placental amino acid transport in folate deficient dams are a result of mTORC1 and 2 inhibition.
Folate deficiency resulted in a decreased phosphorylation of specific functional readouts of mTORC1 and mTORC2 signaling in maternal muscle, liver, heart tissues and fetal liver, heart, consistent with the possibility that mTOR is highly sensitive to folate availability in cells other than trophoblasts and that mTOR folate sensing has broad biological significance. For example, antifolates, which are drugs that inhibit the cellular actions of folate by blocking the function of the enzyme dihydrofolate reductase, have been used in cancer treatment for a long time. The presumed action of antifolates is to limit the availability of nucleotides for DNA synthesis in rapidly proliferation cancer cells. In addition, emerging evidence suggest that high folate intake also promotes carcinogenesis73, 74, however the underlying molecular mechanism remains elusive. Given the importance of mTOR signalling in promoting cancer cell growth and proliferation and our identification of mTOR as a folate sensor, we speculate that mTOR inhibition contributes to the mechanism of action of antifolates and mTOR activation may link folate excess to carcinogenesis.
Recent advances in the field of mTORC1 amino acid sensing have placed this cellular process at the surface of the lysosome60,61,62. Specifically it is believed that amino acids regulate the recruitment of mTORC1 to the lysosomal surface, where mTORC1 is activated. Using confocal microscopy and proximity ligation assay we provided evidence that mTOR is localized at the lysosomal surface in placentas of control animals. However in placentas collected from folate deficient dams, mTOR was no longer associated to the lysosomes, confirming our previous in vitro data in primary human trophoblast cells43. These preliminary observations suggest that mTOR amino acid and folate sensing share common mechanisms, however additional studies are required to unravel the molecular mechanisms linking folate to mTOR signaling.
In conclusion, we have identified a novel specific molecular link between maternal folate availability and fetal growth, involving regulation of placental mTOR signaling by folate resulting in changes in placental nutrient transport. We propose that placental mTOR folate sensing cells matches placental nutrient transport and fetal growth to maternal folate status. These findings are clinically relevant because we show that maternal serum folate levels are positively correlated to placental mTOR signaling and nutrient transport and fetal weight in women. mTOR folate sensing may have broad biological significance because of the critical role of folate in normal cell function and the wide range of disorders, including cancer, that have been linked to folate availability.
References
Tamura, T. & Picciano, M. F. Folate and human reproduction. Am J Clin Nutr 83, 993–1016, doi:83/5/993 [pii] (2006).
Fekete, K. et al. Effect of folate intake on health outcomes in pregnancy: a systematic review and meta-analysis on birth weight, placental weight and length of gestation. Nutr J 11, 75, doi:10.1186/1475-2891-11-75 (2012).
Smithells, R. W., Sheppard, S. & Schorah, C. J. Vitamin dificiencies and neural tube defects. Arch Dis Child 51, 944–950 (1976).
Force, U. S. P. S. T. Folic acid for the prevention of neural tube defects: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med 150, 626–631 (2009).
Blencowe, H., Cousens, S., Modell, B. & Lawn, J. Folic acid to reduce neonatal mortality from neural tube disorders. Int J Epidemiol 39(Suppl 1), i110–121, doi:10.1093/ije/dyq028 (2010).
Czeizel, A. E. & Dudas, I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med 327, 1832–1835, doi:10.1056/NEJM199212243272602 (1992).
Relton, C. L., Pearce, M. S. & Parker, L. The influence of erythrocyte folate and serum vitamin B12 status on birth weight. Br J Nutr 93, 593–599 (2005).
Scholl, T. O. & Johnson, W. G. Folic acid: influence on the outcome of pregnancy. Am J Clin Nutr 71, 1295S–1303S (2000).
van Uitert, E. M. & Steegers-Theunissen, R. P. Influence of maternal folate status on human fetal growth parameters. Mol Nutr Food Res 57, 582–595, doi:10.1002/mnfr.201200084 (2013).
Greenberg, J. A., Bell, S. J., Guan, Y. & Yu, Y. H. Folic Acid supplementation and pregnancy: more than just neural tube defect prevention. Rev Obstet Gynecol 4, 52–59 (2011).
Czeizel, A. E., Puho, E. H., Langmar, Z., Acs, N. & Banhidy, F. Possible association of folic acid supplementation during pregnancy with reduction of preterm birth: a population-based study. Eur J Obstet Gynecol Reprod Biol 148, 135–140, doi:10.1016/j.ejogrb.2009.10.016 (2010).
Papadopoulou, E. et al. The effect of high doses of folic acid and iron supplementation in early-to-mid pregnancy on prematurity and fetal growth retardation: the mother-child cohort study in Crete, Greece (Rhea study). Eur J Nutr 52, 327–336, doi:10.1007/s00394-012-0339-z (2013).
Gluckman, P. D., Hanson, M. A., Cooper, C. & Thornburg, K. L. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359, 61–73, doi:10.1056/NEJMra0708473 (2008).
Gluckman, P. D. & Hanson, M. A. Living with the past: evolution, development, and patterns of disease. Science 305, 1733–1736, doi:10.1126/science.1095292 (2004).
Barker, D. J. The developmental origins of well-being. Philos Trans R Soc Lond B Biol Sci 359, 1359–1366, doi:10.1098/rstb.2004.1518 (2004).
Barker, D. J. Developmental origins of adult health and disease. J Epidemiol Community Health 58, 114–115 (2004).
Yajnik, C. S. & Deshmukh, U. S. Fetal programming: maternal nutrition and role of one-carbon metabolism. Rev Endocr Metab Disord 13, 121–127, doi:10.1007/s11154-012-9214-8 (2012).
Efstathiou, S. P., Skeva, I. I., Zorbala, E., Georgiou, E. & Mountokalakis, T. D. Metabolic syndrome in adolescence: can it be predicted from natal and parental profile? The Prediction of Metabolic Syndrome in Adolescence (PREMA) study. Circulation 125, 902–910, doi:10.1161/CIRCULATIONAHA.111.034546 (2012).
Stewart, C. P. et al. Antenatal micronutrient supplementation reduces metabolic syndrome in 6- to 8-year-old children in rural Nepal. J Nutr 139, 1575–1581, doi:10.3945/jn.109.106666 (2009).
Burdge, G. C., Lillycrop, K. A., Jackson, A. A., Gluckman, P. D. & Hanson, M. A. The nature of the growth pattern and of the metabolic response to fasting in the rat are dependent upon the dietary protein and folic acid intakes of their pregnant dams and post-weaning fat consumption. Br J Nutr 99, 540–549, doi:10.1017/S0007114507815819 (2008).
Stover, P. J. Physiology of folate and vitamin B12 in health and disease. Nutr Rev 62, S3-12; discussion S13 (2004).
Zoncu, R., Efeyan, A. & Sabatini, D. M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12, 21–35, doi:10.1038/nrm3025 (2011).
Kim, D. H. et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 (2002).
Jacinto, E. et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6, 1122–1128, doi:10.1038/ncb1183 (2004).
Roos, S. et al. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J Physiol 582, 449–459, doi:10.1113/jphysiol.2007.129676 (2007).
Roos, S., Kanai, Y., Prasad, P. D., Powell, T. L. & Jansson, T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am J Physiol Cell 296, C142–C150 (2009).
Rosario, F. J., Kanai, Y., Powell, T. L. & Jansson, T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J Physiol 591, 609–625, doi:10.1113/jphysiol.2012.238014 (2013).
Rosario, F. J., Dimasuay, K. G., Kanai, Y., Powell, T. L. & Jansson, T. Regulation of Amino Acid Transporter Trafficking by mTORC1 in Primary Human Trophoblast cells is Mediated by the Ubiquitin Ligase Nedd4-2. Clin Sci (Lond), doi:10.1042/CS20150554 (2015).
Chen, Y. Y. et al. Increased ubiquitination and reduced plasma membrane trafficking of placental amino acid transporter SNAT-2 in human IUGR. Clin Sci (Lond), doi:10.1042/CS20150511 (2015).
Yung, H. W. et al. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol 173, 451–462, doi:10.2353/ajpath.2008.071193 (2008).
Jansson, N. et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab 98, 105–113, doi:10.1210/jc.2012-2667 (2013).
Arroyo, J. A., Brown, L. D. & Galan, H. L. Placental mammalian target of rapamycin and related signaling pathways in an ovine model of intrauterine growth restriction. Am J Obstet Gynecol 201(616), e611–617, doi:10.1016/j.ajog.2009.07.031 (2009).
Rosario, F. J. et al. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology 152, 1119–1129, doi:10.1210/en.2010-1153 (2011).
Kavitha, J. V. et al. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J 28, 1294–1305, doi:10.1096/fj.13-242271 (2014).
Aye, I. L., Rosario, F. J., Powell, T. L. & Jansson, T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc Natl Acad Sci USA 112, 12858–12863, doi:10.1073/pnas.1515484112 (2015).
Rosario, F. J., Powell, T. L. & Jansson, T. Activation of placental insulin and mTOR signaling in a mouse model of maternal obesity associated with fetal overgrowth. Am J Physiol Regul Integr Comp Physiol 310, R87–93, doi:10.1152/ajpregu.00356.2015 (2016).
Ross, J. C., Fennessey, P. V., Wilkening, R. B., Battaglia, F. C. & Meschia, G. Placental transport and fetal utilization of leucine in a model of fetal growth retardation. Am J Physiol 270, E491–503 (1996).
Anderson, A. H., Fennessey, P. V., Meschia, G., Wilkening, R. B. & Battaglia, F. C. Placental transport of threonine and its utilization in the normal and growth-restricted fetus. Am J Physiol 272, E892–900 (1997).
de Vrijer, B., Regnault, T. R., Wilkening, R. B., Meschia, G. & Battaglia, F. C. Placental uptake and transport of ACP, a neutral nonmetabolizable amino acid, in an ovine model of fetal growth restriction. Am J Physiol Endocrinol Metab 287, E1114–1124, doi:10.1152/ajpendo.00259.2004 (2004).
Gulati, P. & Thomas, G. Nutrient sensing in the mTOR/S6K1 signalling pathway. Biochem Soc Trans 35, 236–238, doi:10.1042/BST0350236 (2007).
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293, doi:10.1016/j.cell.2012.03.017 (2012).
Efeyan, A., Comb, W. C. & Sabatini, D. M. Nutrient-sensing mechanisms and pathways. Nature 517, 302–310, doi:10.1038/nature14190 (2015).
Rosario, F. J., Aye, I., Powell, T. & Jansson, T. Folate sensing by placental mTOR signaling: A novel mechanism linking maternal folate levels, placental function and fetal development. Placenta 36, 471 (2015).
Rosario, F. J. et al. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J Physiol 590, 1495–1509 (2012).
Kusinski, L. C., Jones, C. J., Baker, P. N., Sibley, C. P. & Glazier, J. D. Isolation of plasma membrane vesicles from mouse placenta at term and measurement of system A and system beta amino acid transporter activity. Placenta 31, 53–59, doi:10.1016/j.placenta.2009.11.006 (2010).
Schlabritz-Loutsevitch, N. E. et al. Development of a system for individual feeding of baboons maintained in an outdoor group social environment. J Med Primatol 33, 117–126, doi:10.1111/j.1600-0684.2004.00067.x (2004).
McDonald, T. J. et al. Effect of 30% nutrient restriction in the first half of gestation on maternal and fetal baboon serum amino acid concentrations. Br J Nutr 109, 1382–1388, doi:10.1017/S0007114512003261 (2013).
Jansson, T., Ekstrand, Y., Bjorn, C., Wennergren, M. & Powell, T. L. Alterations in the activity of placental amino acid transporters in pregnancies complicated by diabetes. Diabetes 51, 2214–2219 (2002).
Pantham, P. et al. Reduced placental amino acid transport in response to maternal nutrient restriction in the baboon. Am J Physiol Regul Integr Comp Physiol 309, R740–746, doi:10.1152/ajpregu.00161.2015 (2015).
van Uitert, E. M. & Steegers-Theunissen, R. P. Influence of maternal folate status on human fetal growth parameters. Mol Nutr Food Res, doi:10.1002/mnfr.201200084 (2012).
Hara, K. et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 (2002).
Chiang, G. G. & Abraham, R. T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J Biol Chem 280, 25485–25490, doi:10.1074/jbc.M501707200 (2005).
Fadden, P., Haystead, T. A. & Lawrence, J. C. Jr. Identification of phosphorylation sites in the translational regulator, PHAS-I, that are controlled by insulin and rapamycin in rat adipocytes. J Biol Chem 272, 10240–10247 (1997).
Sarbassov, D. D. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14, 1296–1302, doi:10.1016/j.cub.2004.06.054 (2004).
Wen, S. Y. et al. Rictor is an independent prognostic factor for endometrial carcinoma. Int J Clin Exp Pathol 7, 2068–2078 (2014).
Griner, E. M. & Kazanietz, M. G. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer 7, 281–294, doi:10.1038/nrc2110 (2007).
Ikenoue, T., Inoki, K., Yang, Q., Zhou, X. & Guan, K. L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J 27, 1919–1931, doi:10.1038/emboj.2008.119 (2008).
Lang, F. et al. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 86, 1151–1178, doi:10.1152/physrev.00050.2005 (2006).
Sarbassov, D. D., Guertin, D. A., Ali, S. M. & Sabatini, D. M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101, doi:10.1126/science.1106148 (2005).
Sancak, Y. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501, doi:10.1126/science.1157535 (2008).
Sancak, Y. et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303, doi:10.1016/j.cell.2010.02.024 (2010).
Efeyan, A., Zoncu, R. & Sabatini, D. M. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 18, 524–533, doi:10.1016/j.molmed.2012.05.007 (2012).
Roos, S., Lagerlöf, O., Wennergren, M., Powell, T. L. & Jansson, T. Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mTOR signaling Am J Physiol. Cell 297, C723–C731 (2009).
Rosario, F. J., Powell, T. L. & Jansson, T. Mechanistic target of rapamycin (mTOR) regulates trophoblast folate uptake by modulating the cell surface expression of FR-alpha and the RFC. Sci Rep 6, 31705, doi:10.1038/srep31705 (2016).
Rosario, F. J. et al. mTOR regulation of trophoblast transcriptome and mitochondrial respiration. FASEB J 28 (2014).
Dennis, P. B. et al. Mammalian TOR: a homeostatic ATP sensor. Science 294, 1102–1105 (2001).
Wiczer, B. M. & Thomas, G. Phospholipase D and mTORC1: nutrients are what bring them together. Sci Signal 5, pe13, doi:10.1126/scisignal.2003019 (2012).
Dann, S. G., Selvaraj, A. & Thomas, G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med 13, 252–259 (2007).
Jansson, T. & Powell, T. L. Role of placental nutrient sensing in developmental programming. Clin Obstet Gynecol 56, 591–601, doi:10.1097/GRF.0b013e3182993a2e (2013).
Gaccioli, F., Lager, S., Powell, T. L. & Jansson, T. Placental transport in response to altered maternal nutrition. J DOHaD 4, 101–115 (2013).
Desforges, M. & Sibley, C. P. Placental nutrient supply and fetal growth. The International journal of developmental biology 54, 377–390, doi:10.1387/ijdb.082765md (2010).
Gaccioli, F., Lager, S., Powell, T. L. & Jansson, T. Placental transport in response to altered maternal nutrition. J Dev Orig Health Dis 4, 101–115, doi:10.1017/S2040174412000529 (2013).
Ulrich, C. M. Folate and cancer prevention: a closer look at a complex picture. Am J Clin Nutr 86, 271–273 (2007).
Wien, T. N. et al. Cancer risk with folic acid supplements: a systematic review and meta-analysis. BMJ Open 2, e000653, doi:10.1136/bmjopen-2011-000653 (2012).
Acknowledgements
This study was supported by grants from NIH (R01HD078376).
Author information
Authors and Affiliations
Contributions
F.J.R., T.J. and T.L.P. made contribution to conception and design of the experiments, performed collection, analysis and interpretation of data. F.J.R., P.W.N., T.J. and T.L.P. wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosario, F.J., Nathanielsz, P.W., Powell, T.L. et al. Maternal folate deficiency causes inhibition of mTOR signaling, down-regulation of placental amino acid transporters and fetal growth restriction in mice. Sci Rep 7, 3982 (2017). https://doi.org/10.1038/s41598-017-03888-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-03888-2
- Springer Nature Limited