
Abstract
We sequenced the mitochondrial (mt) genome of the grass thrips, Anaphothrips obscurus, which is highly rearranged and differs from the four thrips species reported previously in the arrangement of both tRNA genes and a protein-coding gene, nad3, and in the copy number of the control region (CR). We reconstructed the phylogeny of the thrips with mt genome sequences, and used it as a framework to gain insights into mt genome evolution in thrips. It is evident that A. obscurus is less rearranged in mt genome organization than the other four known thrips. nad3 is in its ancestral location in A. obscurus but was translocated in other four thrips. Also, A. obscurus has one CR, which is ancestral to hexapods whereas other thrips have two or three CRs. All of the five thrips whose mt genomes have been sequenced to date are from the subfamily Thripinae, which represents about a quarter of the species richness in the order Thysanoptera. The high variation in mt genome organization observed in a subfamily challenges our knowledge about animal mt genomes. It remains to be investigated why mt genomes evolved so fast in the subfamily Thripinae and how mt genomes evolved in other lineages of thrips.
Similar content being viewed by others

Introduction
Most bilateral animals have the typical circular mitochondrial (mt) genome, 13–17 kb in size, containing 37 genes (i.e. 13 proteins, two rRNAs and 22 tRNAs) and a control region1, 2. Variation in the length of mt genomes is usually due to the length variation in the control region, which ranges from 70 bp (Orthoptera: Ruspoliadubia)3 to 4.6 kb (Diptera: Drosophila melanogaster) in insects4. Due to the small size, abundance in copy number, maternal inheritance mode, and fast evolutionary rate (up to 10 times faster than nuclear genomes5, 6), mt genomes have been explored widely in systematic, phylogenetic, diagnostic, and evolutionary studies7,8,9,10,11,12,13.
To date, more than 1,100 species from 28 of the 32 orders of insects (from RefSeq database) have been sequenced for complete mt genomes. Most of the insect mt genomes are highly conserved in genome organization and retain the ancestral condition for hexapods, or derive slightly from the ancestral condition1, 14. Major changes in mt genome organization, however, have been found in three paraneopteran orders: Pthiraptera (parasitic lice)15, 16, Psocoptera (booklice and barklice)17, 18, and Thysanoptera (thrips)19, 20. Fragmented mt genomes, which comprise 9–20 minichromosomes, have been found in parasitic lice21, 22; bipartite mt genomes have been found in booklice (Liposcelis)23 and the yellow tea thrips20. In both the parasitic lice and the booklice, the mt genomes are highly rearranged relative to the ancestral condition of hexapods.
The order Thysanoptera contains nearly 6,000 species24. A large number of these species (mostly in the family Thripidae) are phytophagous, feeding on plant tissues; some of them are crop pests or vectors of viral diseases25, 26. Also, many species of thrips (mostly in the family Phlaeothripidae) are fungivorous, feeding on fungi. Most of the other thrips are omnivorous, feeding on a wide range of food resources including mosses, gymnosperms, pollen, and arthropod prey27. Four species of thrips have been sequenced for mt genomes so far; all of them are highly rearranged and differ from each other but share the same arrangement of the protein-coding and rRNA genes except in one strain of the yellow tea thrips, Scirtothrips dorsalis 19, 20, 28, 29. The mt genomes of two strains of S. dorsalis have been sequenced; the typical single circular chromosome was found in the East Asia strain (EA1) while the South Asia strain (SA1) has the mt genome consisting of a large circle with 35 genes and a small circle with only three genes20. In addition, duplication or triplication of control region was found in the mt genomes of these thrips reported previously19, 20, 28, 29.
In this study, we sequenced the complete mt genome of the grass thrips, Anaphothrips obscurus, which is a cosmopolitan pest of cereal crops in many parts of the world30. Like the four other thrips species reported previously, A. obscurus also has a highly rearranged mt genome and furthermore, a different arrangement of protein-coding genes from the four thrips species reported previously. We reconstructed the phylogeny of A. obscurus with other four thrips species using mt genome sequences, and used the phylogeny as a framework to gain insights into mt genome evolution in thrips.
Results and Discussion
The mitochondrial genome of the grass thrips, A. obscurus
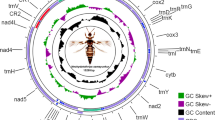
The full-length mt genome of A. obscurus was obtained by assembling the sequence reads from the two long PCR fragments, which were 7,188 bp and 7,409 bp in size respectively, together with the sequenced short PCR fragments, cox1 (759 bp) and rrnS (450 bp). Like in most animals, the mt genome of A. obscurus is circular, 14,890 bp in size and contains 37 genes (for 13 proteins, two rRNAs, 22 tRNAs) and a CR (Fig. 1). The mt genome of A. obscurus is A and T rich with 38.4% A, 39.8% T, 10.6% C and 11.3% G. Thirty-one genes are on one strand (majority strand) and six genes are on the other strand (minority strand). Ten pairs of genes appear to overlap by 1 to 10 bp: atp8-atp6, atp6-trnS 1 , cox1-trnL 2 , trnH-nad5, trnD-trnR, trnR-trnG, trnN-trnE, trnQ-trnI, trnA-trnF, and trnL 1 -trnC. Most of the tRNA genes can be folded into the conventional cloverleaf structure except for trnS 1 , trnV and trnN, in which the DHU arms form a loop, instead of a stem-loop. The CR is 174 bp long between trnS 1 and nad5, and is highly A and T rich (83.3%). Several features commonly found in the mt CRs of insects are present in A. obscurus, including a stem-loop, an AT-rich segment, a T-stretch, and three G[A]nT motifs31 (Fig. 2). Like other four species of thrips reported previously19, 20, 28, 29, A. obscurus also has a highly rearranged mt genome. Only three ancestral mt gene clusters of insects, containing nine genes in total, are retained in A. obscurus: cox1-trnL 2 -cox2, atp8-atp6, and nad4L-nad4-trnH-nad5; all other genes have rearranged relative to the ancestral mt genome organization of hexapods (Fig. 3).

The mitochondrial genome of the grass thrips, Anaphothrips obscurus. Direction of gene transcription is indicated by arrows. Protein-coding genes are shown as dark blue arrows, rRNA genes as orchid arrows, tRNA genes as coral arrows and large NC regions (>100 bp) as grey rectangles. tRNAs are labeled according to their single-letter abbreviations. The GC content is plotted using a black sliding window, as the deviation from the average GC content of the entire sequence. GC-skew is plotted using a colored sliding window (green and orchid color), as the deviation from the average GC-skew of the entire sequence. In the very inner cycle, the nucleotide positions in the mitochondrial genome start from the 5′-end of cox1.

Sequence features in the mitochondrial control region of the grass thrips, Anaphothrips obscurus. T-stretch is shown as double line, stem-loop as thin line, AT-rich sequence as thick line and G(A)nT motifs as boxes with blue color. The secondary structure of stem-loop is shown in the right of the figure. The numbers above sequences in the upper row indicate nucleotide positions starting from the 5′-end of control region; the number in the lower row indicate nucleotide positions starting from the 5′-end of cox1 in the mitochondrial genome.

The phylogenetic tree inferred from the mitochondrial genome sequences of five species of thrips (left) and linearized mitochondrial genome organization of each thrips (right). Numbers at the nodes are ML bootstrap values and Bayesian posterior probabilities from the PCGR (upper) and PCG12R (lower) datasets. tRNA genes are abbreviated using their corresponding one-letter amino acid names. The CRs were marked as red boxes. The tRNA genes that are conserved among the five species of thrips in their location relative to their upstream and/or downstream genes were in yellow. Genes are transcribed from left to right except those underlined, which are transcribed from right to left. A grey dot at the node indicates the MRCA of the four thrips (F. intonsa, F. ocidentalis, A. obscurus and S. dorsalis) and the origin of the translocated nad3.
Phylogenetic relationships among five species of thrips inferred from mt genome sequences
To help understand the evolution of mt genome organization in thrips, we inferred the phylogenetic relationships among the five thrips species whose mt genomes have been sequenced. The maximum likelihood (ML) and Bayesian inference (BI) trees have an identical topology with strong support for each of the nodes (Fig. 3). As expected, the two species of the genus Frankliniella, F. intonsa and F. ocidentalis, were grouped together. The two strains of S. dorsalis (EA1 and SA1) formed a group and were most closely related to Thrips imagini; together, they were more closely related to the two Frankliniella species than to A. obscurus.
Evolution of mt genome organization in thrips
Thysanoptera is one of the least studied orders among the 32 orders of insects for mt genomes. Prior to the current study, only four species of thrips have been sequenced for mt genomes19, 20, 28, 29. All of these thrips have highly rearranged mt genomes relative to the inferred ancestral mt genome organization of hexapods1, 32. Furthermore, the four species of thrips also differ from each other in mt genome organization, even between the two strains of the yellow tea thrips, S. dorsalis 19, 20, 28, 29. The mt genome of the East Asia strain (EA1) of S. dorsalis has a single circular chromosome whereas the South Asia strain (SA1) has two circular chromosomes: the small one is 921 bp in length and contains three genes, nad6, trnC and trnS 1 , and a CR; the larger one is 14,283 bp in length and contains all other mt genes, trnS 1 , and two CRs20. The variation among the EA1 strain of S. dorsalis and other three species of thrips is limited only to tRNA genes and CRs; the arrangement of protein-coding and rRNA genes are conserved among the four species of thrips (Fig. 4). A. obscurus, however, differs from the four species of thrips reported previously in the arrangement of both tRNA genes and one protein-coding gene, nad3. In the four species of thrips reported previously, nad3 was translocated to between cox1 and trnL 2 -cox2. In A. obscurus, however, nad3, cox1 and trnL 2 -cox2 retained their ancestral positions: nad3 is after cox3; cox1 and trnL 2 -cox2 are in one cluster with no genes in between (Fig. 3). As the four thrips species reported previously are more closely related to each other than either of them is to A. obscurus, the most plausible explanation is that the translocation of nad3 occurred in the most recent common ancestor (MRCA) of these four thrips species (Fig. 3).

The arrangement of protein-coding and rRNA genes in the mitochondrial genomes of thrips. Genes are transcribed from left to right except those underlined, which are transcribed from right to left. tRNA genes are excluded.
A number of studies have shown that in insects mt tRNA genes are much more mobile than protein-coding genes and rRNA genes33,34,35,36,37,38,39,40,41,42,43,44. Thrips are no exceptions and appear to go much further than other insects in tRNA-gene rearrangement. A. obscurus and the four thrips species reported previously all differ from each other in the arrangement of tRNA genes; only 10 of the 22 tRNA genes, trnL 2 , trnG-trnK, trnY, trnW, trnA-trnF, trnH, trnC and trnV, are conserved among the five thrips species in their locations relative to a protein-coding or rRNA gene upstream or downstream (Fig. 3). Even the two species of the same genus Frankliniella differ in the arrangement of three tRNA genes, trnI, trnT and trnQ (Fig. 3).
In addition to the genes, the CR (also called large non-coding region) also varies among the five thrips species. First, A. obscurus has one CR, T. imaginis and S. dorsalis EA1 have two CRs, whereas the two Frankliniella species and S. dorsalis SA1 have three CRs respectively (Fig. 3). Second, the two Frankliniella species and S. dorsalis SA1 differ in the location of one of their three CRs. T. imaginis and S. dorsalis EA1 also differ in the location of one of their two CRs. Third, the two CRs of T. imaginis have similar length and nearly identical sequence, so do the three CRs of each Frankliniella species. The two CRs of S. dorsalis (EA1), however, differ substantially in length (623 and 197 bp, respectively) and have no detectable sequence similarity to each other. In S. dorsalis SA1, the two CRs in the large mt chromosome differ in length (181 bp and 242 bp respectively) and there is no detectable sequence similarity between them. The CR3 in the small mt chromosome has high sequence similarity (98.35%) to part of CR1 on the large mt chromosome but differ in overall length (181 bp and 344 bp respectively). The CR upstream nad5 is present in all of the five thrips species and thus can be inferred to be ancestral to these thrips. A duplication of CR likely occurred in the MRCA of Frankliniella species, T. imaginis and S. dorsalis, which produced CR2 between cox3 and cob; the different location of CR2 in S. dorsalis EA1 and S. dorsalis SA1 can be accounted for by a translocation event of CR2. Another duplication of CR occurred likely in the MRCA of the two Frankliniella species, which produced CR3 between cob and nad2; in S. dorsalis SA1 strain, CR3 is likely due to the duplication of CR1 when the small chromosome was generated (Fig. 3).
It should be pointed out that the five thrips species whose mt genomes have been sequenced in previous studies and the current study are all from the same subfamily Thripinae of the family Thripidae. The order Thysanoptera contains nearly 6,000 described species in nine families of two suborders. The suborder Terebrantia contains eight families whereas the suborder Tubulifera contains only a single family45,46,47. Therefore, the unusual high variation in mt genome organization revealed so far from the five thrips species is essentially only from a small section of the order Thysanoptera. It remains to be investigated how mt genomes evolved in the thrips outside the subfamily Thripinae.
In summary, we sequenced the mt genome of the grass thrips, A. obscurus. The mt genome is highly rearranged and differs from the four thrips reported previously in the arrangement of both tRNA genes and a protein-coding gene, nad3, and in the copy number of CR. We reconstructed the phylogeny of the five thrips species with mt genome sequences, and used the phylogeny as a framework to gain insights into mt genome evolution and gene rearrangement in thrips. It is clear that A. obscurus is less rearranged in mt genome organization than the other four thrips species reported previously. nad3 is in its ancestral location in A. obscurus but was translocated in other four thrips. Also, A. obscurus has one CR, which is ancestral to hexapods whereas other thrips species have two or three CRs. Finally, all of the five thrips species whose mt genomes have been sequenced to date are from the subfamily Thripinae, which represents about a quarter of the order Thysanoptera in terms of species richness. The high variation in mt genome organization observed so far in the subfamily Thripinae is very unusual and goes against what we know in most other animals. It remains to be investigated why mt genomes evolved so fast in the subfamily Thripinae and how mt genomes evolved in other thrips.
Methods
Sample collection and amplification of mt genome
Specimens of A. obscurus were collected at Northwest A&F University, Yangling, China. Thrips specimens were stored at −80 °C in 95% ethanol. Genomic DNA was extracted from individual thrips using DNeasy Tissue kit (QIAGEN). Fragments of mt cox1 (759 bp) and rrnS (450 bp) were amplified by PCR with conserved primer pairs mtd6-mtd11 and 12SF-12SR (see Supplementary Table S1). These amplicons were sequenced at the Australian Genome Research Facility (AGRF). Two pairs of A. obscurus specific primers, RS45C1F-RS45rrnSF and RS45C1R-RS45rrnSR (see Supplementary Table S1), were then designed from the sequences of cox1 and rrnS fragments. Long PCR with these specific primers amplified the entire mt genome of A. obscurus with two gaps filled by the cox1 and rrnS fragments (42 bp and 251 bp respectively).
PrimeSTAR MAX DNA Polymerase (Takara) was used in the short PCR; the cycling conditions were: 94 °C for 1 min; 35 cycles of 98 °C for 10 sec, 50 °C for 15 sec, 68 °C for 30 sec, and 72 °C for 2 min. EmeraldAmp HS PCR Master (Takara) was used in the long PCR; the cycling conditions were: 94 °C for 1 min; 40 cycles of 98 °C for 10 sec, 60 °C for 30 sec, 68 °C for 8 min and 72 °C for 15 min. Negative controls were executed with each PCR experiment to detect DNA contamination and false positive amplicons. PCR amplicons were checked with 1.5% agarose-gel electrophoresis and were purified with Wizard® SV Gel and PCR Clean-Up System for sequencing.
Next-generation sequencing, mt genome assembly and gene identification
The two long PCR amplicons, 7,409 bp and 7,188 bp in size, were obtained at different time and were sequenced separately. The 7,409-bp amplicon was sequenced with Illumina Hiseq 2000 platform at the BGI, Hong Kong. The 7,188-bp amplicon was sequenced with Illumina Hiseq 2500 platform at the Berry Genomics, Beijing. Illumina sequence-reads obtained from the long PCR amplicons were checked for quality and then assembled into contigs with Geneious 6.0.648; the assembly parameters were: minimum overlap identity 98%; no gaps; maximum mismatches per read 2%; maximum ambiguity 2; and minimum overlap 100 bp.
We identified tRNA genes using tRNAscan-SE 1.2.149 and ARWEN50. A few tRNA genes that could not be identified by these programs were found by manual inspection for predicted anti-codon sequences and secondary structure found in other thrips. Protein-coding and rRNA genes were identified by BLAST searches of GenBank51. The annotated mt genome sequence of A. obscurus has been deposited in GenBank under accession numbers KY498001.
Phylogenetic analysis
We inferred the phylogenetic relationship of A. obscurus with four other species of thrips whose mt genomes have been reported (see Supplementary Table S2). The damsel bug, Alloeorhychus bakeri 52, which retained the ancestral mt genome organization of insects, was used as the outgroup. Each protein-coding gene was aligned individually by codons using MAFFT algorithm implemented in TranslatorX with L-INS-i strategy and default settings53. Poorly aligned sites were removed from the amino acid alignment before translating back to nucleotides using GBlocks in TranslatorX with default settings. The rRNA genes were individually aligned using MAFFT 7.0 online server with G-INS-i strategy54. Ambiguous positions in the rRNA gene alignments were filtered using GBlocks v0.9155 with default settings. Alignments of individual genes were concatenated as two datasets: 1) PCGR dataset, containing all three codon positions of 13 protein-coding genes, and two rRNA genes (11,767 bp in total); and 2) PCG12R dataset, which is the same as the PCGR dataset except the third codon positions of protein-coding genes are excluded (8,646 bp in total).
The two concatenated datasets were analyzed using maximum likelihood (ML) method implemented in RAxML-HPC2 8.1.1156, and Bayesian inference (BI) method implemented in MrBayes 3.2.357. Multiparametric bootstrapping analysis of 1,000 replicates was performed in RAxML based on the optimal tree with the best likelihood score and the GTRGAMMA model. For MrBayes analyses, two simultaneous runs of 10 million generations were conducted for the dataset using GTR + I + G model and trees were sampled every 1,000 generations, with the first 25% discarded as burn-in. Stationarity was considered to be reached when the average standard deviation of split frequencies was below 0.0158.
References
Boore, J. L. Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780 (1999).
Lavrov, D. V. Key transitions in animal evolution: a mitochondrial DNA perspective. Integr. Comp. Biol. 47, 734–743 (2007).
Zhou, Z., Huang, Y. & Shi, F. The mitochondrial genome of Ruspolia dubia (Orthoptera: Conocephalidae) contains a short A+ T-rich region of 70 bp in length. Genome 50, 855–866 (2007).
Lewis, O. L., Farr, C. L. & Kaguni, L. S. Drosophila melanogaster mitochondrial DNA: completion of the nucleotide sequence and evolutionary comparisons. Insect Mol. Boil. 4, 263–278 (1995).
Lin, C. P. & Danforth, B. N. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol. Phylogenet. Evol. 30, 686–702 (2004).
Wolfe, K. H., Li, W. H. & Sharp, P. M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA. 84, 9054–9058 (1987).
Cameron, S. L. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117 (2014).
Ma, C. et al. Mitochondrial genomes reveal the global phylogeography and dispersal routes of the migratory locust. Mol. Ecol. 21, 4344–4358 (2012).
Nelson, L. A. et al. Beyond barcoding: A mitochondrial genomics approach to molecular phylogenetics and diagnostics of blowflies (Diptera: Calliphoridae). Gene 511, 131–142 (2012).
Hurst, G. D. & Jiggins, F. M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. Biol. Sci. 272, 1525–1534 (2005).
Castro, L. R., Austin, A. D. & Dowton, M. Contrasting rates of mitochondrial molecular evolution in parasitic Diptera and Hymenoptera. Mol. Biol. Evol. 19, 1100–1113 (2002).
Shao, R., Dowton, M., Murrell, A. & Barker, S. C. Rates of gene rearrangements and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol. Biol. Evol. 20, 1612–1619 (2003).
Li, H. et al. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5, 8527 (2015).
Hassanin, A. Phylogeny of Arthropoda inferred from mitochondrial sequences: strategies for limiting the misleading effects of multiple changes in pattern and rates of substitution. Mol. Phylogenet. Evol. 38, 100–116 (2006).
Shao, R., Kirkness, E. F. & Barker, S. C. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 19, 904–912 (2009).
Cameron, S. L., Yoshizawa, K., Mizukoshi, A., Whiting, M. F. & Johnson, K. P. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genomics 12, 394 (2011).
Wei, D. D. et al. The multipartite mitochondrial genome of Liposcelis bostrychophila: insights into the evolution of mitochondrial genomes in bilateral animals. PLoS ONE 7, e33973 (2012).
Chen, S. C. et al. Evolution of multipartite mitochondrial genomes in the booklice of the genus Liposcelis (Psocoptera). BMC Genomics 15, 1 (2014).
Shao, R. & Barker, S. C. The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol. Biol. Evol. 20, 362–370 (2003).
Dickey, A. M. et al. A novel mitochondrial genome architecture in thrips (Insecta: Thysanoptera): extreme size asymmetry among chromosomes and possible recent control region duplication. BMC Genomics 16, 439 (2015).
Song, S. D., Barker, S. C. & Shao, R. Variation in mitochondrial minichromosome composition between blood-sucking lice of the genus Haematopinus that infest horses and pigs. Parasit. Vectors 7, 1 (2014).
Shao, R. et al. Fragmented mitochondrial genomes in two suborders of parasitic lice of eutherian mammals (Anoplura and Rhynchophthirina, Insecta). Sci. Rep. 5, 17389 (2015).
Shi, Y. et al. The mitochondrial genome of booklouse, Liposcelis sculptilis (Psocoptera: Liposcelididae) and the evolutionary timescale of Liposcelis. Sci. Rep. 6, 30660 (2016).
Mound, L. A. Thysanoptera (thrips) of the world – a checklist http://www.ento.csiro.au/thysanoptera/worldthrips.html (2007).
Jones, D. R. Plant viruses transmitted by thrips. Eur. J. Plant Pathol. 113, 119–157 (2005).
Zhao, M., Ho, H., Wu, Y., He, Y. & Li, M. Western flower thrips (Frankliniella occidentalis) transmits Maize chlorotic mottle virus. J. Phytopathol. 162, 532–536 (2014).
Mound, L. A. & Teulon, D. A. J. Thysanoptera as phytophagous opportunists In Thrips Biology and Management (eds Parker, B. L., Skinner, M. & Lewis T.) 3–20 (Plenum, 1995).
Yan, D. et al. The mitochondrial genome of Frankliniella intonsa: insights into the evolution of mitochondrial genomes at lower taxonomic levels in Thysanoptera. Genomics 104, 306–312 (2014).
Yan, D. et al. The complete mitochondrial genome sequence of the western flower thrips Frankliniella occidentalis (Thysanoptera: Thripidae) contains triplicate putative control regions. Gene 506, 117–124 (2012).
Mirab-Balou, M. & Chen, X. X. First description of the male of the wheat thrips, Anaphothrips obscurus (Thysanoptera: Thripidae). Zootaxa 2540, 65–68 (2010).
Zhang, D. X. & Hewitt, G. M. Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 25, 99–120 (1997).
Wolstenholme, D. R. Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 141, 173–216 (1992).
Carapelli, A., Comandi, S., Convey, P., Nardi, F. & Frati, F. The complete mitochondrial genome of the Antarctic springtail Cryptopygus antarcticus (Hexapoda: Collembola). BMC Genomics 9, 315 (2008).
Zhang, J., Zhou, C., Gai, Y., Song, D. & Zhou, K. The complete mitochondrial genome of Parafronurus youi (Insecta: Ephemeroptera) and phylogenetic position of the Ephemeroptera. Gene 424, 18–24 (2008).
Fenn, J. D., Song, H., Cameron, S. L. & Whiting, M. F. A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol. Phylogenet. Evol. 49, 59–68 (2008).
Cameron, S. L. et al. Mitochondrial genome organization and phylogeny of two vespid wasps. Genome 51, 800–808 (2008).
Kim, I. et al. The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect. Mol. Biol. 15, 217–225 (2006).
Lee, E. S. et al. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae). Gene 373, 52–57 (2006).
Hu, J. et al. The complete mitochondrial genome of the yellow coaster, Acraea issoria (Lepidoptera: Nymphalidae: Heliconiinae: Acraeini): sequence, gene organization and a unique tRNA translocation event. Mol. Biol. Rep. 37, 3431–3438 (2010).
Junqueira, A. C. M. et al. The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene 339, 7–15 (2004).
Beckenbach, A. T. & Joy, J. B. Evolution of the mitochondrial genomes of gall midges (Diptera: Cecidomyiidae): rearrangement and severe truncation of tRNA genes. Genome Biol. Evol. 1, 278–287 (2009).
Chen, Z. T., Mu, L. X., Wang, J. R. & Du, Y. Z. Complete mitochondrial genome of the citrus spiny whitefly Aleurocanthus spiniferus (Quaintance) (Hemiptera: Aleyrodidae): implications for the phylogeny of whiteflies. PLoS ONE 11, e0161385 (2016).
Li, H. et al. The complete mitochondrial genome and novel gene arrangement of the unique-headed bug Stenopirates sp. (Hemiptera:Enicocephalidae). PLoS ONE 7, e29419 (2012).
Song, F. et al. Rearrangement of mitochondrial tRNA genes in flat bugs (Hemiptera: Aradidae). Sci. Rep. 6, 25725 (2016).
Mound, L. A. Thysanoptera biodiversity in the neotropics. Rev. Biol Trop. 50, 477–484 (2002).
Hoddle, M. S., Mound, L. A. & Nakahara, S. Thysanoptera recorded from California, USA: a checklist. Fla. Entomol. 87, 317–323 (2004).
Mound, L. A. & Morris, D. C. The insect order Thysanoptera: classification versus systematics. Zootaxa 1668, 395–411 (2007).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Laslett, D. & Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Li, H. et al. The complete mitochondrial genome of the damsel bug Alloeorhynchus bakeri (Hemiptera: Nabidae). Int. J. Biol. Sci. 8, 93–107 (2012).
Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, W7–W13 (2010).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Stamatakis, A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690 (2006).
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Huelsenbeck, J. P., Ronquist, F., Nielsen, R. & Bollback, J. P. Bayesian inference of phylogeny and its impact on evolutionary biology. Science 294, 2310–2314 (2001).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos 31420103902, 31372229, 31401991), the Beijing Natural Science Foundation (Nos 6152016, 6144027), the Australia-China Science & Research Fund Group Mission (ACSRF00980) and the Australian Research Council (DP120100240).
Author information
Authors and Affiliations
Contributions
H.R.L., R.S., and W.C. designed and performed the research. H.R.L. and F.S. performed the molecular work. H.R.L., H.L., F.S. and R.S. analyzed the data. All authors discussed results and implications. H.R.L., H.L., R.S. and W.C. wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, H., Li, H., Song, F. et al. Novel insights into mitochondrial gene rearrangement in thrips (Insecta: Thysanoptera) from the grass thrips, Anaphothrips obscurus . Sci Rep 7, 4284 (2017). https://doi.org/10.1038/s41598-017-04617-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04617-5
- Springer Nature Limited
This article is cited by
-
The complete mitochondrial genome and novel gene arrangement in Nesodiprion zhejiangensis Zhou & Xiao (Hymenoptera: Diprionidae)
Functional & Integrative Genomics (2023)
-
The mitochondrial genome of Grapsus albolineatus (Decapoda: Brachyura: Grapsidae) and phylogenetic associations in Brachyura
Scientific Reports (2022)
-
Two complete mitochondrial genomes in Scolopendra and a comparative analysis of tRNA rearrangements in centipedes
Molecular Biology Reports (2022)
-
Two New Strains of Wolbachia Affecting Natural Avocado Thrips
Indian Journal of Microbiology (2021)
-
Rearrangement and evolution of mitochondrial genomes in Thysanoptera (Insecta)
Scientific Reports (2020)