
Abstract
Amelogenesis imperfecta (AI), characterized by a deficiency in the quantity and/or quality of dental enamel, is genetically heterogeneous and phenotypically variable. The most severe type, hypocalcified AI, is mostly caused by truncating mutations in the FAM83H gene. This study aimed to identify genetic mutations in four Chinese families with hypocalcified AI. We performed mutation analysis by sequencing the candidate FAM83H gene. Three novel mutations (c.931dupC, p.V311Rfs*13; c.1130_1131delinsAA, p.S377X; and c.1147 G > T, p.E383X) and one previously reported mutation (c.973 C > T, p.R325X) in the last exon of FAM83H gene were identified. Furthermore, constructs expressing Green fluorescent protein (GFP)-tagged wild-type and three novel mutant FAM83Hs were transfected into rat dental epithelial cells (SF2 cells). Wild-type FAM83H-GFP was localized exclusively in the cytoplasm, especially in the area surrounding the nucleus, while the mutant FAM83H-GFPs (p.V311Rfs*13, p.S377X, and p.E383X) were localized predominantly in the nucleus, with lower levels in the cytoplasm.
Similar content being viewed by others

Introduction
Dental enamel is the most highly mineralized tissue in the human body. Unique among the mineralized tissues, it is produced by ameloblasts, which have an epithelial origin. Enamel formation is a remarkably complex biomineralization process that is controlled by the regulated expression of many genes1. Studies have indicated that mutations in many of the genes can lead to amelogenesis imperfecta2, 3.
Amelogenesis imperfecta (AI) represents a group of inherited disorders that are clinically heterogeneous and exhibit enamel defects with or without systemic manifestation4, 5. Hypocalcified AI is the most severe form, in which the malformed enamel is cheesy-soft and stained, often abrading soon after tooth eruption. Mutations in FAM83H gene have been shown to cause autosomal-dominant hypocalcified AI (ADHCAI)6,7,8. Originally, the genes encoding enamel extracellular matrix proteins and proteases were primary candidates for AI, such as AMELX, ENAM, AMBN, AMTN, MMP20, and KLK4 9,10,11,12,13. FAM83H is the first-known gene encoding an intracellular protein that is associated with AI6. To date, more than 20 genes have been proven to be candidate genes in AI14,15,16,17,18,19,20,21,22. However, defects in FAM83H account for more AI cases than any other single gene23.
FAM83H is not specifically expressed in teeth, but is expressed in many tissues, such as the larynx, cervix, and bladder. Despite the relatively high prevalence of AI cases caused by FAM83H mutations, its function in enamel formation remains obscure. Based on bioinformatic analysis and structure and domain predictions, the characteristics of FAM83H show little indication of its potential function. Recently, some studies demonstrated that FAM83H might act as a scaffold and could regulate keratin cytoskeleton organization24, 25.
In the present study, we recruited four families with ADHCAI and identified three novel mutations (p.V311Rfs*13, p.S377X, and p.E383X) and one previously reported (p.R325X) FAM83H mutation causing enamel hypocalcification. The intracellular localization of the three novel mutant FAM83Hs were altered.
Results
Clinical and genetic findings
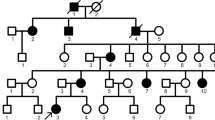
The proband of Family 1 was an 11-year-old girl who presented to our department with dental discolouration. Her enamel was generally rough and yellowish-brown, but normal-looking enamel could be identified in two lower incisors and two upper first permanent molars. According to her mother’s report, the enamel defects were inherited from the maternal grandmother (I:1). The proband’s mother (II:1), one aunt (II:3), one uncle (II:6), and one cousin (III:4) were all affected, indicating that the dental malformations were caused by a dominant mutation (Fig. 1A,B). Two affected family members (II:1, II:6) had restored their teeth with many dental crowns, but their unrestored teeth showed defective enamel (Fig. S1). Radiographically, the enamel layers of the proband were generally thin and less radio-opaque than the underlying dentin. Based on the enamel phenotype and the dominant pattern of inheritance, a diagnosis of hypocalcified AI was made, which implicated FAM83H as the possible pathogenic gene. By direct sequencing, we discovered a novel FAM83H frameshift mutation (c.931dupC, p.V311Rfs*13) that segregated with the disease phenotype (Fig. 1C–E).

Clinical and mutational analysis of Family 1. (A) Pedigree of Family 1, Proband (III:1). Black dots indicate members recruited for this study. (B) Oral photographs showing enamel malformations categorized as hypocalcified AI. (C,D) FAM83H exon 5 sequencing chromatogram of an unaffected family member (II:7) (C), and the proband (III:1) (D), revealing a novel frameshift mutation: c.931dupC, p.V311Rfs*13. (E) Panoramic radiograph of the proband taken at the age of 11.
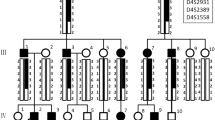
The proband of family 2 was an 8-year-old girl who was the only affected individual in the family (Fig. 2A). The enamel of her mixed dentition was yellow-brown discoloured and cheesy-soft. The defective enamel chipped off two erupting upper incisors, which made them lack a normal contour. Despite the irregular rough tooth surfaces, proband 2 showed relatively sound gingival condition (Fig. 2B). The panoramic radiograph showed that the enamel of unerupted teeth appeared to be of normal thickness but did not contrast with the dentin. We screened the entire coding region and adjacent intron boundaries of the FAM83H gene directly, and identified a novel heterozygous nonsense mutation (c.1130_1131delinsAA, p.S377X) (Fig. 2C–E). The mutational analysis confirmed the clinical diagnosis and revealed that the enamel defects in the proband were caused by a spontaneous de novo mutation in FAM83H.

Clinical and mutational analysis of Family 2. (A) Pedigree of Family 2, Proband (III:1). Black dots indicate members recruited for this study. (B) Oral photographs showing enamel malformations categorized as hypocalcified AI. (C,D) FAM83H exon 5 sequencing chromatogram of an unaffected family member (II:1) (C), and the proband (III:1) (D), revealing a novel nonsense mutation: c.1130_1131delinsAA, p.S377X. (E) Panoramic radiograph of the proband taken at the age of 8.
The proband of family 3, a 16-year-old boy, presented to our department with dental discolouration and sensitivity to thermal changes. He was also the only affected individual in the non-consanguineous family (Fig. 3A). His enamel was dark brown, and had chipped off from most of the incisors, whose incisal 1/2 was left particularly thin. Large amounts of dental calculus were accumulated on the irregular rough tooth surfaces, resulting in severe generalized gingivitis (Fig. 3B). The proband had an anterior cross bite and an impacted upper left canine, which was shown on the radiograph. Target gene mutational analysis revealed a novel FAM83H nonsense mutation (c.1147 G > T, p.E383X), which confirmed the diagnosis of hypocalcified AI (Fig. 3C–E).

Clinical and mutational analysis of Family 3. (A) Pedigree of Family 3, Proband (III:1). Black dots indicate members recruited for this study. (B) Oral photographs showing enamel malformations categorized as hypocalcified AI. (C,D) FAM83H exon 5 sequencing chromatogram of an unaffected family member (II:1) (C), and the proband (III:1) (D), revealing a novel nonsense mutation: c.1147 G > T, p.E383X. (E) Panoramic radiograph of the proband taken at the age of 16. The arrow indicates the impacted upper left canine.
The enamel of proband 4 showed similar defects to those of the probands from the above three families. However, she exhibited a relatively complex malocclusion, including anterior cross bite, severe dental crowding, and ectopic eruption (Fig. 4A,B). The radiographic density of the enamel was lower than the underlying dentin. Therefore, mutational analysis was performed by amplifying all of the coding regions and splice junctions of the candidate FAM83H gene. One previously reported disease-causing mutation (c.973 C > T, p.R325X) was identified (Fig. 4C–E).

Clinical and mutational analysis of Family 4. (A) Pedigree of Family 4, Proband (II:1). Black dots indicate members recruited for this study. (B) Oral photographs showing enamel malformations categorized as hypocalcified AI. (C,D) FAM83H exon 5 sequencing chromatogram of an unaffected family member (I:1) (C), and the proband (II:1) (D), revealing a previously reported nonsense mutation: c.973 C > T, p.R325X. (E) Panoramic radiograph of the proband taken at the age of 14.
Intracellular localization
The wild-type FAM83H-GFP fusion protein, expressed from vector pEGFP-C1, was localized exclusively in the cytoplasm, especially the area surrounding the nucleus. The expression patterns around the nucleus and in the cytoplasm were observed as filamentous and speckle-like shapes (Fig. 5). However, three mutant FAM83H-GFP proteins (p.V311Rfs*13, p.S377X, and p.E383X) were accumulated predominantly in the nucleus with lower levels in the cytoplasm, and their expression patterns were changed into speckle-like shapes (Fig. 5).

Subcellular localization of wild-type and mutant FAM83H-GFP in SF2 cells. Wild-type FAM83H-GFP was localized exclusively in the cytoplasm, especially the area surrounding the nucleus. The expression patterns around the nucleus and in the cytoplasm were present as filamentous and speckle-like shapes. The mutant FAM83H-GFPs (p.V311Rfs*13, p.S377X, p.E383X) were accumulated predominantly in the nucleus with lower levels in the cytoplasm, and their expression pattern was changed into speckle-like shapes. Nuclei were stained with Hoechst 33342.
Discussion
In the present study, we reported four FAM83H mutations, among which three (c.931dupC, p.V311Rfs*13; c.1130_1131delinsAA, p.S377X; c.1147 G > T, p.E383X) were newly identified. Consistent with previous studies, the novel mutations were all in the last exon of the FAM83H gene leading to a premature termination codon26. Among the genes that cause AI, FAM83H causes the highest percentage of cases and the most severe enamel defects. The genotype-phenotype correlation in predicting the causative gene in such cases has proved useful because of the association of hypocalcified enamel with mutations in FAM83H.
Clinically, dental and skeletal malocclusion in patients with AI has been observed in different populations. Open bite is the most commonly reported malocclusion27. In this study, the investigated AI patients showed various degrees of crowding, cross-bite, and tooth impaction, but no signs of obvious anterior open bite. It is notable that the nonsense mutation (c.973 C > T, p.R325X) identified in family 4 has been reported previously to cause hypocalcified AI in one Korean and two Chinese families, and is presumably an AI mutation hotspot in Asian population6, 8, 28. According to previous reports, several affected individuals in the Korean pedigree exhibited an anterior open bite; however, those in the Chinese pedigrees showed no signs of severe malocclusion. Although having the same AI-causing mutation, the proband in family 4 exhibited severe skeletal cross-bite and anterior crowding, which was quite different from the phenotypes of the reported cases.
To date, FAM83H has been discovered as the contributor to the aetiology of all reported hypocalcified AI cases. It is expressed in many tissues, while its mutations only cause phenotypes for dental enamel. The occurrence of truncating-only mutations in the last exon of FAM83H (except for one missense mutation) highly suggests a dominant negative effect or a gain of function effect as the underlying mechanism that triggers AI7, 29. Unlike the enamel matrix proteins and proteases important for enamel formation, FAM83H is a non-secreted protein because of the absence of a signal peptide26. Previous studies showed that wild-type FAM83H is localized in the cytoplasm, whereas the truncated FAM83H is expressed mainly in the nucleus7. It was shown that Fam83h overexpression did not cause an overt abnormality in enamel30. Moreover, inactivation of Fam83h expression in mouse models showed no apparent enamel defects25. These results further supported the pathological mechanism for hypocalcified AI in humans as a gain-of-function rather than a dominant negative effect25.
Recently, it has been revealed that FAM83H interacts with casein kinase I (CK-1) via an N-terminal motif, and has many phosphorylation sites in its C-terminus, which might be phosphorylated by CK-125. In vivo, FAM83H and CK-1ε are co-localized on keratin filaments in mouse dental enamel cells. In vitro, FAM83H also showed preferential localization to keratin filaments around the nucleus, which often extended to cell-cell junctions in human ameloblastoma cells24. Furthermore, two AI-causing mutants of FAM83H (p.S287X, p.Y297X) were found to disturb the keratin cytoskeleton and disrupt desmosomes. The importance of keratins and desmosomal proteins during amelogenesis, taken together with our results, suggested that FAM83H might function as a scaffold protein to guide CK-1 to its physiological sites, where it is involved in the formation of enamel by regulating the organization of the keratin cytoskeleton24. In this study, similar results were found whereby the three novel FAM83H truncations exhibited a prominent nuclear localization in rat dental epithelial SF2 cells; however, wild-type FAM83H was distributed mainly around the nucleus and in the cytoplasm, with expression patterns similar to filamentous shapes. The alteration in the subcellular localization of FAM83H probably increased its concentration in the nucleus, while reducing the amount of FAM83H in the cytoplasm to a level below what is required for its normal function. The nuclear localization might prevent the proper organization of the keratin cytoskeleton and the formation of desmosomes via failing to recruit CK-1 to keratin filaments, thus leading to disturbed enamel mineralization. Otherwise, the ectopic location of mutant FAM83H in the nucleus might cause it to interact with nuclear proteins and interfere with their functions7.
The involvement of FAM83H in colorectal cancer was reported recently31. FAM83H-knockout mice model showed a slightly scurfy skin25. However, neither colorectal cancer nor skin problems have been reported in AI patients with FAM83H mutations, including the affected individuals in our study. The predicted structures of the N- and C-termini of FAM83H are quite different, indicating that the two parts may play different roles in different tissues8. Further studies are needed to understand the functions of FAM83H during enamel calcification and its bioactivities in other tissues.
Materials and Methods
Subjects and DNA extraction
The study protocol was reviewed and approved by the Ethics Committee of Peking University School and Hospital of Stomatology (PKUSSIRB-201311083). Informed consent was obtained from all participants, including the guardians on behalf of the minors enrolled, and all methods were in accordance with the Declaration of Helsinki. Four unrelated probands, without any other health problems, and their available family members, were recruited. Of these families, one showed dominant transmission, and three had only a single affected individual. There were no co-segregating systemic diseases reported in these families. According to previous studies, FAM83H is involved in colorectal cancer31. In addition, mild skin problems were also found in Fam83h null mice25. However, no history of such diseases was reported by the affected individuals, either. Each of the probands underwent comprehensive dental and radiological examinations. A 4 mL peripheral blood sample was obtained from each participant. Genomic DNA was extracted from the peripheral blood using the TIANamp Blood DNA mini kit (Tiangen, Beijing, China), according to the manufacturer’s instructions.
Mutation analysis
The entire coding region and adjacent intron boundaries of the FAM83H gene were amplified by polymerase chain reaction (PCR) using Takara Ex-Taq (Takara Bio, Kyoto, Japan). Primers were designed using Primer 3 on the Web (http://bioinfo.ut.ee/primer3-0.4.0/). The products were purified and sequenced using an ABI 377 Automatic Sequencer (Applied Biosystems, Foster City, CA, USA). We analysed the insertion/deletion mutations with the help of Mutation Surveyor® (SoftGenetics, State College, PA, USA).
Cloning and mutagenesis of expression vectors
To transiently express green fluorescent protein fusions, the FAM83H coding sequencing was amplified from pCR2.1-FAM83H plasmid (kindly provided by Prof. Jan C-C. Hu, Department of Biologic and Materials Sciences, University of Michigan School of Dentistry) by PCR using forward (5′-GGAGATCTATGGCCCGTCGCTCTCAGAGCT-3′) and reverse (5′-GGAATTCACTTCTTGCTTTTGAACGTG-3′) primers containing BglII and EcoRI sites (underlined). The amplified product was subcloned into BglII and EcoRI sites of the pEGFP-C1 vector. The construct was completely sequenced to exclude random mutagenesis and was used as template for all other subcloning strategies. The mutants carrying V311Rfs*13 (forward, 5′-CGGGCCTCTCGTGGGCCGTCCCTGGGGTCGGGG-3′, reverse, 5′-CCCCGACCCCAGGGACGGCCCACGAGAGGCCCG-3′), S377X (forward, 5′-GGGCTGCGGCCGCTCTAACGGCGCCTGGAGGCCG-3′, reverse, 5′-CGGCCTCCAGGCGCCGTTAGAGCGGCCGCAGCCC-3′) and E383X (forward, 5′-GCGGCGCCTGGAGGCCTAGGCCGGGCCGGCTGG-3′, reverse, 5′-CCAGCCGGCCCGGCCTAGGCCTCCAGGCGCCGC-3′) mutations were constructed using the PrimeMut XL Site-directed Mutagenesis Kit (ExCell Biology, Shanghai, China), following the manufacturer’s instructions. Correct clones were confirmed by direct sequencing.
Intracellular localization
Rat dental epithelial SF2 cells (kindly provided by Prof. Fukumoto S, Department of Oral Health and Development Sciences, Tohoku University Graduate School of Dentistry, Japan) were cultured in Dulbecco’s Modified Eagle Medium/F12 medium (DMEM-F12) supplemented with 10% foetal bovine serum (FBS) and 1% penicillin and streptomycin at 37 °C under 5% CO2. For transient transfection, SF2 cells were trypsinised, counted, and seeded on a coverglass in 6-well plates at a density of 8 × 104 cells per well. After overnight incubation, 1.5 μg of plasmid DNA was transfected into the cells using Lipofectamine LTX (Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. Twenty-four hours after transfection, the cells were fixed with 4% paraformaldehyde, and nuclear DNA was stained in Hoechst 33342 (C1026, Beyotime, China) for 5 minutes. The samples were washed with phosphate buffered saline (PBS) three times for 5 minutes each. Confocal laser scanning was performed using a ZEISS-LSM 5 EXCITER fluorescence microscope. The experiment was performed in triplicate.
References
Paine, M. L. et al. Regulated gene expression dictates enamel structure and tooth function. Matrix biology 20, 273–292 (2001).
Hu, J. C., Chun, Y. H., Al Hazzazzi, T. & Simmer, J. P. Enamel formation and amelogenesis imperfecta. Cells, tissues, organs 186, 78–85, doi:10.1159/000102683 (2007).
Wright, J. T., Carrion, I. A. & Morris, C. The molecular basis of hereditary enamel defects in humans. Journal of dental research 94, 52–61, doi:10.1177/0022034514556708 (2015).
Witkop, C. J. Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. Journal of oral pathology 17, 547–553 (1988).
Aldred, M. J., Savarirayan, R. & Crawford, P. J. Amelogenesis imperfecta: a classification and catalogue for the 21st century. Oral diseases 9, 19–23 (2003).
Kim, J. W. et al. FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. American journal of human genetics 82, 489–494, doi:10.1016/j.ajhg.2007.09.020 (2008).
Lee, S. K. et al. FAM83H mutations cause ADHCAI and alter intracellular protein localization. Journal of dental research 90, 377–381, doi:10.1177/0022034510389177 (2011).
Song, Y. L., Wang, C. N., Zhang, C. Z., Yang, K. & Bian, Z. Molecular characterization of amelogenesis imperfecta in Chinese patients. Cells, tissues, organs 196, 271–279, doi:10.1159/000334210 (2012).
Cho, E. S. et al. Alteration of conserved alternative splicing in AMELX causes enamel defects. Journal of dental research 93, 980–987, doi:10.1177/0022034514547272 (2014).
Kida, M., Ariga, T., Shirakawa, T., Oguchi, H. & Sakiyama, Y. Autosomal-dominant Hypoplastic Form of Amelogenesis Imperfecta Caused by an Enamelin Gene Mutation at the Exon-Intron Boundary. Journal of dental research 81, 738–742, doi:10.1177/154405910208101103 (2002).
Poulter, J. A. et al. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Human molecular genetics 23, 5317–5324, doi:10.1093/hmg/ddu247 (2014).
Smith, C. E. et al. Deletion of amelotin exons 3-6 is associated with amelogenesis imperfecta. Human molecular genetics 25, 3578–3587, doi:10.1093/hmg/ddw203 (2016).
Seymen, F. et al. Novel MMP20 and KLK4 Mutations in Amelogenesis Imperfecta. Journal of dental research 94, 1063–1069, doi:10.1177/0022034515590569 (2015).
O’Sullivan, J. et al. Whole-Exome sequencing identifies FAM20A mutations as a cause of amelogenesis imperfecta and gingival hyperplasia syndrome. American journal of human genetics 88, 616–620, doi:10.1016/j.ajhg.2011.04.005 (2011).
Wang, S. et al. STIM1 and SLC24A4 Are Critical for Enamel Maturation. Journal of dental research 93, 94S–100S, doi:10.1177/0022034514527971 (2014).
Lee, S. K. et al. Novel WDR72 mutation and cytoplasmic localization. Journal of dental research 89, 1378–1382, doi:10.1177/0022034510382117 (2010).
Parry, D. A. et al. Mutations in C4orf26, encoding a peptide with in vitro hydroxyapatite crystal nucleation and growth activity, cause amelogenesis imperfecta. American journal of human genetics 91, 565–571, doi:10.1016/j.ajhg.2012.07.020 (2012).
Poulter, J. A. et al. Whole-exome sequencing, without prior linkage, identifies a mutation in LAMB3 as a cause of dominant hypoplastic amelogenesis imperfecta. European journal of human genetics: EJHG 22, 132–135, doi:10.1038/ejhg.2013.76 (2014).
Wang, S. K. et al. ITGB6 loss-of-function mutations cause autosomal recessive amelogenesis imperfecta. Human molecular genetics 23, 2157–2163, doi:10.1093/hmg/ddt611 (2014).
Dong, J. et al. DLX3 mutation associated with autosomal dominant amelogenesis imperfecta with taurodontism. American journal of medical genetics. Part A 133A, 138–141, doi:10.1002/ajmg.a.30521 (2005).
Parry, D. A. et al. Mutations in the pH-Sensing G-protein-Coupled Receptor GPR68 Cause Amelogenesis Imperfecta. American journal of human genetics 99, 984–990, doi:10.1016/j.ajhg.2016.08.020 (2016).
Seymen, F. et al. Recessive Mutations in ACPT, Encoding Testicular Acid Phosphatase, Cause Hypoplastic Amelogenesis Imperfecta. American journal of human genetics 99, 1199–1205, doi:10.1016/j.ajhg.2016.09.018 (2016).
Chan, H. C. et al. Target gene analyses of 39 amelogenesis imperfecta kindreds. European journal of oral sciences 119(Suppl 1), 311–323, doi:10.1111/j.1600-0722.2011.00857.x (2011).
Kuga, T. et al. FAM83H and casein kinase I regulate the organization of the keratin cytoskeleton and formation of desmosomes. Scientific reports 6, 26557, doi:10.1038/srep26557 (2016).
Wang, S. K. et al. Fam83h null mice support a neomorphic mechanism for human ADHCAI. Molecular genetics & genomic medicine 4, 46–67, doi:10.1002/mgg3.178 (2016).
Lee, S. K. et al. Mutational spectrum of FAM83H: the C-terminal portion is required for tooth enamel calcification. Human mutation 29, E95–99, doi:10.1002/humu.20789 (2008).
Poulsen, S. et al. Amelogenesis imperfecta - a systematic literature review of associated dental and oro-facial abnormalities and their impact on patients. Acta odontologica Scandinavica 66, 193–199, doi:10.1080/00016350802192071 (2008).
Zhang, C., Song, Y. & Bian, Z. Ultrastructural analysis of the teeth affected by amelogenesis imperfecta resulting from FAM83H mutations and review of the literature. Oral surgery, oral medicine, oral pathology and oral radiology 119, e69–76, doi:10.1016/j.oooo.2014.09.002 (2015).
Urzua, B. et al. Novel missense mutation of the FAM83H gene causes retention of amelogenin and a mild clinical phenotype of hypocalcified enamel. Archives of oral biology 60, 1356–1367, doi:10.1016/j.archoralbio.2015.06.016 (2015).
Kweon, Y. S. et al. Effects of Fam83h overexpression on enamel and dentine formation. Archives of oral biology 58, 1148–1154, doi:10.1016/j.archoralbio.2013.03.001 (2013).
Kuga, T. et al. A novel mechanism of keratin cytoskeleton organization through casein kinase Ialpha and FAM83H in colorectal cancer. Journal of cell science 126, 4721–4731, doi:10.1242/jcs.129684 (2013).
Acknowledgements
We thank the patients and their family members for their participation in this study. We are grateful to Prof. Jan C-C. Hu (Department of Biologic and Materials Sciences, University of Michigan School of Dentistry) and Prof. Fukumoto S (Department of Oral Health and Development Sciences, Tohoku University Graduate School of Dentistry) for providing FAM83H cDNA and SF2 cells respectively. This study was supported by the Beijing Natural Science Foundation (7164311) and the Young Scholars Foundation of Peking University School and Hospital of Stomatology (PKUSS20160104).
Author information
Authors and Affiliations
Contributions
W.X. initiated the study, W.X. and W.-W.J. performed the experiments, analyzed the data, and drafted the manuscript. Q.M. analyzed the data and revised the manuscript. Z.-Y.M. supervised the project, advised with regard to the experimental design and revised the manuscript. All authors read and proofed the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xin, W., Wenjun, W., Man, Q. et al. Novel FAM83H mutations in patients with amelogenesis imperfecta. Sci Rep 7, 6075 (2017). https://doi.org/10.1038/s41598-017-05208-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05208-0
- Springer Nature Limited