
Abstract
Transdifferentiation of hypertrophic chondrocytes into bone-forming osteoblasts has been reported, yet the underlying molecular mechanism remains incompletely understood. SHP2 is an ubiquitously expressed cytoplasmic protein tyrosine phosphatase. SHP2 loss-of-function mutations in chondroid cells are linked to metachondromatosis in humans and mice, suggesting a crucial role for SHP2 in the skeleton. However, the specific role of SHP2 in skeletal cells has not been elucidated. To approach this question, we ablated SHP2 in collagen 2α1(Col2α1)-Cre- and collagen 10α1(Col10α1)-Cre-expressing cells, predominantly proliferating and hypertrophic chondrocytes, using “Cre-loxP”-mediated gene excision. Mice lacking SHP2 in Col2α1-Cre-expressing cells die at mid-gestation. Postnatal SHP2 ablation in the same cell population caused dwarfism, chondrodysplasia and exostoses. In contrast, mice in which SHP2 was ablated in the Col10α1-Cre-expressing cells appeared normal but were osteopenic. Further mechanistic studies revealed that SHP2 exerted its influence partly by regulating the abundance of SOX9 in chondrocytes. Elevated and sustained SOX9 in SHP2-deficient hypertrophic chondrocytes impaired their differentiation to osteoblasts and impaired endochondral ossification. Our study uncovered an important role of SHP2 in bone development and cartilage homeostasis by influencing the osteogenic differentiation of hypertrophic chondrocytes and provided insight into the pathogenesis and potential treatment of skeletal diseases, such as osteopenia and osteoporosis.
Similar content being viewed by others


Introduction
Skeletal development occurs through two distinct processes: intramembranous ossification, which generates craniofacial bones and the lateral part of clavicles, and endochondral ossification, which produces the long bones of the limbs, the base of the skull, the vertebrae, the ribs and medial part of the clavicles1. Beginning with the condensation of undifferentiated mesenchymal cells, intramembranous ossification forms flat bones in mesenchymal tissue without a cartilaginous anlagen2. By contrast, during endochondral ossification, cells within the center of the condensation differentiate into chondrocytes that secrete extracellular matrix (ECM) rich in aggrecan and collagen type II (COL2α1)3,4. As endochondral ossification progresses, cells in the center of the condensation exit the cell cycle, undergo hypertrophic differentiation, and begin to produce ECM rich in collagen type X (COL10α1), which is calcified during skeletal development5. Terminally differentiated hypertrophic chondrocytes are ultimately removed from the cartilage template as the mineralized cartilage matrix is replaced by bone. It is well established that many of the hypertrophic chondrocytes are removed via programmed (apoptotic or autophagic) cell death6,7. However, there is increasing evidence that a substantial fraction of the hypertrophic chondrocytes in the growth plate transdifferentiate into osteoblasts that persist to produce trabecular bone and maintain mineral homeostasis8,9,10.
The formation of the cartilage anlage, its subsequent differentiation into mature chondrocytes, and the ultimate transdifferentiation of hypertrophic chondrocytes into functioning osteoblasts all depend on signals evoked by growth factors and other regulatory peptides11,12, though cell-cell, and cell-matrix interactions are also important13,14. Besides producing ECM proteins, terminally differentiated hypertrophic chondrocytes release matrix metalloproteases, such as MMP13, MMP9, and cathepsins15,16,17, and growth factors (e.g. VEGF, IGF1, RANKL, and FGFs), which influence matrix remodeling, osteoclast precursor recruitment and subsequent osteoclastogenesis at the chondro-osseous front, respectively18,19. The coordinated coupling of these events is critical for skeletal development and longitudinal bone growth. Although the exact molecular signals controlling these processes remain incompletely understood, the coordinated activation of sequential signaling pathways involving FGF, WNT/β-CATENIN, hedgehog, and BMP have been shown to be crucial20,21,22,23,24,25,26,27. Understanding how these signaling pathways are regulated will provide insight into skeletal development and the treatment of disease.
SHP2, encoded by PTPN11, is a widely expressed SH2 domain-containing non-receptor protein tyrosine phosphatase. Its orthologs are shared by nearly all vertebrates, functioning to regulate the viability, proliferation, differentiation, and migration of a wide variety of cells28,29. Accumulating evidence suggests a crucial role for SHP2 in skeletal development and maintenance. In humans, SHP2 gain-of-function (GOF) mutations cause Noonan Syndrome (NS), whose skeletal manifestations include short stature, scoliosis, pectus malformation and craniofacial abnormalities (macrocephaly, oral malformation and hypertelorism)30,31. Mice bearing SHP2 GOF mutations are small, and have increased skull length and hypertelorism32. Hyperactivation of ERK1/2 signaling reportedly causes these developmental abnormalities, and inhibition of ERK1/2 can rescue the skull defects in mice with NS32,33,34. Conversely, SHP2 loss-of-function (LOF) mutations have been linked to the benign cartilage tumor syndrome metachondromatosis, in both humans and mice35,36,37,38. Thus, alterations in the expression of SHP2 can be associated with both osteogenic and chondrogenic phenotypes. The specific role of SHP2 in skeletal development and disease is an important question that has yet to be explored.
The SOX9 and WNT/β-CATENIN signaling pathways are important regulators of chondrogenesis and osteoblastogenesis, respectively39. SOX9, a member of the high-mobility group (HMG) DNA-binding proteins, is considered a master chondrogenic transcription factor. SOX9 expression commences in mesenchymal precursors and persists through chondrocyte hypertrophy40,41,42. SOX9 promotes the expression of critical chondrocytic genes, including Col2α1, Col10α1, Matn3 and Acan 41,43, and inhibits the terminal differentiation and Vegfα expression of hypertrophic chondrocytes42,44. Downregulation of SOX9 in the hypertrophic layer of growth plate cartilage disinhibits vascular invasion and endochondral ossification42. β-CATENIN and RUNX2, on the other hand, are crucial for osteoblastogenesis and ossification45,46,47,48. Removal of β-CATENIN from hypertrophic chondrocytes impairs their osteogenic differentiation and trabecular bone formation, while sustained β-CATENIN activation leads to enhanced bone mineralization49. This suggests that β-CATENIN signaling is crucial for the transdifferentiation of hypertrophic chondrocytes.
SHP2 has been implicated in the regulation of SOX950,51, which is an antagonist of β-CATENIN45,52. However, it is unknown whether the abundance and transcriptional activity of SOX9 and β-CATENIN in hypertrophic chondrocytes, and whether the osteogenic differentiation of hypertrophic chondrocytes, are modulated by SHP2. To begin addressing these questions, we generated chondrocyte-stage-specific SHP2 deficient mice. Characterization of the skeletal phenotype of mice lacking SHP2 in COL2α1- and COL10α1-expressing cells led to the discovery of SHP2 as an important regulator of chondrocyte proliferation, maturation, and differentiation to bone forming osteoblasts.
Results
SHP2 deletion in COL2α1- but not COL10α1-expressing cells causes dwarfism, exostoses, and chondrodysplasia
A “Cre-loxP”-mediated gene deletion approach was used to study the function of SHP2 in cartilage and circumvent the embryonic lethality of global SHP2 deletion (Fig. S1)53,54. Control and SHP2 knockout mice were generated by crossing homozygous Ptpn11 floxed (Ptpn11 fl/fl) mice36 with SHP2 heterozygous (Ptpn11 fl/+) mice carrying Col2α1 or Col10α1 promoter-driven Cre recombinase55,56 (Fig. S1A). Breeders for the SHP2 ablation in chondrocytes were generated by crossing mice bearing a single Ptpn11 floxed allele (Ptpn11 fl/+) to transgenic mice in which Cre expression is under the control of Col2α1 55,57 or Col10α1 promoter56. Deleting SHP2 in proliferating chondrocytes required tamoxifen-inducible Tg(Col2α1-CreERt2) 55, since SHP2 deletion via Tg(COL2α1-Cre) 57 resulted in intrauterine death by day E11.5 (Fig. S1C,D ). The final breeding strategy yielded Ptpn11 fl/+ ;Tg(Col2α1-CreER T2), Ptpn11 fl/fl ;Tg(Col2α1-CreER T2), Ptpn11 fl/+ ;Tg(Col10α1-Cre), and Ptpn11 fl/fl ;Tg(Col10α1-Cre) compound mice, abbreviated as SHP2Col2α1ERCTR, SHP2Col2α1ERKO, SHP2Col10α1CTR, and SHP2Col10α1KO, respectively (Fig. S1B ). Tg(Col2α1-CreER T2) and Tg(Col10α1-Cre) mice express a functional Cre in COL2α1- and COL10α1-expressing chondrocytes as revealed by Rosa26 LacZ (R26lacZ) reporter58 (Fig. S2A–C ).
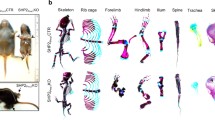
Age- and sex-matched SHP2Col2α1ERCTR and SHP2Col2α1ERKO mice received three doses of tamoxifen (TM) at the end of postnatal week 2 and cohorts of animals from SHP2Col2α1ERCTR, SHP2Col2α1ERKO, SHP2Col10α1CTR and SHP2Col10α1KO strains were euthanized at weeks 6, 8 and 10 for skeletal development evaluation. By 8 weeks SHP2Col2α1ERKO mice were significantly smaller (−17%) than the corresponding SHP2Col2α1ERCTR mice (13.63 ± 0.48 cm vs. 16.38 ± 0.75 cm, n = 4) (Fig. 1Ai,ii; Fig. S3A). Other features of the skeletal phenotype in adult SHP2Col2α1ERKO mice included scoliosis, small rib cages, and multiple joint dysplasias affecting the phalanges, metatarsals, knees, hips, vertebrae and chondrocostal junctions (Fig. 1Ai–x,Bi–iv ). Micro-CT (µ-CT) and x-ray imaging revealed the existence of exostoses at the knees and hips, deformed pelvises, shallow acetabulums and misshapen femoral heads and greater trochanters (Fig. 1Bi–iv and Fig. 2D). Of note, the bone mineral density in the SHP2Col2α1ERKO mice was reduced compared to sex- and age-matched SHP2Col2α1ERCTR mice (Fig. 1Aix,x), and the growth plates in the caudal vertebrae (Fig. 1Av,vi, arrows) and other tubular bones of the SHP2Col2α1ERKO mice were much wider than those of the SHP2Col2α1ERCTR mice. Similar morphological analysis was performed on the SHP2Col10α1KO and SHP2Col10α1CTR mice. In contrast to the SHP2Col2α1ERKO mice, there were no gross skeletal defects in the SHP2Col10α1KO mice through 10 weeks of age. However, the SHP2Col10α1KO mice did exhibit the same apparent reduction in bone mineralization compared to corresponding sex- and age-matched SHP2Col2α1ERCTR mice (Fig. S3B).

Mice with SHP2 deletion in COL2α1- and COL10α1-expressing cells have osteopenia, and SHP2 deletion in COL2α1- but not COL10α1-expressing chondrocytes causes dwarfism, exostoses, and multiple joint dysplasia. (A) X-ray images demonstrate dwarfism and scoliosis (i, ii), and chondrodysplasia of the metatarsophalangeal joints (iii, iv), caudal vertebrae (v, vi), hip (vii, viii) and knee joints (ix, x) in 8-week-old SHP2Col2α1ERKO mice, compared to age and sex-matched SHP2Col2α1ERCTR mice. Note that SHP2Col2α1ERKO mice exhibited slightly reduced bone mineral density, exostoses, and a broadened growth plate cartilage in the caudal vertebrae (v, vi, bars and arrows). (B) Representative µ-CT images show the shallow acetabular sockets, misshapen femoral heads and greater trochanters (i, ii) and exostoses (iii, iv) in SHP2Col2α1ERKO mice, compared to SHP2Col2α1ERCTR mice. (C) X-ray images demonstrate comparable morphology in 10-week-old SHP2Col10α1CTR and SHP2Col10α1KO mice. Note the slight decrease in bone mineral density in SHP2Col10α1KO mice compared to SHP2Col10α1CTR controls (n = 4).

SHP2 modulates the proliferation and hypertrophic differentiation of growth plate chondrocytes. (A) Representative images of mouse knee joint coronal sections stained with Safranin O/fast green demonstrating broad, disorganized growth plate cartilage, affecting both proliferating (P) and hypertrophic (H) chondrocytes (double arrow lines) in 6-week-old SHP2Col2α1ERKO mice compared to SHP2Col2α1ERCTR mice. SHP2Col2α1ERKO and SHP2Col2α1ERCTR mice received TM injection at the end of week 2. Note that the articular cartilage in the SHP2Col2α1ERCTR and SHP2Col2α1ERKO mice is grossly similar at this age. Images iii to vi are enlarged views of the color-bounded areas in images i and ii. n = 4. (B) Images of 8-week-old mouse proximal tibia sections stained with H&E, Safranin O/fast green demonstrating comparable growth plate and articular cartilage morphology in SHP2Col10α1CTR and SHP2Col10α1KO mice (n = 5). (C) Images of vertebral sections stained with Safranin O demonstrate enchondromas (arrows) developed in 10-week-old SHP2Col2α1ERKO mice (SHP2Col2α1ERCTR served as controls). Both mice received TM injection at the end of week 2 (n = 5). (D) Representative x-ray images of femur and tibia harvested from 10-week-old SHP2Col2a1ERKO mice demonstrating exostoses in the proximal femur and an enchondroma in the tibia (arrow) (n = 5).
Taken together, these data suggest that functional SHP2 is crucial for normal cartilage development and bone mineralization in COL2α1-expressing chondrocytes, but that it only influences bone mineral homeostasis in COL10α1-expressing chondrocytes.
Cartilage and bone development require SHP2
Having established the importance of SHP2 in both cartilage and bone development, we sought to explore the cellular mechanisms by which SHP2 regulates skeletogenesis. To start, knee joints from TM-treated 6- and 8-week-old SHP2Col2α1ERCTR and SHP2Col2α1ERKO mice were harvested and examined histologically. By 6 weeks of age the SHP2Col2α1ERCTR mice had well-organized growth plates and articular cartilage stained strongly positive for proteoglycan (Fig. 2Ai,iii,v ). In contrast, the columnar structure of the growth plate cartilage in the SHP2Col2α1ERKO mice was dysregulated, featuring significantly expanded proliferating and hypertrophic zones (Fig. 2Aii,iv; Fig. S4A ). Similar morphologic changes were observed in SHP2Col2α1ERKO mice bearing a R26mTG reporter (Fig. S4B). These drastic changes in the growth plate cartilage of SHP2Col2α1ERKO mice were not observed in SHP2Col10α1KO mice, which had normal-appearing cartilage with the exception of a slight increase in height of the hypertrophic layer of chondrocytes at 8 weeks (Fig. 2B ). Surprisingly, the articular cartilage in the knee joints in the SHP2Col2α1ERKO, SHP2Col10α1KO, and littermate control mice were comparable at 6 and 8 weeks of age. (Fig. 2Av,vi; Bv,vi and Fig. S4Biii,iv ), though the adult SHP2Col2α1ERKO mice developed exostoses and enchondromas at the metaphyseal regions of the long bones and vertebrae (Fig. 2C and D ).
To investigate whether SHP2 was required for cell proliferation in COL2α1-expressing physeal cartilage cells as it is in many other cell types28,29, pregnant females that had received 2 doses of TM injections at embryonic (E) day 13.5 and E15.5 were administered one dose of 5-ethynyl-2-deoxyuridine (EdU) at E16.5, and sacrificed at E17.5 to collect embryos. EdU staining of tibia frozen sections from these mice revealed an increased number of EdU+ cells in SHP2Col2α1ERKO mice, compared to SHP2Col2α1ERCTR mice, indicating that SHP2 deletion in COL2α1-expressing cells promoted EdU uptake and that under normal condition SHP2 functions as a negative regulator of chondrocyte proliferation (Fig. S4C ).
Given the apparent reduction in x-ray bone mineral density in the COL2α1+ and COL10α1+ cell-specific SHP2 knockout mice, we performed histomorphometric analysis on von Kossa-stained femoral sections from 10-week-old SHP2Col2α1ERKO and SHP2Col10α1KO mice. SHP2 deletion in both knockout strains was associated with a significant reduction of calcified trabecular bone compared to age- and sex-matched controls (Fig. 3A ). These observations were generally supported by microcomputed tomographic analysis (μCT), which revealed significant decreases in volumetric density (bone volume/total volume or BV/TV) and trabecular thickness (Tb.th.) in SHP2Col2α1ERKO and SHP2Col10α1KO mice compared to their corresponding controls (SHP2Col2α1ERCTR and SHP2Col10α1CTR, respectively) (Fig. 3B ). The structure model index (SMI) increased in the SHP2Col2α1ERKO mice. Trabecular number (Tb.N), space (Tb.S), and connectivity density (conn dens) were not significantly affected at this time point. Collectively, the histology, cell proliferation, and morphometric results demonstrate that SHP2 has a developmental stage-specific role in chondrogenesis and that SHP2 regulates bone mineral homeostasis in SHP2Col2α1ERKO and SHP2Col10α1KO through its effect on chondroid cells.

SHP2 deletion in COL2α1- and COL10α1-expressing cells compromised endochondral ossification. (A) Representative images of von Kossa/Fast red stained mouse femoral sections demonstrating a reduction of cancellous bones in 10-week-old SHP2Col2α1ERKO and SHP2Col10α1KO mice, compared to SHP2Col2α1ERCTR and SHP2Col10α1CTR mice. Mineralized bone was segmented from an 800 µm × 1200 µm rectangular region interested below the growth plate cartilage and quantified using NIH ImageJ software (n = 4, *p < 0.05, Student’s t test). (B) µ-CT analysis of proximal tibia morphology demonstrating reduced BV/TV and Tb.Th in 10-week-old SHP2Col2α1ERKO and SHP2Col10α1KO mice, compared to SHP2Col2α1ERCTR and SHP2Col10α1CTR controls (n = 4, *p < 0.05, Student’s t test). BV/TV: bone volume/total volume; Tb.Th: trabeculae thickness; Tb.N: trabeculae number; Tb.Sp: trabecule space; Conn.Dens: connectivity density; SMI: Structure model index.
SHP2 regulates the osteogenic differentiation of growth plate hypertrophic chondrocytes
The transdifferentiation of COL10α1+ growth plate chondrocytes into osteoblasts and osteocytes has been reported8,9,10 and has now been established as a process crucial for endochondral ossification and mineral homeostasis49,59. Our observation that mineralization was reduced in mice lacking SHP2 in Col10α1-expressing chondrocytes (radiographically and via μCT) prompted us to investigate whether SHP2 might influence the differentiation of hypertrophic chondrocytes into osteoblasts. To start, we performed fluorescent reporter-based cell lineage tracing using SHP2Col10α1CTR;R26ZsG and SHP2Col10α1KO;R26ZsG mice crossed to Sp7 mCherry reporter mice60 (SHP2Col10α1CTR;R26ZsG;Sp7mCherry and SHP2Col10α1KO;R26ZsG;Sp7mCherry, respectively). In these mice COL10α1(R26ZsG)-positive cells fluoresce green, OSTERIX (SP7mCherry)-positive cells fluoresce red, and COL10α1(R26ZsG)/OSTERIX(Sp7mCherry)-double positive cells fluoresce yellow. Yellow fluorescence identifies cells of COL10α1-expressing origin that subsequently began expressing OSTERIX, and thus were capable of participating in osteoblastogenesis and endochondral ossification.
Frozen sections from P0.5 day-old pups revealed COL10α1+/OSTERIX+ double positive (yellow) cells in the developing metaphyseal cancellous bone of both SHP2Col10α1CTR;R26ZsG;Sp7mCherry and SHP2Col10α1KO;R26ZsG;Sp7mCherry mice, but not in the growth plate and articular cartilage. Importantly, there were fewer, sparsely scattered COL10α1+/OSTERIX+ double positive cells in the SHP2Col10α1KO;R26ZsG; Sp7mCherry mice compared to the SHP2Col10α1CTR;R26ZsG;Sp7mCherry controls (mean± stdv of percentage: 56.75 ± 10.58 vs. 43.17 ± 13.05. *p < 0.05, Student’s t test) (Fig. 4 A–C). Similar results were seen in P9.5 SHP2Col10α1CTR;R26mTG and SHP2Col10α1KO; R26mTG newborns. Intriguingly, GFP+ cells accumulated in the hypertrophic layer of growth plate cartilage in SHP2Col10α1KO;R26mTG mice (Fig. S5 ). Collectively, our cell lineage tracing studies suggest that SHP2 plays an important role in regulating the differentiation of terminal hypertrophic chondrocytes into osteoblasts, primarily affecting the metaphyseal trabecular bone formation; it has minimal effect on cortical bone.

SHP2 deletion in COL10α1-expressing chondrocytes arrests their osteogenic differentiation. (A) Representative fluorescent images of mouse tibia sections demonstrating the abundance and distribution of COL10α1-expressing chondrocytes (ZsG+), OSTERIX+ osteoblasts (RFP+) and osteoblasts derived from COL10α1-expressing chondrocytes (ZsG+/RFP+, yellow) in P0.5-day-old SHP2Col10α1CTR;R26ZsG;Sp7mCherry and SHP2Col10α1KO;R26ZsG;Sp7mCherry mice (n = 3). (B) Enlarged views of the corresponding boxed areas in A demonstrating the reduction of ZsG+/RFP+ double positive (yellow) cells on trabecular bone surfaces in SHP2Col10α1KO;R26ZsG;Sp7mCherry mice, compared to SHP2Col10α1CTR;R26ZsG;Sp7mCherry controls (n = 3). The scale bar on the right is 500 µm. Only cells within the 500 µm × 50 µm rectangular region of interest were counted. (C) Bar graphs depicting the number of COL10α1+-cell-derived osteoblasts. SHP2 deletion in COL10a1-expressing chondrocytes compromised their osteogenic differentiation, compared to the controls (n = 3, *p < 0.05, Student’s t test).
SHP2 deletion in COL10α1-expressing hypertrophic chondrocytes promotes chondrocytic but represses osteogenic gene expression
After establishing that SHP2 influences the osteogenic differentiation of hypertrophic chondrocytes, we next investigated SHP2-related changes in gene and protein expression. To do so, we examined the effect of SHP2 deletion on osteogenic and chondrogenic marker expression in Col10α1-expressing chondrocytes using in situ hybridization and immunohistochemistry. We found that Col2α1 was upregulated in the metaphyseal region of the tibias from P1.5 SHP2Col10α1KO;R26mTG mice compared to SHP2Col10α1CTR;R26mTG controls, and that the osteogenic genes Ibsp, Mmp13, Runx2, and Ctnnb1 were all down-regulated (Fig. 5 ). Staining of sections from P1.5 newborns revealed that SOX9 protein was significantly increased in the hypertrophic chondrocytes of SHP2Col10α1KO mice (Fig. 6A, red bar; Fig. S9 ), and that the transcript for Sox9 was increased in the upper hypertrophic chondrocytes (Fig. 6B ). Both Sox9 and SOX9 were expressed in similar quantities in the physeal proliferating chondrocytes in the SHP2Col10a1CTR and SHP2Col10a1KO mice (Fig. 6A,B). Immunostaining for β-CATENIN was non-informative as the protein levels were sufficiently low that they were beyond detection.

SHP2 deletion in COL10α1-expressing chondrocytes sustains the expression of chondrocytic but represses the expression of osteogenic genes. (A) Representative images of P0.5-day-old mouse tibia sections hybridized in situ with the probes indicated to assess the abundance of gene transcripts. (B) Enlarged views of corresponding boxed areas in A. (C) Bar graphs demonstrating an increase of Col2α1 and a decrease of Ibsp, Mmp13, Runx2 and Ctnnb1 in the SHP2Col10α1KO; R26mTG mice, compared to the controls (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001, Student’s t test.). Scale bar:100 µm.

SHP2 deletion in hypertrophic chondrocytes increases SOX9 abundance and reducing Sox9 in hypertrophic chondrocytes restores osteogenic gene expression in SHP2Col10α1KO mice. (A) Representative images of P1.5 mouse tibia sections immunostained with SOX9 antibody (left), with enlarged views of the boxed areas (middle). Quantification of SOX9 expression in hypertrophic chondrocytes is shown on the right. SOX9 expression was elevated in the hypertrophic chondrocytes of SHP2Col10α1KO (KO) mice, compared to SHP2Col10α1CTR (CTR). (n = 3, **p < 0.01, Student’s t test). (B) Images of the proximal tibia sections demonstrating the abundance of Sox9 in P0.5 SHP2Col10α1CTR and SHP2Col10α1KO mice. In situ hybridization was carried out using RNAscope technology. Transcript abundance was visualized by DAB staining of HRP-conjugated DNA probes. Note that the height of the hypertrophic zone was increased in SHP2Col10α1KO mice, accompanied by an expansion of the Sox9-expressing top layer (between doted lines) of the hypertrophic zone. Expression of Sox9 and SOX9 was comparable in proliferating chondrocytes between SHP2Col10α1CTR and SHP2Col10α1KO mice, n = 3. (C) Western blots (left) and bar graphs (right) demonstrating the expression of SHP2 and SOX9 in immortalized SHP2WTSOX9WT, SHP2WTSOX9cKO, SHP2KDSOX9WT and SHP2KDSOX9cKO chondrocytes (See Supplementary Fig. 10 for the full-length blots). SHP2 was efficiently knocked down in SHP2KDSOX9WT and SHP2KDSOX9cKO chondrocytes, and SOX9 was robustly deleted in SHP2WTSOX9cKO and SHP2KDSOX9cKO chondrocytes with tamoxifen treatment. Importantly, SHP2 knockdown in SHP2KDSOX9WT cells significantly increases the level of SOX9. Note that SHP2 was markedly knocked down in SHP2KDSOX9WT and SHP2KDSOX9cKO chondrocytes and SOX9 was robustly deleted in SHP2WTSOX9cKO and SHP2KDSOX9cKO chondrocytes upon TM treatment. Importantly SHP2 knockdown in SHP2KDSOX9WT chondrocytes significantly increased SOX9 abundance (red arrow) (*p < 0.05, Student’s t test; n = 3). (D) qRT-PCR data show the increased transcript abundance of chondrocytic genes Acan and Col10α1 in SHP2KDSOX9WT chondrocytes in which SOX9 was upregulated upon SHP2 knockdown. SHP2WTSOX9WT chondrocytes served as controls. Note that the elevated abundance of Acan and Col10α1 in SHP2KDSOX9WT chondrocytes was rescued by SOX9 deletion in SHP2KDSOX9cKO chondrocytes (n = 3, *p < 0.05, Student’s t test).
To corroborate our in situ and immunostaining data, we sought further validation of the regulation of SOX9 and chondrocyte gene expression by SHP2 in vitro. We were forced to use proliferating rather than hypertrophic chondrocytes for this signaling study due to the technical difficulty of obtaining homogenous populations of hypertrophic chondrocytes. To do so, we established ribcage chondrocyte cell lines that expressed either normal (SHP2WT) or reduced (SHP2KD) levels of SHP2 (via shRNA against murine SHP2) that could be induced (or not) to ablate SOX9 expression by tamoxifen administration (SOX9cKO and SOX9WT). Our nomenclature for the cell lines was SHP2WTSOX9WT, SHP2WTSOX9cKO, SHP2KDSOX9WT, and SHP2KDSOX9cKO for the wild type, SOX9 conditional knockout, SHP2 knockdown, and dual SHP2 knockdown/SOX9 conditional knockout, respectively (Fig. S7A,B ). SOX9 knockout was effected by 96 hours of exposure to 4-OH tamoxifen (TM) to induce Sox9 deletion, and total cell lysates were analyzed by western blotting and total RNA was used for qRT-PCR.
SOX9 was robustly deleted upon TM treatment and SHP2 was effectively knocked down by the shRNA (Figs 6C; S10 ). As anticipated, the transcript level for the chondrocytic genes Acan and Col10α1 decreased significantly in the tamoxifen-mediated SOX9 deleted SHP2KDSOX9cKO and SHP2WTSOX9cKO chondrocytes (Fig. 6D ). Importantly, the abundance of both SOX9 and the transcripts for the chondrocytic genes Acan and Col10α1 increased significantly in the SHP2KDSOX9WT chondrocytes compared to the SHP2WTSOX9WT controls (Fig. 6C,D ), providing compelling evidence that SHP2 modifies Acan and Col10α1 expression via SOX9. Similar results were also obtained on E17.5 embryos in which SHP2 was deleted in COL2α1-expressing cells (Fig. S6 ).
Taken together, our cell lineage tracing and in situ hybridization studies suggest that SHP2 influences osteoblastogenesis and endochondral ossification, at least in part, by promoting the differentiation of hypertrophic chondrocytes into osteoblasts, and that SHP2 modulates the osteogenic differentiation of hypertrophic chondrocytes by indirectly influencing the expression and activity of osteogenic transcription factors via SOX9.
Haploinsufficiency of Sox9 rescues the osteogenic differentiation of hypertrophic chondrocytes in SHP2Col10α1KO mice
To confirm our finding that SHP2 acts through SOX9 to modulate the osteogenic differentiation of hypertrophic chondrocytes, we carried out a genetic rescue experiment in which Sox9 haploinsufficiency would offset the chondrogenic effect of SHP2 deletion. Sox9 wild type (Sox9 +/+) and heterozygous Sox9 floxed mice (Sox9 fl/+) were crossed to SHP2Col10α1CTR;R26ZsG and SHP2Col10α1KO;R26ZsG mice, to yield mice in which both SHP2 and Sox9 were modulated in Col10α1-expressing cells. Characterization of the mice in which Sox9 was halved in the hypertrophic chondrocytes demonstrated that the transcript levels for the osteogenic genes Ctnnb1, Ibsp and Mmp13, were significantly increased (in situ hybridization and quantification using NIH ImageJ), as were the number of GFP+ cells in the metaphyseal cancellous bone (Fig. S8 ). Presumably these GFP+ cells are osteoblasts that had transdifferentiated from hypertrophic chondrocytes labeled with the R26ZsG reporter. Lineage tracing using tibias from P0.5 newborns showed that loss of one allele of Sox9 did not significantly affect the number of ZsGreen+ cells (Fig. S8A top) or/and the abundance of osteogenic gene transcripts, Ctnnb1, Ibsp, and Mmp13 (Fig. S8B top) in the metaphyseal cancellous bone of the SHP2Col10α1CTR;SOX9fl/+;R26ZsG mice compared to SHP2Col10α1CTR;SOX9+/+;R26ZsG controls. In contrast, both the number of ZsGeen+ cells (Fig. S8 bottom) and the abundance of osteogenic marker genes markedly increased in the corresponding regions of the SHP2Col10α1KO;SOX9fl/+;R26ZsG mice, compared to SHP2Col10α1KO;SOX9+/+;R26ZsG controls (Fig. 7B, bottom, S8 bottom). Collectively these data provide convincing evidence that SHP2 regulates osteogenic differentiation in the primary spongiosa, a process that has been shown to include transdifferentiation of hypertrophic chondrocytes. Importantly, they also show that this regulation is mediated in part by SOX9.

Haploinsufficiency of Sox9 in SHP2 deficient hypertrophic chondrocytes restores osteogenic marker gene expression. (A) Representative images of P1.5 mouse tibia frozen sections hybridized in situ to the probes indicated demonstrating the abundance of Ctnnb1, Ibsp, and Mmp13. The expression of Ctnnb1, Ibsp, and Mmp13 was reduced in SHP2Col10α1KO;Sox9+/+ (SHP2−SOX9+) mice, compared to SHP2Col10α1CTR;Sox9+/+ (SHP2+ SOX9+) controls. Removal of one allele of Sox9 from the hypertrophic chondrocytes in SHP2Col10a1KO;Sox9+/− mice (SHP2−SOX9−) restored the expression of Ctnnb1, Ibsp, and Mmp13 comparable to SHP2+ SOX9 + controls. (B) In situ hybridization data quantified using NIH ImageJ. n = 3, **p < 0.01, Student’s t test. (C) Diagram depicting the working model by which SHP2 modifies the signals evoked by receptor tyrosine kinases (RTK), extracellular matrix proteins (ECM) and cytokines, and the expression of SOX9 and β-CATENIN. Tilting the expression of SOX9 and β-CATENIN in the hypertrophic chondrocytes due to SHP2 deletion favors chondrogenic but represses osteogenic differentiation. S:Serine, Y:Tyrosine.
Discussion
SHP2 is ubiquitously expressed, but little is known about its function in the skeletal system. Recently SHP2 loss-of-function mutations have been linked to the cartilage tumor syndrome metachondromatosis35,36,37,38 and scoliosis37, suggesting a crucial role for SHP2 in the skeleton. To further study the role of SHP2 in cartilage, we adopted a genetic loss of function approach and ablated SHP2 expression in COL2α1+ (proliferating) and COL10α1+ (hypertrophic) chondrocytes respectively in mice. Phenotypic characterization showed that mice lacking SHP2 in the hypertrophic chondrocytes appeared normal through 10 weeks of age with the exception of a slight decrease in bone mineral density. In contrast, mice deficient SHP2 in the proliferating chondrocytes had a drastic skeletal phenotype. Developmental SHP2 deletion in COL2α1+ chondrocytes caused midgestation lethality (around E11.5), but postnatal SHP2 deletion in the same cell population led to scoliosis, expansion of the growth plate cartilage affecting both proliferating and hypertrophic chondrocytes, chondrodysplasia, enchondromas, exostoses, and reduced bone mineral density. These findings are consistent with published work showing the formation of scoliosis and enchondroma-like lesions on the vertebrae of SHP2Col2α1ERKO mice and altered chondrocyte maturation and disorganized vertebral growth plates37,50. The distinct bone and cartilage phenotypes in the mice lacking SHP2 in proliferating and hypertrophic chondrocytes suggest that SHP2 has a developmental stage-specific role in chondrogenesis and bone mineral homeostasis and that the cellular signaling networks wired in the proliferating and hypertrophic chondrocytes are complex.
It’s not surprising that SHP2 deletion in COL2α1-expressing cells during development caused embryonic lethality in mice. It’s well known that SHP2 is essential for early embryogenesis and for multiple organs/tissues development53,54; Col2α1 promoter is reportedly active not only in chondroid cells, but also in the mesenchyme of the frontal nasal mass, spinal neural tube, and the most ventral and dorsal parts of the forebrain during early embryogenesis61,62,63. The midgestation lethality of SHP2Col2α1KO mice and the survival of mice with postnatal SHP2 deletion in the COL2α1-expressing cells suggest that the lethality likely resulted from SHP2 deletion in COL2α1+ non-chondroid cells.
Although the transdifferentiation of hypertrophic chondrocytes into osteoblasts8,9,10 and their involvement in endochondral ossification and mineral homeostasis have been reported49,59, the molecular mechanism(s) that regulates this process remains incompletely understood. Our data suggest that SHP2 is a key regulator for the osteogenic differentiation of hypertrophic chondrocytes. Mice deficient in SHP2 in COL10α1+ chondrocytes had a reduction of bone mineral density, BV/TV and Tb.Th, and an increased layer of hypertrophic chondrocytes within the growth plate. Indeed, our cell lineage tracing studies revealed that SHP2 deletion in COL10α1+ chondrocytes significantly decreased the number of osteoblasts marked by both Col10α1-Cre;R26ZsG and Sp7 mCherry dual reporters in the cancellous bone region, which was accompanied by a reduction of osteogenic marker gene transcripts Ibsp, Runx2 and Ctnnb1. Collectively these data indicate that SHP2 is a key regulator for the differentiation of hypertrophic chondrocytes into osteoblasts.
Commitment of condensed mesenchymal cells into chondrocytes and osteoblasts requires several fate decisions, which are modified by multiple signaling pathways and transcription factors, including SOX9 and β-CATENIN39,40,41,42. SOX9 is one of the earliest makers for mesenchymal condensation and for the commitment of mesenchymal progenitors to osteochondroprogenitors43. With the progression of skeletal development, SOX9 is mainly restricted to the epiphyseal proliferating chondrocytes, maintains chondrocyte columnar proliferation41,43 and drives cell hypertrophy42. Although SOX9 persists after Sox9 expression ceases in the hypertrophic chondrocytes in the growth plate, it was reported that Sox9 is expressed in the upper layer of the COL10α1+ hypertrophic zone42. In hypertrophic chondrocytes, SOX9 keeps β-CATENIN in check and inhibits osteoblastic differentiation42. Conversely β-CATENIN promotes the osteogenic commitment of osteochondroprogenitors39,52,64 and hypertrophic chondrocytes; deletion of β-CATENIN in COL10α1+ cells impairs trabecular bone formation49,59,65. Importantly, SOX9 and β-CATENIN reciprocally regulate one another in an antagonistic manner. Disrupting the balance of SOX9 and β-CATENIN signaling in skeletal cells modifies the chondrogenic or osteogenic fate decision45,66,67.
We examined the effect of SHP2 deletion in COL10+ chondrocytes via the expression of SOX9, β-CATENIN, Sox9 and Ctnnb1. Although β-CATENIN and Ctnnb1 were below the level of detection in these cells, SOX9 and Sox9 was significantly upregulated in the hypertrophic zone and in the upper layer of the hypertrophic zone, respectively. Given that β-CATENIN signaling is essential for the osteogenic differentiation of hypertrophic chondrocytes and the reciprocally antagonistic regulatory relationship of β-CATENIN and SOX9, we therefore proposed a model (Fig. 7C ) wherein SHP2 regulates the osteogenic differentiation of hypertrophic chondrocytes by balancing SOX9 and β-CATENIN signaling. This model is supported by the results of our rescue experiment, where removal of one allele of Sox9 in the SHP2 deficient hypertrophic chondrocytes restored the expression of osteogenic marker genes Ctnnb1 and Ibsp. However, this experiment didn’t exclude the possibility that the increase of SOX9 is due to the reduction of β-CATENIN as the consequence of SHP2 deletion in COL10α1+ chondrocytes. Indeed, SHP2 is reported to regulate the tyrosyl phosphorylation of β-CATENIN and its stability in other types of cells68. The work of evaluating how SHP2 regulates the expression of SOX9 and β-CATENIN in chondrocytes is ongoing in our laboratory.
The distinct skeletal phenotypes in mice lacking SHP2 in the COL2α1- and COL10α1-expressing cells suggest that SHP2 has a development-stage-specific effect in chondrogenesis. Proliferating and hypertrophic chondrocytes are distinct in several aspects. First, the proliferation potential of chondrocytes markedly declines when they gradually transition from the proliferating to the hypertrophic stage. And second, this morphological switch is accompanied by the qualitative and quantitative change of gene expression profiles, which affect certain ligands69,70, receptors71,72, extracellular matrix proteins and transcription factors. Some of them or their downstream effectors may serve as the target of SHP236,66,73. This may explain why the phenotypic outcomes are different in mice with SHP2 ablation in the COL2α1- and COL10α1-expressing cells73,74,75.
The etiology of enchondromas and exostoses, common benign cartilaginous lesions, remain elusive. Anatomically, exostoses and enchondromas arise adjacent to the growth plate cartilage and resemble it morphologically. This suggests that growth plate chondrocytes may be candidate cells-of-origin and that dysregulation of cellular signaling pathways that modulate growth plate chondrocytes could contribute to the pathogenesis of these lesions76. The findings from this study support these views. Mice with SHP2 deletion in COL2α1+ chondrocytes grew exostoses and enchondromas at the metaphysis of the long bones, affecting hip, tibia, phalanges and vertebrae; chondrocytes with SHP2 knockout or knockdown had an elevated cell proliferation and chondrocytic gene expression. Most importantly, SHP2 knockout or knockdown in chondrocytes in vivo and in vitro significantly increases the expression of SOX9, a master chondrogenic transcription factor43,77 that has also been shown as the driving force of tumorigeneses in multiple organs and tissues78,79,80. Our results and published work indicate that SHP2 normally represses the proliferation and/or maturation of chondrocytes, functioning as a tumor suppressor in cartilage. However, SHP2 is historically considered as an oncogene and essential for the development and/or homeostasis of multiple organs and tissues28,29. The double-edged sword effect of SHP2 (tumorigenic and anti-tumorigenic) in different types of cells reflects the complexity of cellular signaling networks and the particular “wiring” of the signaling pathways therein.
In sum, we found that SHP2 regulates the differentiation of hypertrophic chondrocytes to an osteoblastic fate, a transition that is critical for endochondral bone formation, bone mineral and cartilage homeostasis. Our study also illustrates an important role for SHP2 in the embryonic and postnatal cartilage development. Its effect on osteogenesis is mediated by SOX9-mediated β-CATENIN signaling. Therefore, manipulating SHP2 and SHP2-regulated signaling pathways can potentially facilitate the development of novel therapeutics to treat cartilage and bone developmental and degenerative diseases.
Methods
Animals
Ptpn11 floxed (Ptpn11 fl/+)36, Sox9 floxed (Sox9 fl/+)41,81, Tg(Col2α1-Cre)57, Tg(Col2α1-CreER T2)55, Tg(Col10α1-Cre) 56 , Tg(CMV-CreER T2 ) 82, Tg(Sp7/mCherry) (Sp7 mCherry ) 60, Rosa26 lacZ (R26lacZ)58, Rosa26 ZsG (R26ZsG)83 and Rosa26 mTmG (R26mTG)84 mice were reported previously. PCR genotyping conditions for Ptpn11 fl and Sox9 fl alleles, R26lacZ and R26mTG reporters and Cre transgenes have been described in the original publications and are available upon request. To delete SHP2 in chondrocytes that express collagen type II, alpha-1 (COL2α1) and type X, alpha-1 (COL10α1), a Ptpn11 floxed allele was interbred to Tg(Col2α1-Cre), Tg(Col2α1-CreER T2 ) and Tg(Col10α1-Cre) mice to generate offspring with the following nomenclature: SHP2Col2α1CTR, SHP2Col2α1KO, SHP2Col2α1ERCTR, SHP2Col2α1ERKO, SHP2Col10α1CTR, and SHP2Col10α1KO, respectively (Fig. S1A,B ). To delete SOX9 in chondrocytes in vitro, a Sox9 floxed allele was interbred to Tg(CMV-CreER T2) mice to generate Sox9 fl/fl and Sox9 fl/fl ;Tg(CMV-CreER T2) offspring, abbreviated respectively as SOX9WT and SOX9cKO mice (Fig. S7 ). To trace COL2α1 and COL10α1-expressing cells in vivo, SHP2Col2α1ERCTR, SHP2Col2α1ERKO, SHP2Col10α1CTR and SHP2Col10α1KO mice were also bred with R26lacZ or R26mTG reporters, respectively. R26mTG reporter expresses fluorescent protein Tomato Red ubiquitously before Cre recombination and GFP following recombination. To induce Tg(Col2α1-CreER) activity, 4-OH tamoxifen (TM; Sigma, MO) was dissolved in DMSO-ethanol-corn oil (4:6:90) mixture at a concentration of 10 mg/mL and injected intraperitoneally into SHP2Col2α1ERCTR, SHP2Col2α1ERKO mice (1 mg/per mouse/each dose)55,85. All transgenic mice were maintained on C57BL/6 J background.
Control and SHP2 mutant animals were sacrificed at the indicated time points and used for x-ray, histological, biochemical and biological analyses. All animal work was reviewed and approved by the Rhode Island Hospital Institutional Animal Care and Use Committee (Assurance No. A3922-01) and performed in accordance with PHS policy on the humane care and use of laboratory animals.
Chondrocyte isolation and cultures
Primary chondrocytes were derived from 1- to 3-day-old pups with modified procedures86. Briefly, the ventral parts of the rib cages from newborn mice were collected and incubated with trypsin-EDTA (0.25%, Invitrogen) for 1 hour at 37 °C. After washing with PBS, the rib cages were further incubated with hyaluronidase (2 mg/ml; Sigma) for 2 hours and hyaluronidase/collagenase D mixture (1 mg/ml, Roche) for 4 hours in DMEM at 37 °C. Undigested bony tissues were discarded by filtration, chondrocytes were collected by centrifugation and cultured in DMEM/F12 medium (1:1) (Invitrogen) supplemented with 10% of FBS, and 1% of ampicillin and streptomycin.
To immortalize primary chondrocytes, retrovirus expressing SV40 large T antigen were prepared from 293 T cells36,87 and used to infect chondrocytes (passage 1) overnight in the presence of 4 µg/ml polybrene. Infected cells were cultured for 48 hours and then selected with neomycin for 7 days. Neomycin-resistant clones were pooled, expanded, and used for this study. To knock down SHP2 expression in SOX9WT and SOX9cKO chondrocytes, retrovirus expressing a control (SHP2WT) or short hairpin RNAi against murine SHP2 (SHP2KD) were prepared and used to infect SOX9WT and SOX9cKO chondrocytes as described previously36,88. Puromycin-resistant chondrocytes were selected, expanded and used for this study. To induce SOX9 deletion in vitro, SOX9WT and SOX9cKO chondrocytes were exposed to TM (1 µM) in the culture medium for 72 hours50. TM-treated chondrocytes were then used for biological and biochemical studies.
Antibodies and Reagents
Polyclonal antibodies against murine SHP2 and SOX9 were purchased from Santa Cruz and EMD Millipore, respectively. Monoclonal antibody against murine ERK2 was purchased from Santa Cruz. Alcian blue and Safranin O staining solutions were purchased from Poly Scientific.
Histology analysis and von Kossa staining
Femurs and tibias from control and SHP2 mutant mice were fixed in 4% formaldehyde for 3 days, decalcified, paraffin-embedded, and sectioned to stain with hematoxylin and eosin (H&E), alcian blue and Safranin O /fast green. To trace the fate of COL2α1- and COL10α1-expressing chondrocytes in vivo, femurs and tibias were collected from SHP2Col2α1ERCTR;R26mTG, SHP2Col2α1ERKO;R26mTG, SHP2Col10α1CTR;R26mTG and SHP2Col10α1KO;R26mTG mice at the indicated time points and fixed in 4% formaldehyde overnight. Frozen sections were examined microscopically to visualize green (GFP) and red (RFP) fluorescent protein-positive cells. DAPI was used for nucleus counterstaining. All florescent and phase contrast images were taken using a Nikon digital fluorescence microscope and Aperio slide scanner (Vista, CA). Immunostaining was carried out using Vectorstain ImmPACT/DAB kit following the manufacture’s instruction. von Kossa staining was carried out using commercially-available kits, per the manufacturer’s instructions (Millipore).
In situ hybridization
Femors and tibias were collected from neonates at the indicated time points, fixed in 4% formaldehyde overnight, and then embedded in OCT compound. 10 µm frozen sections were cut using a cryostat for in situ hybridization with probes against murine Sox9, Acan, Col2α1, Col1α1, Ctnnb1, Ibsp, Runx2 and Mmp13. Hybridization and detection of hybridization signals were achieved using RNAscope HD-Brown kit per the manufacture’s instruction (Advanced Cell Diagnostics).
Quantitative RT-PCR analyses
Total RNA was extracted from chondrocytes using RNeasy kit (Qiagen). cDNA was synthesized using 1 µg of total RNA with iScript™ cDNA Synthesis Kit (Bio-Rad) and qRT-PCR was performed with RT2SYBR® Green kit on a Bio-Rad CFX machine. All samples were normalized to Gapdh and Actin; gene expression was presented as fold increases or decreases compared with that of controls. All primer sequences used for this study are listed in the Supplementary Table 1.
µ-CT and X-ray radiograph analysis
X-ray imaging analysis of mice skeletons was done immediately after euthanasia using a digital radiography system (MX-20, Faxitron Bioptics, LLC, Tucson, AZ, USA). High resolution 3D volume images were generated using a desktop µ-CT40 system (Scanco Medical AG, CH) after fixation of bone specimens in 4% formaldehyde.
Western blot analysis
Cells were lysed into modified NP-40 lysis buffer (0.5% NP40, 150 mM NaCl, 1 mM EDTA, 50 mM Tris [pH 7.4]) supplemented with a protease inhibitor cocktail (1 mM PMSF, 10 mg/ml aprotinin, 0.5 mg/ml antipain, and 0.5 mg/ml pepstatin)53. For immunoblotting, cell lysates (30–50 µg) were resolved by SDS-PAGE, transferred to PVDF membranes, and incubated with primary antibodies for 2 hours or overnight at 4 °C (according to the manufacturer’s instructions), followed by incubation with HRP-conjugated secondary antibodies (Bio-Rad).
Statistical analysis
Statistical differences between groups were evaluated by student t and x 2 tests. A p value of <0.05 was considered to be significant. Analyses were performed by using Prism 3.0 (GraphPad, San Diego, CA) and Excel (Microsoft).
References
Nakashima, K. & de Crombrugghe, B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet 19, 458–466 (2003).
Long, F. & Ornitz, D. M. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol 5, a008334, https://doi.org/10.1101/cshperspect.a008334 (2013).
de Crombrugghe, B. et al. Transcriptional mechanisms of chondrocyte differentiation. Matrix Biol 19, 389–394, doi:S0945-053X(00)00094-9 [pii] (2000).
Olsen, B. R., Reginato, A. M. & Wang, W. Bone development. Annu Rev Cell Dev Biol 16, 191–220, doi:10.1146/annurev.cellbio.16.1.191 16/1/191 [pii] (2000).
Kronenberg, H. M. Developmental regulation of the growth plate. Nature 423, 332-336, doi:10.1038/nature01657 nature01657 [pii] (2003).
Bianco, P., Cancedda, F. D., Riminucci, M. & Cancedda, R. Bone formation via cartilage models: the “borderline” chondrocyte. Matrix Biol 17, 185–192 (1998).
Shapiro, I. M., Adams, C. S., Freeman, T. & Srinivas, V. Fate of the hypertrophic chondrocyte: microenvironmental perspectives on apoptosis and survival in the epiphyseal growth plate. Birth Defects Res C Embryo Today 75, 330–339, https://doi.org/10.1002/bdrc.20057 (2005).
Ono, N., Ono, W., Nagasawa, T. & Kronenberg, H. M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol 16, 1157–1167, https://doi.org/10.1038/ncb3067 (2014).
Yang, L., Tsang, K. Y., Tang, H. C., Chan, D. & Cheah, K. S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci USA 111, 12097–12102, https://doi.org/10.1073/pnas.1302703111 (2014).
Zhou, X. et al. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet 10, e1004820, https://doi.org/10.1371/journal.pgen.1004820 (2014).
Nishimura, R. et al. Regulation of endochondral ossification by transcription factors. Frontiers in bioscience (Landmark edition) 17, 2657–2666 (2012).
Mackie, E. J., Ahmed, Y. A., Tatarczuch, L., Chen, K. S. & Mirams, M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 40, 46–62, https://doi.org/10.1016/j.biocel.2007.06.009 (2008).
Goldring, M. B., Tsuchimochi, K. & Ijiri, K. The control of chondrogenesis. J Cell Biochem 97, 33–44, https://doi.org/10.1002/jcb.20652 (2006).
Zuscik, M. J., Hilton, M. J., Zhang, X., Chen, D. & O’Keefe, R. J. Regulation of chondrogenesis and chondrocyte differentiation by stress. J Clin Invest 118, 429–438, https://doi.org/10.1172/JCI34174 (2008).
Uusitalo, H., Hiltunen, A., Soderstrom, M., Aro, H. T. & Vuorio, E. Expression of cathepsins B, H, K, L, and S and matrix metalloproteinases 9 and 13 during chondrocyte hypertrophy and endochondral ossification in mouse fracture callus. Calcified tissue international 67, 382–390 (2000).
Stickens, D. et al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 131, 5883–5895, https://doi.org/10.1242/dev.01461 (2004).
Zhou, Z. et al. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci USA 97, 4052–4057, https://doi.org/10.1073/pnas.060037197 (2000).
Nakashima, T. et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17, 1231–1234, https://doi.org/10.1038/nm.2452 (2011).
Xiong, J. et al. Matrix-embedded cells control osteoclast formation. Nat Med 17, 1235–1241, https://doi.org/10.1038/nm.2448 (2011).
Naski, M. C., Colvin, J. S., Coffin, J. D. & Ornitz, D. M. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 125, 4977–4988 (1998).
Ornitz, D. M. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev 16, 205–213, https://doi.org/10.1016/j.cytogfr.2005.02.003 (2005).
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J. & Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGFreceptor (Igf1r). Cell 75, 59–72, doi:0092-8674(93)90679-K [pii] (1993).
Wang, J., Zhou, J. & Bondy, C. A. Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J 13, 1985–1990 (1999).
Ehlen, H. W., Buelens, L. A. & Vortkamp, A. Hedgehog signaling in skeletal development. Birth Defects Res C Embryo Today 78, 267–279, https://doi.org/10.1002/bdrc.20076 (2006).
Yoon, B. S. et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA 102, 5062–5067, doi:0500031102 [pii] 10.1073/pnas.0500031102 (2005).
Day, T. F. & Yang, Y. Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am 90(Suppl 1), 19–24, https://doi.org/10.2106/jbjs.g.01174 (2008).
Baron, R., Rawadi, G. & Roman-Roman, S. Wnt signaling: a key regulator of bone mass. Curr Top Dev Biol 76, 103–127, https://doi.org/10.1016/s0070-2153(06)76004-5 (2006).
Grossmann, K. S., Rosario, M., Birchmeier, C. & Birchmeier, W. The tyrosine phosphatase Shp2 in development and cancer. Adv Cancer Res 106, 53–89, doi:S0065-230X(10)06002-1 [pii] 10.1016/S0065-230X(10)06002-1 (2010).
Neel, B. G., Chan G., Dhanji S. SH2 Domain-Containing Protein-Tyrosine Phosphatases. Handbook of Cell Signaling, 771–809 (2009).
Kosaki, K. et al. PTPN11 (protein-tyrosine phosphatase, nonreceptor-type 11) mutations in seven Japanese patients with Noonan syndrome. J Clin Endocrinol Metab 87, 3529–3533 (2002).
Tartaglia, M. et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29, 465–468 (2001).
Araki, T. et al. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10, 849–857, https://doi.org/10.1038/nm1084 (2004).
Nakamura, T., Gulick, J., Pratt, R. & Robbins, J. Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc Natl Acad Sci USA 106, 15436–15441, doi:0903302106 [pii]10.1073/pnas.0903302106 (2009).
Nakamura, T. et al. Mediating ERK 1/2 signaling rescues congenital heart defects in a mouse model of Noonan syndrome. J Clin Invest 117, 2123–2132, https://doi.org/10.1172/JCI30756 (2007).
Bowen, M. E. et al. Loss-of-function mutations in PTPN11 cause metachondromatosis, but not Ollier disease or Maffucci syndrome. PLoS Genet 7, e1002050, https://doi.org/10.1371/journal.pgen.1002050 (2011).
Yang, W. et al. Ptpn11 deletion in a novel progenitor causes metachondromatosis by inducing hedgehog signalling. Nature 499, 491–495, https://doi.org/10.1038/nature12396 (2013).
Kim, H. K. et al. Induction of SHP2-deficiency in chondrocytes causes severe scoliosis and kyphosis in mice. Spine, doi:https://doi.org/10.1097/BRS.0b013e3182a3d370 (2013).
Sobreira, N. L. et al. Whole-genome sequencing of a single proband together with linkage analysis identifies a Mendelian disease gene. PLoS Genet 6, e1000991, https://doi.org/10.1371/journal.pgen.1000991 (2010).
Day, T. F., Guo, X., Garrett-Beal, L. & Yang, Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 8, 739–750, https://doi.org/10.1016/j.devcel.2005.03.016 (2005).
Harley, V. & Lefebvre, V. Twenty Sox, twenty years. Int J Biochem Cell Biol 42, 376–377, https://doi.org/10.1016/j.biocel.2009.12.004 (2010).
Akiyama, H., Chaboissier, M. C., Martin, J. F., Schedl, A. & de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 16, 2813–2828, https://doi.org/10.1101/gad.1017802 (2002).
Dy, P. et al. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell 22, 597–609, https://doi.org/10.1016/j.devcel.2011.12.024 (2012).
Bi, W., Deng, J. M., Zhang, Z., Behringer, R. R. & de Crombrugghe, B. Sox9 is required for cartilage formation. Nat Genet 22, 85–89, https://doi.org/10.1038/8792 (1999).
Hattori, T. et al. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 137, 901–911, https://doi.org/10.1242/dev.045203 (2010).
Akiyama, H. et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev 18, 1072–1087, doi:https://doi.org/10.1101/gad.117110418/9/1072 [pii] (2004).
Lefebvre, V., Behringer, R. R. & de Crombrugghe, B. L-Sox5, Sox6 and Sox9 control essential steps of the chondrocyte differentiation pathway. Osteoarthritis Cartilage 9(Suppl A), S69–75 (2001).
Zorn, A. M. et al. Regulation of Wnt signaling by Sox proteins: XSox17 alpha/beta and XSox3 physically interact with beta-catenin. Mol Cell 4, 487–498 (1999).
Kormish, J. D., Sinner, D. & Zorn, A. M. Interactions between SOX factors and Wnt/beta-catenin signaling in development and disease. Dev Dyn 239, 56–68, https://doi.org/10.1002/dvdy.22046 (2010).
Houben, A. et al. beta-catenin activity in late hypertrophic chondrocytes locally orchestrates osteoblastogenesis and osteoclastogenesis. Development, doi:https://doi.org/10.1242/dev.137489 (2016).
Bowen, M. E., Ayturk, U. M., Kurek, K. C., Yang, W. & Warman, M. L. SHP2 Regulates Chondrocyte Terminal Differentiation, Growth Plate Architecture and Skeletal Cell Fates. PLoS Genet 10, e1004364, https://doi.org/10.1371/journal.pgen.1004364 (2014).
Lapinski, P. E., Meyer, M. F., Feng, G. S., Kamiya, N. & King, P. D. Deletion of SHP-2 in mesenchymal stem cells causes growth retardation, limb and chest deformity and calvarial defects in mice. Dis Model Mech 6, 1448–1458, https://doi.org/10.1242/dmm.012849 (2013).
Hill, T. P., Spater, D., Taketo, M. M., Birchmeier, W. & Hartmann, C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell 8, 727–738, https://doi.org/10.1016/j.devcel.2005.02.013 (2005).
Yang, W. et al. An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell 10, 317–327, https://doi.org/10.1016/j.devcel.2006.01.002 (2006).
Saxton, T. et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J 16, p2352–2364 (1997).
Zhu, M., Chen, M., Lichtler, A. C., O’Keefe, R. J. & Chen, D. Tamoxifen-inducible Cre-recombination in articular chondrocytes of adult Col2a1-CreER(T2) transgenic mice. Osteoarthritis Cartilage 16, 129–130, doi:S1063-4584(07)00279-8 [pii]10.1016/j.joca.2007.08.001 (2008).
Gebhard, S. et al. Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter. Matrix Biol 27, 693–699, doi:S0945-053X(08)00104-2 [pii]10.1016/j.matbio.2008.07.001 (2008).
Ovchinnikov, D. A., Deng, J. M., Ogunrinu, G. & Behringer, R. R. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 26, 145–146, doi:10.1002/(SICI)1526-968X(200002)26:2<145::AID-GENE14>3.0.CO;2-C [pii] (2000).
Soriano, P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21, 70–71, https://doi.org/10.1038/5007 (1999).
Golovchenko, S. et al. Deletion of beta catenin in hypertrophic growth plate chondrocytes impairs trabecular bone formation. Bone 55, 102–112, https://doi.org/10.1016/j.bone.2013.03.019 (2013).
Strecker, S., Fu, Y., Liu, Y. & Maye, P. Generation and characterization of Osterix-Cherry reporter mice. Genesis 51, 246–258, https://doi.org/10.1002/dvg.22360 (2013).
Cheah, K. S., Lau, E. T., Au, P. K. & Tam, P. P. Expression of the mouse alpha 1(II) collagen gene is not restricted to cartilage during development. Development 111, 945–953 (1991).
Wood, A., Ashhurst, D. E., Corbett, A. & Thorogood, P. The transient expression of type II collagen at tissue interfaces during mammalian craniofacial development. Development 111, 955–968 (1991).
Aszodi, A., Chan, D., Hunziker, E., Bateman, J. F. & Fassler, R. Collagen II is essential for the removal of the notochord and the formation of intervertebral discs. J Cell Biol 143, 1399–1412 (1998).
Hill, T. P., Taketo, M. M., Birchmeier, W. & Hartmann, C. Multiple roles of mesenchymal beta-catenin during murine limb patterning. Development 133, 1219–1229, https://doi.org/10.1242/dev.02298 (2006).
Park, J. et al. Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol Open 4, 608–621, https://doi.org/10.1242/bio.201411031 (2015).
Shung, C. Y., Ota, S., Zhou, Z. Q., Keene, D. R. & Hurlin, P. J. Disruption of a Sox9-beta-catenin circuit by mutant Fgfr3 in thanatophoric dysplasia type II. Hum Mol Genet 21, 4628–4644, https://doi.org/10.1093/hmg/dds305 (2012).
Topol, L., Chen, W., Song, H., Day, T. F. & Yang, Y. Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J Biol Chem 284, 3323–3333, https://doi.org/10.1074/jbc.M808048200 (2009).
Timmerman, I. et al. The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of beta-catenin phosphorylation. Mol Biol Cell 23, 4212–4225, https://doi.org/10.1091/mbc.E12-01-0038 (2012).
Haigh, J. J., Gerber, H. P., Ferrara, N. & Wagner, E. F. Conditional inactivation of VEGF-A in areas of collagen2a1 expression results in embryonic lethality in the heterozygous state. Development 127, 1445–1453 (2000).
Kiepe, D., Ciarmatori, S., Haarmann, A. & Tonshoff, B. Differential expression of IGF system components in proliferating vs. differentiating growth plate chondrocytes: the functional role of IGFBP-5. Am J Physiol Endocrinol Metab 290, E363–371, doi:00363.2005 [pii] 10.1152/ajpendo.00363.2005 (2006).
Karolak, M. R., Yang, X. & Elefteriou, F. FGFR1 signaling in hypertrophic chondrocytes is attenuated by the Ras-GAP neurofibromin during endochondral bone formation. Hum Mol Genet 24, 2552–2564, https://doi.org/10.1093/hmg/ddv019 (2015).
Iwata, T. et al. A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Hum Mol Genet 9, 1603–1613 (2000).
Ahmed, Z. et al. Grb2 controls phosphorylation of FGFR2 by inhibiting receptor kinase and Shp2 phosphatase activity. J Cell Biol 200, 493–504, https://doi.org/10.1083/jcb.201204106 (2013).
Leung, V. Y. et al. SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet 7, e1002356, https://doi.org/10.1371/journal.pgen.1002356 (2011).
Hart, K. C. et al. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene 19, 3309–3320 (2000).
Bovee, J. V., Hogendoorn, P. C., Wunder, J. S. & Alman, B. A. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer 10, 481–488, doi:nrc2869 [pii] 10.1038/nrc2869 (2010).
Akiyama, H. Control of chondrogenesis by the transcription factor Sox9. Modern rheumatology/the Japan Rheumatism Association 18, 213–219, https://doi.org/10.1007/s10165-008-0048-x (2008).
Larsimont, J. C. et al. Sox9 Controls Self-Renewal of Oncogene Targeted Cells and Links Tumor Initiation and Invasion. Cell Stem Cell 17, 60–73, https://doi.org/10.1016/j.stem.2015.05.008 (2015).
Matheu, A. et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res 72, 1301–1315, https://doi.org/10.1158/0008-5472.CAN-11-3660 (2012).
Pritchett, J., Athwal, V., Roberts, N., Hanley, N. A. & Hanley, K. P. Understanding the role of SOX9 in acquired diseases: lessons from development. Trends Mol Med 17, 166–174, https://doi.org/10.1016/j.molmed.2010.12.001 (2011).
Kist, R., Schrewe, H., Balling, R. & Scherer, G. Conditional inactivation of Sox9: a mouse model for campomelic dysplasia. Genesis 32, 121–123 (2002).
Hayashi, S. & McMahon, A. P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244, 305–318, https://doi.org/10.1006/dbio.2002.0597 (2002).
Madisen, L. et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience 13, 133–140, https://doi.org/10.1038/nn.2467 (2010).
Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. & Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605, https://doi.org/10.1002/dvg.20335 (2007).
Chen, M. et al. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis 45, 44–50, https://doi.org/10.1002/dvg.20261 (2007).
Lefebvre, V. et al. Characterization of primary cultures of chondrocytes from type II collagen/beta-galactosidase transgenic mice. Matrix Biol 14, 329–335 (1994).
Hahn, W. C. et al. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol 22, 2111–2123 (2002).
Hidaka, K. et al. Involvement of the phosphoinositide 3-kinase/protein kinase B signaling pathway in insulin/IGF-I-induced chondrogenesis of the mouse embryonal carcinoma-derived cell line ATDC5. Int J Biochem Cell Biol 33, 1094–1103, doi:S1357-2725(01)00067-X [pii] (2001).
Acknowledgements
We would like to thank Scott McAllister and Paul Monfils for X-ray radiographs and histology analysis. This publication was made possible by the NIGMS Grant #8P20GM103468 and NIAMS Grant RO1AR066746 (to W.Y). This work was also supported by the Rhode Island Hospital Orthopaedic Foundation and a grant from Arthritis National Research Foundation (W.Y.).
Author information
Authors and Affiliations
Contributions
L.W., C.Z., J.H., D.M. and W.Y. contributed to data collection; J.H. and Q.W. carried out histological staining and data interpretation; L.X. conducted µCT data collection and analysis; K.V.D.M., D.C. and X.Y. provided key reagents for this study; M.G.E. and M.L.W. contributed important intellectual content; D.A.M. and W.Y. wrote the manuscript. All authors read and approve the final version of this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., Huang, J., Moore, D.C. et al. SHP2 Regulates the Osteogenic Fate of Growth Plate Hypertrophic Chondrocytes. Sci Rep 7, 12699 (2017). https://doi.org/10.1038/s41598-017-12767-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-12767-9
- Springer Nature Limited
This article is cited by
-
Compensatory growth and recovery of cartilage cytoarchitecture after transient cell death in fetal mouse limbs
Nature Communications (2024)
-
Protein tyrosine phosphatases in skeletal development and diseases
Bone Research (2022)
-
From Stem to Sternum: The Role of Shp2 in the Skeleton
Calcified Tissue International (2022)
-
A comprehensive review of SHP2 and its role in cancer
Cellular Oncology (2022)
-
SHP-2 deletion in CD4Cre expressing chondrocyte precursors leads to tumor development with wrist tropism
Scientific Reports (2021)