
Abstract
Congenital cataract (CC) is a clinical and genetically heterogeneous eye disease that primarily causes lens disorder and even amblyopic blindness in children. As the mechanism underlying CC is genetically inherited, identification of CC-associated gene mutations and their role in protein distribution are topics of both pharmacological and biological research. Through physical and ophthalmic examinations, two Chinese pedigrees with autosomal dominant congenital cataract (ADCC) were recruited for this study. Mutation analyses of CC candidate genes by next-generation sequencing (NGS) and Sanger sequencing revealed a novel missense mutation in CRYBB2 (p.V146L) and a deletion mutation in CRYAA (p.116_118del). Both mutations fully co-segregated were not observed in unaffected family members or in 100 unrelated healthy controls. The CRYBB2 missense mutation disrupts the distribution of CRYBB2 in human lens epithelial cells (HLEpiCs), and the CRYAA deletion mutation causes hyperdispersion of CRYAA. Furthermore, these two crystallin mutations result in aberrant expression of unfolded protein response (UPR) marker genes as well as apoptosis in HLEpiCs. Collectively, these findings broaden the genetic spectrum of ADCC.
Similar content being viewed by others

Introduction
Congenital cataract (CC) is a major cause of infant blindness and remains a significant health-care burden in children worldwide1,2. CC is characterized by impaired and abnormal expression of crystallin, resulting in lens protein aggregation, which blocks light as it passes through the lens3,4. Globally, nearly 0.01–0.15% of newborns suffer from CC. One-third of cases are inherited, and despite reports of a few cases of autosomal recessive and x-linked inheritance, the vast majority of CC is attributed to autosomal dominant inheritance with high clinical and genetic heterogeneity4,5,6. To date, more than 20 genes have been identified as being responsible for autosomal dominant cataracts; among these, crystallin genes are the most common cause of CC, accounting for 50% of autosomal dominant cataracts7. Crystallin proteins can be divided into two categories based on their characteristics: α-crystallins and β/γ-crystallins. The most abundant soluble protein in the lens, α-crystallin prevents lens cell apoptosis and protects protein stability; α-crystallin can be further divided into two sub-classes, αA- and αB-crystallin, which are encoded by CRYAA and CRYAB, respectively8. Some missense mutations in the CRYAA gene that result in the substitution of an arginine with a neutral or hydrophobic amino acid occur in the core domain of α-crystallin9,10,11,12,13,14,15, and several missense mutations sites in CRYAA have been linked to CC. These mutations may result in the loss of the α-crystallin protein, leading to increased light scattering and lens opacification16,17. Predominantly structural protein, β-crystallin contain four key Greek motifs and are involved in lens development and the maintenance of lens transparency18. Although various CC-causing mutations have been reported in CRYAB, mutations in the key Greek motifs appear to enhance protein-protein interactions or aggregation and protein denaturation, ultimately leading to cataracts19,20,21,22. Despite the association of numerous mutations with cataracts, missense mutations in crystallin genes, particularly CRYAA and CRYBB2, are considered the primary cause of autosomal dominant cataracts. Indeed, according to Cat-Map statistics, approximately 70% of autosomal dominant cataracts may be related to missense mutations in crystallin genes23. Previous studies have reported that the apoptosis triggered by cataract-related mutant proteins is a result of the unfolded protein response (UPR). UPR, which is caused by unfold protein or oxidative damage, comprises a set of intracellular signaling pathways that were recently reported to be activated in the lens during development and endoplasmic reticulum stress24,25. For example, Ma et al. found that splicing mutations in the human βA3/A1-crystallin gene CRYBA1 result in severe misfolding of the protein and activate the UPR stress pathway and eventually apoptosis26. In keeping with these findings, the R49C missense mutation in αA-crystallin is related to upregulation of the PERK UPR pathway in the mouse lens, ultimately leading to apoptosis16, and variable activation of UPR is observed with the Cx50 mutant (S50P, G22R) in mice27. Moreover, induction of UPR with successive apoptosis in lens epithelial cells is expected to be involved in CC formation28.
Regardless, there is no consensus regarding the role of crystallin mutations that result in apoptosis or its molecular mechanism in CC development. In this study, we performed genetic analysis in an attempt to identify causative genes in two Chinese families affected by autosomal dominant congenital cataract (ADCC) through next-generation sequencing (NGS) and Sanger sequencing. Two novel mutations, including one missense mutation in CRYBB2 (c. 436 G > C) that exchanges a valine for a leucine and one homozygous deletion mutation in CRYAA that leads to an in-frame deletion of three amino acids, are likely the dominant cause of cataracts in these two families. Functional analysis showed that the CRYBB2 mutation appears to abolish βB2-crystallin solubility and stabilization, leading to protein aggregation in human lens epithelial cells, whereas the CRYAA deletion mutation causes abnormal protein distribution. Furthermore, our results demonstrate that the CRYBB2 and CRYAA mutations lead to apoptosis in human lens epithelial cells due to UPR. These findings extend the mutation spectrum of crystallin genes in the Chinese CC population and provide clues for exploring the genetic mechanism of CC.
Results
Clinical examination
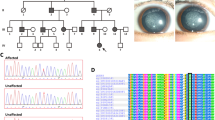
Two Chinese families from Henan Province were enrolled in this study. As shown in Fig. 1A, the pedigrees of the two families revealed an autosomal dominant inheritance pattern. Family I is a three-generation family with two affected members and five unaffected members. According to clinical diagnosis, the four-month-old proband in Family I suffered from a total cataract. As shown in Fig. 1B,C, opacities were observed in the entire lens in both eyes but were more severe in the lens nucleus. Family II was also a three-generation family, including five affected patients and six unaffected individuals. The three-month-old proband was afflicted with a nuclear cataract; the infant suffered from uneven and gray opacification in the lenticular nucleus, though no opacity was found in the peripheral cortex of the lens (Fig. 1D). None of the participating members of both families suffered from other related ophthalmic or systemic syndromes.

Family pedigree, clinical features, mutation screening and multiple sequence alignment analysis. (A) Family history in three-generation families I and II. Dark symbols indicate affected members; clear symbols represent unaffected members. Squares indicate males and circles females. The black arrow represents the proband. (B,C) Slit-lamp photographs of both eyes retrieved from the proband of Family I. (D) Slit-lamp photograph of the affected eye from the proband of Family II. (E,F) Screening of the mutation sites in the two families by sequence analysis. The red frame represents mutation sites. (G,H) Multiple sequence alignment of CRYBB2 and CRYAA from different organisms. The blue frame represents mutation sites.
Two novel mutation sites were identified by NGS and Sanger sequencing
Nearly 134 known candidate genes were evaluated by sequencing, and 64 cataract genes were detected by NGS in the probands. Overall, 560.25 Mb of raw data and 548.39 Mb of processed data were retrieved. The mean coverage of the target region was observed to be more than 95%, with an average sequencing depth of >400X. In addition, the coverage of the targeted base for the N10 and N20 reads was 82.9% and 74.3%, respectively. Two variants remained after filtering of existing variants with a minor allele frequency (MAF) greater than 0.05 in databases (dbSNP138, 1000 Genomes, and in-house Asia database).
In silico analysis of the NGS data revealed a novel missense and a deletion mutation in CRYBB2 and CRYAA, respectively. In Family I, a missense mutation in the exon of CRYBB2 (c.436 G > C) that leads to substitution of the conserved valine to a leucine at codon 146 (p.V146L) was identified (Fig. 1E). A heterozygous deletion mutation, c.344_352del, in CRYAA that results in an in-frame deletion of three residues from codons 116–118 (p.116_118del) was found in the proband of Family II (Fig. 1F). Moreover, Sanger sequencing analysis revealed the lack of these mutations in all unaffected family members and healthy controls. These results show that the mutations were not observed in unaffected relatives and healthy controls from the same ethnic background.
Bioinformatic analysis
Four online prediction tools, Mutation Taster, PolyPhen-2, PROVEAN and SIFT, were employed to evaluate the structural and functional effects of the missense mutation p.V146L in βB2-crystallin, and Mutation Taster and PROVEAN were used to assess the CRYAA deletion mutation. The CRYBB2 mutation was predicted to be ‘probably benign’ (score: 0.005), with no damage, according to PolyPhen-2, and PROVEAN predicted a neutral effect. However, the other two predictions indicated that the missense mutation would be deleterious. Similarly, PROVEAN and Mutation Taster predicted the CRYAA deletion to be deleterious.
To evaluate the effects of mutations in crystallin, the simulation program SWISS-MODEL was utilized to predict the 3-D structure of both the mutant and wild-type proteins. As shown in Supplementary Fig. S1, we observed a difference between the structure of the mutated and wild-type αA-crystallin protein, which in turn resulted in structural variation. In contrast, the missense mutation in βB2-crystallin appears to have a lower structural impact.
Furthermore, multiple sequence alignment was performed to explore the conserved nature of these mutations. As shown in Fig. 1G, a valine at the 146th position of βB2-crystallin is highly conserved among several mammalian species. Arginine-arginine-tyrosine are also highly conserved in most of the species examined (Fig. 1H).
These results demonstrate that the missense p.V146L and the deletion p.116_118del might be deleterious mutations, resulting in CC.
Functional verification of transfected cells
Transfected HLEpiCs were grown in DMEM supplemented with G418. CRYBB2 and CRYAA mRNA was assessed by quantitative polymerase chain reaction (qPCR) analysis, which revealed a significantly higher transcript abundance than in non-transfected and enhanced green fluorescent protein (EGFP)-transfected cells (Fig. 2A,B). Expression of the proteins was further evaluated by western blotting using an anti-EGFP antibody. As shown in Fig. 2,D, a specific cross-reactive band was observed in transfected cells, whereas no such band was detected in non-transfected cells. These results reveal that both genes, with or without mutation, were successfully expressed in transfected cells.

Molecular verification of transfected cells. (A) CRYBB2 relative expression level in untransfected and transfected cells. WT, wild-type cells; WT-E, cells expressing EGFP; Crybb2-WT, cells expressing normal CRYBB2; Crybb2-mut, cells expressing mutant CRYBB2. (B) CRYAA relative expression level in normal cells and transfected cells. WT, wild-type cells; WT-E, cells expressing EGFP; Cryaa-WT, cells expressing normal CRYAA; Cryaa-mut, cells expressing mutant CRYAA. GAPDH was used as a housekeeping gene. (C) Western blot analysis of βB2-crystallin in normal cells and transfected cells. 1, wild-type cells; 2, cells expressing EGFP; 3, cells expressing normal βB2-crystallin; 4, cells expressing mutant βB2-crystallin; GAPDH was used as the internal control. The original western blot images are shown in Supplementary Fig. S2 and S3. (C) Western blot analysis of αA-crystallin in normal cells and transfected cells. 1, wild-type cells; 2, cells expressing EGFP; 3, cells expressing normal αA-crystallin; 4, cells expressing mutant αA-crystallin, GAPDH was used as the internal control. The original western blot images are shown in Supplementary Fig. S4 and S5.
Fluorescence observation of protein distribution and aggregation
Fluorescence observation was used to determine the impact of the mutant proteins in HLEpiCs. As shown in Fig. 3, normal βB2-crystallin was detected and uniformly distributed in the cytoplasm, which is consistent with the results of previous studies. In contrast, βB2-crystallin-V146L was observed to be aggregated in the nuclear peripheral membrane. Wild-type αA-crystallin was also equally distributed in the cytoplasm, whereas the deletion mutation accumulated at the nuclear peripheral membrane, with less inside the nucleus (Fig. 4). These results indicate that the p.V146L mutation in βB2-crystallin and p.116_118del in αA-crystallin disrupt protein transport and localization and promote protein aggregation at target sites.

Representative fluorescence microscopy images of HLEpiCs expressing wild-type βB2-crystallin fused to EGFP or mutant βB2-crystallin fused to GFP. (A) Cells expressing wild-type βB2-crystallin displayed hypodispersion. (B) Mutant βB2-crystallin aggregated at the nuclear peripheral membrane. The white arrow indicates the location of protein aggregation. Left, GFP fluorescence; middle, DAPI (4′,6-diamidino-2-phenylindole) nuclear fluorescence; right, overlay.

Representative fluorescence microscopy images of HLEpiCs expressing wild-type αA-crystallin fused to GFP or mutant αA-crystallin fused to GFP. (A) Cells expressing wild-type αA-crystallin displayed hypodispersion; (B) Mutant αA-crystallin aggregated at the nuclear peripheral membrane. The white arrow indicates the location of protein aggregation. Left, GFP fluorescence; middle, DAPI (4′,6-diamidino-2-phenylindole) nuclear fluorescence; right, overlay.
Mutant proteins influence apoptosis
We used Hoechst 33342 staining to determine the influence of the mutations on apoptosis. As shown in Fig. 5, overexpression of wild-type βB2-crystallin in HLEpiCs did not affect viability. Conversely, mutant βB2-crystallin accelerated apoptosis (Fig. 5A), as did mutant αA-crystallin (Fig. 5B).

Representative fluorescence microscopy images of HLEpiCs expressing wild-type βB2-crystallin or αA-crystallin or mutants. (A) Cells expressing wild-type βB2-crystallin and mutant βB2-crystallin. (B) Cells expressing wild-type αA-crystallin and mutant αA-crystallin. The white arrow indicates an apoptotic cell.
Furthermore, we also assessed apoptosis by Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) staining, whereby the increment in Annexin V-positive/PI-negative cells reflect increased apoptosis. As shown in Fig. 6A–C, cells expressing mutant βB2-crystallin displayed a significant increase in apoptosis compared to cells expressing wild-type βB2-crystallin and control cells. Cells overexpressing mutant αA-crystallin also exhibited more apoptosis compared to cells expressing wild-type αA-crystallin and control cells (Fig. 6D–F). These results demonstrate that the βB2-crystallin p.V146L and αA-crystallin p.116_118del mutations potently induce apoptosis.

Apoptotic cells expressing wild-type and mutant crystallins according to flow cytometry (A–C) Non-transfected cells and those expressing wild-type βB2-crystallin and mutant βB2-crystallin, respectively. (B) Non-transfected cells and those expressing wild-type αA-crystallin and mutant αA-crystallin.
Previous studies reported that UPR is important in the pathogenesis of protein aggregation in cataracts16,29,30. To determine the apoptosis mechanism induced by the crystallin mutations, we assessed expression of BiP, an endoplasmic reticulum (ER) protein that strongly promotes UPR activation, in both mutant transfected cells. As shown in Fig. 7A, western blot analysis revealed the BiP expression level to be upregulated in cells expressing both mutants. Transcription of UPR genes, including HSPA5 and DDIT3, was also markedly increased (Fig. 7B,C). During UPR activation, the transmembrane sensor Ire1 is first activated via non-canonical splicing of X-box binding protein 1 (Xbp1) mRNA, and the level of Ire1 transcription was extremely elevated in both mutants compared to cells expressing wild-type crystallin and control cells (Fig. 7D). In addition, as shown in Fig. 7E,F, the level of spliced Xbp1 was higher in cells expressing mutants.

UPR-associated gene expression status in cells. (A) Western blotting analysis confirming the increase in BiP expression in cells expressing the βB2-crystallin and αA-crystallin mutants. 1, cells without transfection; 2, cells expressing wild-type βB2-crystallin; 3, cells expressing mutant βB2-crystallin; 4, cells without transfection; 5, cells expressing wild-type αA-crystallin; 6, cells expressing mutant αA-crystallin. GAPDH was used as an internal control gene. The original western blot image is shown in Supplementary Fig. S6 and S7. (B–D) HSPA5, DDIT3 and Ire1 relative expression levels in normal cells and transfected cells. WT, wild-type cells; Cryaa-WT, cells expressing normal cryaa; Cryaa-mut, cells expressing mutant cryaa; Cryaa-WT, cells expressing normal cryaa; Cryaa-mut, cells expressing mutant cryaa. GAPDH was used as a housekeeping gene. (E,F) Xbp1 splicing detection. 1, wild-type cells; 2, cells expressing wild-type crystallin; 3, cells expressing mutant crystallin.
Discussion
CC is a serious hereditary disease resulting in blindness with clinical and genetic heterogeneity, and autosomal dominant inheritance is reported to be the major cause of CC. With the advent of high-throughput molecular techniques, more research is being conducted on the genetic basis of CC. Shiels et al. reported mutation in crystallin genes among nearly half of CC cases31. In this study, we identified two novel mutations in two three-generation Chinese families by NGS and reconfirmed these results by Sanger sequencing.
Crystallins are the predominant lens structural proteins of mammals and have a significant function in providing transparency and light transmission to the eye lens32. However, mutations in the genes encoding crystallin proteins lead to abnormal expression, which might disrupt lens opacity and even cause blindness12,33,34.
Previous studies have demonstrated that expression of CRYAA is necessary for normal lens development and that knockout of α-crystallins resulted in abnormal differentiation of lens fiber cells in zebrafish35. Similarly, transgenic expression of a mutant crystallin (CRYBA1) protein resulted in abnormal differentiation of lens fibers in transgenic mice and consequently led to lens capsule rupture26. Hence, it is clear that mutated crystallin proteins have a pivotal role in lens development in animal cells; however, there are very few studies reporting the type of mutation in these genes and impacts on lens differentiation in human lens cell lines. The protein αA-crystallin is responsible for preventing apoptosis through chaperone-analogous activities and for protecting lens proteins from precipitation36,37. CRYAA-encoded αA-crystallin has been mapped to chromosome 21q22.3, and several mutations in the CRYAA gene have been associated with various CC types worldwide14,38,39. For instance, the missense mutation p.R116C causes a decrease in positive charge and an increase in mercapto groups in αA-crystallin, leading to polymer hydrophobicity and protein precipitation38. Missense mutations of p.R116H and p.G98R reduce the stability of αA-crystallin, which, in turn, causes cataracts13,15. Notably, in this study, we identified a c.344_352del in-frame mutation in CRYAA that causes deletion of three amino acids (residues 116–118), and this mutation results in significant variation in protein distribution, with notable aggregation at the nuclear peripheral membrane and nucleus based on fluorescence observation (Fig. 4B).
βB2-crystallin encoded by CRYBB2 contains four key Greek sequences; the six exons of CRYBB2 map to chromosome 22q11.2-q13.1. The first two Greek key motifs are in the N-terminal domain, and the other two are located at the COOH-terminal domain. The solubility and stability of βB2-crystallin are crucial for proper function in the lens, with disruption of solubility and stability causing aggregation, which leads to lenticular transparency and diopter damage40. Previous studies have reported that mutations in CRYBB2 are associated with the onset of various types of CC. For example, a missense mutation (p.A2V) in the N-terminal domain influences the renaturation process, resulting in βB2-crystallin aggregation41. At the C-terminal domain, the p.Q155X nonsense mutation impacts formation of the Greek key motif, affecting folding and biophysical properties that lead to congenital cataract42. In the present study study, it was observed that the p.V146L mutation alters the molecular weight and hydrophobic properties of βB2-crystallin. Previous studies showed the significance of βB2-crystallin in β-crystallin aggregation within various contexts43,44. Consistently, our overexpression analysis of the mutant βB2-crystallin in cell lines revealed aggregation of the protein at the nuclear peripheral membrane (Fig. 3B). Altogether, the reported missense and deletion mutations in CRYBB2 and CRYAA might be common and responsible for crystallin protein aggregation.
Our results showed that mutations in CRYAA and CRYBB2 induce expression of UPR-associated genes, such as HSPA5, DDIT3 and Ire1. UPR is an adaptive intracellular signaling mechanism that responds to the accumulation of misfolded proteins by triggering upregulation of a characteristic group of target genes26,45,46,47. Earlier studies have reported a crucial role for UPR genes, such as HSPA5, DDIT3 and Ire1, in response to misfolded proteins45,48,49. HSPA5 has been considered to be the key protein in UPR, as it has dual functions as an ER chaperone and as a sensor of protein misfolding50. In our study, we observed that expression of UPR-associated genes was significantly increased in transgenic cells expressing mutant αA-crystallin and βB2-crystallin (Fig. 7) and the occurrence of apoptosis in cells expressing these mutants (Figs 5 and 6). Expression of mutant G98R αA-crystallin in human epithelial B3 cells induced UPR-mediated apoptosis51. Firtina et al. demonstrated that abnormal expression of collagen IV genes triggers transcriptional activation of UPR genes and consequently induces apoptosis and cataract formation in transgenic fiber cells52. Congruently, higher expression of UPR-specific proteins induced apoptosis in lens epithelial cells as well as cataract formation in rats30. In addition, mutant αA-crystallin R49C triggered UPR, which induced lens cell death in a mutant knock-in mouse model16. Thus, mutant αA-crystallin and βB2-crystallin cause upregulation of characteristic UPR genes and, consequently, apoptosis in transfected cells, which is in accordance with previous reports. In conclusion, two new heterozygous mutations (p.116_118del in CRYAA and p.V146L in CRYBB2) were identified in two Chinese families with ADCC. The deletion mutation p.116_118del was found to influence protein aggregation, and the missense mutation p.V146L was determined to affect protein distribution and induce apoptosis in HLEpiCs. Our findings provide some clues for the vital role of crystallin and the mechanism by which mutation results in CC. Moreover, our results also extend the mutation spectrum of CC causative genes in the Chinese population.
Methods
Ethics statement
This study was performed in adherence with the Declaration of Helsinki and was approved by the Institutional Review Board of the First Affiliated Hospital at Zhengzhou University (Zhengzhou, China). Written informed consent was obtained from all participants (or their legal guardians).
Subjects, clinical examination and DNA isolation
Two Chinese families of the Han ethnicity from Henan Province affected by CC were recruited from the ophthalmology department at First Affiliated Hospital of Zhengzhou University. One hundred unrelated participants without eye disease were also enrolled from the ophthalmology department as healthy controls. Family and medical history were recorded. Complete ophthalmic examinations, including visual acuity, dilated pupil examination, intraocular pressure measurement, and slit-lamp ophthalmoscopy, were performed. The phenotypes obtained by slit-lamp photography were documented. A 5-mL sample of venous blood was obtained from all participants and placed in tubes containing ethylenediaminetetraacetic acid (EDTA). Genomic DNA was extracted using QIAamp DNA Blood Mini Kit (Qiagen, USA) according to the manufacturer’s recommendations.
NGS and Sanger sequencing to validate variants
All candidate genes (134 genes, including 64 cataract genes) implicated in CC were subjected to sequencing. The 64 cataract genes are listed in Supplementary Dataset 1. Libraries were prepared following standard protocols, as previously described53,54. Targeted sequence capture was performed with biotinylated oligo-probes and a disease-related gene panel following the manufacturer’s instructions. Paired-end sequencing for reads of 100 bp was performed using the Illumina HiSeq. 2000 platform (Illumina, USA).
Low-quality sequences in the raw reads were filtered by Trim-Galore, and clean reads were then aligned to the human reference genome using the BWA program. Quality scores of the clean reads were recalibrated and realigned for reference with GATK software. Sequence Alignment/Map tools 3 (SAMtools 3) were used for removing duplicated reads, and only unique mapping reads were further applied for variation detection. Single-nucleotide variants were analyzed and genotyped by GATK UnifiedGenotyper, and indels were analyzed by GATK Indel GenotyperV2. Variants were annotated by an in-house bioinformatic tool with RefSeq (hg19, from UCSC) and UCSC annotation according to the manufacturer’s recommendations.
Two genes acquired by NGS were defined as probable causative genes, and Sanger sequencing was performed for validation. PCR products with mutation were amplified using primers and purified with a gel extraction kit (Omega, USA). An ABI DNA Analyzer (Applied Biosystems, USA) was used to analyze the sequence data. Damage predictions were performed using bioinformatic tools such as PROVEAN, MutationTasting, SIFT and PolyPhen-255,56,57,58.
Cell culture and transfection
Human lens epithelial cells (HLEpiCs) were purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) fortified with 10% fetal bovine serum (FBS, Gibco, USA). The cells were cultivated at 37 °C in a humidified atmosphere containing 5% CO2. Plasmids carrying normal or mutant genes were constructed by restriction enzyme ligation and transfected into cells using Lipofectamine 2000 (Invitrogen, USA) following the manufacturer’s protocols.
Molecular characterizations of transfected cells
To determine the relative expression of mutant and non-mutant CRYBB2 and CRYAA in transfected cells, quantitative real-time PCR (RT-qPCR) was performed following the protocol described by Wang et al.59. Efficiency was determined before performing qPCR, and only primers with an efficiency above 95% were used. Endogenous GAPDH was used for normalization of the threshold cycle (Ct) value detected for both the transfected and normal cells. Western blotting was also carried out to verify expression of the proteins in the transfected cells. Cells transfected with GFP served as controls. Target proteins were blotted using a primary anti-GFP antibody (1:20000, Abcam, USA) and horseradish peroxidase (HRP) Goat anti-Rabbit IgG antibody (1:20000, Boster, USA). Protein expression was normalized to that of GAPDH.
Fluorescence microscopy for determination of aggregation
HLEpiCs transfected for 48 h were used for fluorescence microscopy to observe protein distribution. Briefly, cells were washed three times with phosphate-buffered saline (PBS) and fixed with acetone for 10 min at room temperature. The fixed cells were washed three times with PBS and then stained with DAPI for 5 min at room temperature. The cells were observed by fluorescence microscopy. The percentage of cells with aggregates was calculated from 200 positively transfected cells in 10 random viewing fields.
Apoptosis determination
Apoptosis caused by the mutations was evaluated by nuclear morphology observation of Hoechst 33342-stained cells. In brief, transfected cells were cultivated in 6-well plates and stained with Hoechst 33342, after which the cells were observed by fluorescence microscopy. The numbers of apoptotic nuclei were counted in five random viewing fields.
Transfected cells were first stimulated with 400 μM H2O2 for 24 h, and an apoptosis assay was then performed using Annexin V-FITC/PI Apoptosis Detection Kit (Vazyme, China) according to the manufacturer’s instructions. The cells were assessed by flow cytometry using a BD FACS Aria-IIu apparatus and Cell Quest software (BD Biosciences, USA).
Molecular analysis of apoptosis induced by mutant crystallin
Quantitative real-time PCR was performed as described above. The ribosomal protein gene Rpl19 served as an endogenous control for quantitation of UPR-related genes. For RT-PCR, total RNA was extracted from transfected cells using a Total RNA kit (Omega, USA). Each reaction used 20 ng of total RNA as a template, and the primers used were selected according to a previous study52. Western blot analysis was also performed according to a previous study52, and a rabbit polyclonal antibody against BiP/Grp78 (Abcam, UK) was used as the primary antibody.
References
Pi, L. H. et al. Prevalence of eye diseases and causes of visual impairment in school-aged children in Western China. J Epidemiol 22, 37–44 (2012).
Wu, X., Long, E., Lin, H. & Liu, Y. Prevalence and epidemiological characteristics of congenital cataract: a systematic review and meta-analysis. Sci Rep 6, 28564 (2016).
Fu, L. & Liang, J. J.-N. Alteration of protein–protein interactions of congenital cataract crystallin mutants. Invest Ophth Vis Sci 44, 1155–1159 (2003).
Hejtmancik, J. F. Congenital cataracts and their molecular genetics. Semi Cell Dev Biol 19, 134–149 (2008).
Scott, M. H. et al. Autosomal dominant congenital cataract: interocular phenotypic variability. Ophthalmology 101, 866–871 (1994).
Ionides, A. et al. Clinical and genetic heterogeneity in autosomal dominant cataract. British J Ophthalmol 83, 802–808 (1999).
Shiels, A. & Hejtmancik, J. F. Chapter Twelve-Molecular Genetics of Cataract. Prog Mol Biol Transl Sci 134, 203–218 (2015).
Groenen, P. J., Merck, K. B., Jong, W. W. & Bloemendal, H. Structure and Modifications of the Junior Chaperone α‐Crystallin. FEBS J 225, 1–19 (1994).
Yang, Z. et al. A R54L mutation of CRYAA associated with autosomal dominant nuclear cataracts in a Chinese family. Curr Eye Res 38, 1221–1228 (2013).
Javadiyan, S. et al. Recurrent mutation in the crystallin alpha A gene associated with inherited paediatric cataract. BMC Res Notes 9, 83 (2016).
Laurie, K. J. et al. Identification of a novel oligomerization disrupting mutation in CRYΑA associated with congenital cataract in a South Australian family. Hum Mutat 34, 435–438 (2013).
Kong, X. et al. A novel 3-base pair deletion of the CRYAA gene identified in a large Chinese pedigree featuring autosomal dominant congenital perinuclear cataract. Genet Mol Res 14, 426–432 (2015).
Hansen, L. et al. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophth Vis Sci 48, 3937–3944 (2007).
Khan, A. O., Aldahmesh, M. A. & Meyer, B. Recessive congenital total cataract with microcornea and heterozygote carrier signs caused by a novel missense CRYAA mutation (R54C). Am J Ophthalmol 144, 949–952. e942 (2007).
Santhiya, S. T. et al. Identification of a novel, putative cataract-causing allele in CRYAA (G98R) in an Indian family. Mol Vis 12, 768–773 (2006).
Andley, U. P. & Goldman, J. W. Autophagy and UPR in alpha-crystallin mutant knock-in mouse models of hereditary cataracts. BBA-Gen Subjects 1860, 234–239 (2016).
Datiles, M. B. et al. Clinical detection of precataractous lens protein changes using dynamic light scattering. Arch Ophthalmol 126, 1687–1693 (2008).
Hejtmancik, J. et al. Molecular biology and inherited disorders of the eye lens. The metabolic and molecular basis of inherited disease 8, 6033–6062 (2001).
Litt, M. et al. Autosomal dominant cerulean cataract is associated with a chain termination mutation in the human β-crystallin gene CRYBB2. Hum Mol Genet 6, 665–668 (1997).
Mackay, D. S., Boskovska, O. B., Knopf, H. L., Lampi, K. J. & Shiels, A. A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am J Hum Genet 71, 1216–1221 (2002).
Xi, Y.-B. et al. Cataract-linked mutation R188H promotes βB2-crystallin aggregation and fibrillization during acid denaturation. Biochem Bioph Res Commun 447, 244–249 (2014).
Zhou, Y. et al. A Novel CRYBB2 Stopgain Mutation Causing Congenital Autosomal Dominant Cataract in a Chinese Family. J Ophthalmol 2016 (2016).
Shiels, A., Bennett, T. M. & Hejtmancik, J. F. Cat-Map: putting cataract on the map. Mol Vis 16, 2007 (2010).
Ikesugi, K., Yamamoto, R., Mulhern, M. L. & Shinohara, T. Role of the unfolded protein response (UPR) in cataract formation. Exp Eye Res 83, 508–516 (2006).
Shinohara, T., Ikesugi, K. & Mulhern, M. L. Cataracts: role of the unfolded protein response. Med Hypotheses 66, 365–370 (2006).
Ma, Z. et al. Human βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells. BBA-Mol Basis Dis 1862, 1214–1227 (2016).
Alapure, B. V., Stull, J. K., Firtina, Z. & Duncan, M. K. The unfolded protein response is activated in connexin 50 mutant mouse lenses. Exp Eye Res 102, 28–37 (2012).
Shiels, A. & Hejtmancik, J. F. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res 156, 95–102 (2017).
Malhotra, J. D. & Kaufman, R. J. The endoplasmic reticulum and the unfolded protein response. Semi Cell Dev Biol 18, 716–731 (2007).
Mulhern, M. L. et al. The unfolded protein response in lens epithelial cells from galactosemic rat lenses. Invest Ophth Vis Sci 47, 3951–3959 (2006).
Shiels, A. & Hejtmancik, J. F. Genetic origins of cataract. Arch Ophthalmol 125, 165–173 (2007).
Slingsby, C. & Wistow, G. J. Functions of crystallins in and out of lens: roles in elongated and post-mitotic cells. Prog Biophys Mol Biol 115, 52–67 (2014).
Xiao, X., Mai, G., Li, S., Guo, X. & Zhang, Q. Identification of CYP4V2 mutation in 21 families and overview of mutation spectrum in Bietti crystalline corneoretinal dystrophy. Biochem Bioph Res Commun 409, 181–186 (2011).
Dulle, J. E., Rübsam, A., Garnai, S. J., Pawar, H. S. & Fort, P. E. BetaB2-crystallin mutations associated with cataract and glaucoma leads to mitochondrial alterations in lens epithelial cells and retinal neurons. Exp Eye Res 155, 85–90 (2017).
Zou, P. et al. A conserved role of αA-crystallin in the development of the zebrafish embryonic lens. Exp Eye Res 138, 104–113 (2015).
Hamann, S., Métrailler, S., Schorderet, D. F. & Cottet, S. Analysis of the cytoprotective role of α-crystallins in cell survival and implication of the αA-crystallin C-terminal extension domain in preventing Bax-induced apoptosis. PloS One 8, e55372 (2013).
Nahomi, R. B. et al. Chaperone peptides of α-crystallin inhibit epithelial cell apoptosis, protein insolubilization, and opacification in experimental cataracts. J Biol Chem 288, 13022–13035 (2013).
Litt, M. et al. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet 7, 471–474 (1998).
Gu, F. et al. A novel mutation in AlphaA‐crystallin (CRYAA) caused autosomal dominant congenital cataract in a large Chinese family. Hum Mutat 29, 769–769 (2008).
Graw, J. Congenital hereditary cataracts. Int J Dev Biol 48, 1031–1044 (2004).
Yao, K. et al. Characterization of a novel mutation in the CRYBB2 gene associated with autosomal dominant congenital posterior subcapsular cataract in a Chinese family. Mol Vis 17, 144 (2011).
Li, F. F. et al. Nonsense mutation in the CRYBB2 gene causing autosomal dominant progressive polymorphic congenital coronary cataracts. Mol Vis 14, 750 (2008).
Yao, K. et al. Progressive polymorphic congenital cataract caused by a CRYBB2 mutation in a Chinese family. Mol Vis 11, 758–763 (2005).
Chen, W. et al. A missense mutation in CRYBB2 leads to progressive congenital membranous cataract by impacting the solubility and function of βB2-crystallin. PloS One 8, e81290 (2013).
Gessner, D. K., Schlegel, G., Ringseis, R., Schwarz, F. J. & Eder, K. Up-regulation of endoplasmic reticulum stress induced genes of the unfolded protein response in the liver of periparturient dairy cows. BMC Vet Res 10, 46 (2014).
Høyer-Hansen, M. & Jäättelä, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 14, 1576–1582 (2007).
Cao, S. S. & Kaufman, R. J. Unfolded protein response. Curr Biol 22, R622–R626 (2012).
Oyadomari, S. & Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11, 381–389 (2004).
Hetz, C. et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 312, 572–576 (2006).
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P. & Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2, 326–332 (2000).
Gong, B., Zhang, L.-Y., Pang, C.-P., Lam, D. S.-C. & Yam, G. H.-F. Trimethylamine N-oxide alleviates the severe aggregation and ER stress caused by G98R αA-crystallin. Mol Vis 15, 2829 (2009).
Firtina, Z. et al. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J Biol Chem 284, 35872–35884 (2009).
Gillespie, R. L. et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 121, 2124–2137. e2122 (2014).
Ma, A. S. et al. Sporadic and Familial Congenital Cataracts: Mutational Spectrum and New Diagnoses Using Next‐Generation Sequencing. Hum Mutat 37, 371–384 (2016).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11, 361–362 (2014).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet 76, 7.20.1–27.20.41 (2013).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1081 (2009).
Wang, X. et al. Identification of a putative patatin-like phospholipase domain-containing protein 3 (PNPLA3) ortholog involved in lipid metabolism in microalga Phaeodactylum tricornutum. Algal Res 12, 274–279 (2015).
Acknowledgements
This study was funded by the Science and Technology Research Projects of Henan Province, China (201202010).
Author information
Authors and Affiliations
Contributions
G.Y.Z. conceived and designed the experiments. L.L. and D.B.F. performed the experimental. Y.L., D.Q.K. and F.F.C. analyzed the data. L.L., D.B.F. and G.Y.Z. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Fan, DB., Zhao, YT. et al. Two novel mutations identified in ADCC families impair crystallin protein distribution and induce apoptosis in human lens epithelial cells. Sci Rep 7, 17848 (2017). https://doi.org/10.1038/s41598-017-18222-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18222-z
- Springer Nature Limited
This article is cited by
-
Four mutations identified in Chinese families with autosomal dominant congenital cataracts by next-generation sequencing
Genes & Genomics (2024)
-
Novel cataract-causing variant c.177dupC in c-MAF regulates the expression of crystallin genes for cell apoptosis via a mitochondria-dependent pathway
Molecular Genetics and Genomics (2023)
-
CRYβB2 enhances tumorigenesis through upregulation of nucleolin in triple negative breast cancer
Oncogene (2021)
-
Nonsense variant of NR0B1 causes hormone disorders associated with congenital adrenal hyperplasia
Scientific Reports (2021)
-
GJA8 missense mutation disrupts hemichannels and induces cell apoptosis in human lens epithelial cells
Scientific Reports (2019)