
Abstract
The guanylate cyclase C (GC-C) receptor regulates electrolyte and water secretion into the gut following activation by the E. coli enterotoxin STa, or by weaker endogenous agonists guanylin and uroguanylin. Our previous work has demonstrated that GC-C plays an important role in controlling initial infection as well as carrying load of non-invasive bacterial pathogens in the gut. Here, we use Salmonella enterica serovar Typhimurium to determine whether GC-C signaling is important in host defense against pathogens that actively invade enterocytes. In vitro studies indicated that GC-C signaling significantly reduces Salmonella invasion into Caco2-BBE monolayers. Relative to controls, GC-C knockout mice develop severe systemic illness following oral Salmonella infection, characterized by disrupted intestinal mucus layer, elevated cytokines and organ CFUs, and reduced animal survival. In Salmonella-infected wildtype mice, oral gavage of GC-C agonist peptide reduced host/pathogen physical interaction and diminished bacterial translocation to mesenteric lymph nodes. These studies suggest that early life susceptibility to STa-secreting enterotoxigenic E. coli may be counter-balanced by a critical role of GC-C in protecting the mucosa from non-STa producing, invasive bacterial pathogens.
Similar content being viewed by others


Introduction
Infectious diarrheal disease is a significant cause of morbidity and mortality in the developing world1. Diarrheal disease, such as that caused by enterotoxigenic Escherichia coli (ETEC), kills approximately 500,000 children each year2. Deregulated fluid secretion during ETEC infection can be mediated in part by peptide enterotoxins such as heat stable (STa) peptide variants. STa is thought to bind a single lumen-oriented receptor in the intestine known as guanylate cyclase 2 C (Gucy2C, hereafter referred to as GC-C)3,4,5. GC-C and its endogenously produced ligands (guanylin (Gn) and uroguanylin (Ugn)) regulate cyclic guanosine monophosphate (cGMP) production in the epithelia of the intestine6,7. Release of Gn and Ugn into the gut lumen binds to GC-C, elevates intracellular cGMP, and activates protein kinase G II (PKGII)8,9. In many cell types, studies indicate that cGMP signaling may directly, or by regulating phosphodiesterase activity, cross-activate cyclic adenosine monophosphate (cAMP) signaling pathways as well10,11,12. A critical endpoint of GC-C signaling is secretion of chloride and bicarbonate ions by cystic fibrosis transmembrane conductance regulator (CFTR) and loss of sodium import by Na+/H+ Exchanger 3 (NHE3). The resulting accumulation of extracellular electrolytes pulls water into the gut lumen and has an important hydrating effect on the luminal contents. ETEC STa has a super-agonist effect on GC-C, causing copious electrolyte and water secretion. Importantly, unlike STa, physiological activation of GC-C by Gn or Ugn is well controlled and does not cause secretory diarrhea.
Proper hydration of the intestine by coordinated CFTR and NHE3 activity is essential for homeostatic balance between host and microflora. In addition to persistent lung pathology, patients with CFTR mutation have significant intestinal disease characterized by viscous mucoid obstructions, bacterial overgrowth, mucosal immune activation, and inflammation13. CFTR is necessary for mucin hydration and an effective physical barrier between the epithelial monolayer and luminal bacteria. In CFTR deficiency, a chronic close physical association between microflora and host epithelia induces mucosal inflammation13,14. Similarly, poor sodium absorption due to mutation of human NHE3 causes congenital diarrhea syndrome and intestinal inflammation and deletion of the exchanger in mice leads to pro-inflammatory gene expression, enhanced bacterial adhesion and translocation, and colitis15,16,17.
Several lines of evidence indicate that GC-C signaling, as an upstream cascade leading to CFTR and NHE3, is also essential for intestinal fluid homeostasis. Human kindred having congenital GC-C mutations reveal that it has an important role in linking luminal hydration to infection and inflammation18,19. GC-C loss-of-function mutations cause meconium ileus in infants and these individuals retain an increased susceptibility to bowel infection as they age18. Interestingly, gain-of-function mutations in GC-C also cause significant intestinal pathology including secretory diarrhea, alterations in gut microbiota, and susceptibility to inflammatory bowel disease19,20,21. Using several model systems, our previous work demonstrates that GC-C signaling plays a crucial role in host defense during inflammation and infection22,23,24. Specifically, GC-C is essential during infection with non-invasive attaching/effacing (AE) lesion-forming bacterial pathogens in order to reduce bacterial load and minimize systemic dissemination25. However, little is understood about a broader role for GC-C during bacterial infection. Specifically, a detailed understanding of the impact of electrolyte and fluid regulation by GC-C and differing mechanisms of host-bacteria interactions is lacking. For example, it remains unclear whether cGMP-dependent secretion affects adherence and invasion by more aggressive, cell-penetrating bacteria. Accordingly, we hypothesize that activation of the GC-C signaling pathway and its associated secretory response reduces pathology and systemic spread of enteric invasive bacterial species. In this study, we turn to an invasive bacterial pathogen, Salmonella enterica serovar Typhimurium (hereafter referred to as Salmonella), to investigate a role for GC-C in directly regulating host-pathogen interactions. We demonstrate using cell culture and mouse model systems that GC-C signaling in epithelia of the gut is critical for minimizing bacterial invasion into enterocytes and that it is essential for animal survival during infection by invasive bacterial pathogens.
Results
Cells lacking GC-C are prone to Salmonella invasion
We began by using a well-described in vitro cell culture system to investigate epithelial cell-bacteria interactions. Caco2-BBE cell monolayers, which express GC-C and downstream signaling components, were used to determine the impact of GC-C deletion on Salmonella adherence and invasion. We chose this pathogen for these studies because it provides an effective, well-characterized research model of in vitro and in vivo bacterial infection. This approach allows for direct analysis of bacterial adhesion and invasion of enterocytes and contrasts nicely with our previous work using an AE lesion-forming species. In addition, Salmonella infection is of high clinical relevance, with millions of cases of enteric disease and systemic salmonellosis occurring each year26,27. We transduced cells with virus expressing a scrambled shRNA sequence as control or one of two separate GC-C targeting sites (shRNA605 and shRNA3280). Western blot confirmed efficient GC-C knockdown in puromycin selected cells and further studies were performed with scramble control and GC-C shRNA 3280 (Fig. 1A). Upon Salmonella infection (using a multiplicity of infection (MOI) of 100) for 30 minutes, we measured lower adhesion of bacteria to GC-C knockdown cells (Fig. 1B). Conversely, GC-C knockdown led to elevated numbers of Salmonella that had invaded the cell monolayer after 90 minutes (Fig. 1B). This was confirmed by fluorescence microscopy showing significant Salmonella uptake in GC-C knockdown Caco2-BBE cells (Fig. 1C). Expression of GC-C mRNA was measured four hours after bacterial infection and found to be elevated relative to uninfected control cells and still diminished in shRNA-expressing cells (Fig. 1D). These results suggest that GC-C signaling plays an important role in regulating host-bacteria interactions as well as bacterial invasion into enterocytes.

Elevated Salmonella invasion in GC-C knockdown cells. (A) Immunoblot confirming GC-C knockdown using two lentivirus-based shRNAs (shRNA 605 or shRNA 3280). An shRNA of scrambled sequence was used as control and general transcription factor IIB (TF2B) was used to confirm even loading. (B) Adhesion assays and gentamicin protection invasion assays were performed in confluent shRNA 3280 GC-C knockdown (GC-C KD) and control (scramble) cells. Experiments were conducted in triplicate using three different cell line clones. (C) Immunofluorescence of Caco2-BBE cells showing GC-C expression and invasion of Salmonella in GC-C knockdown cells. Confluent cells were infected for 90 minutes, treated with gentamicin, fixed and stained with GC-C (TRITC) or Salmonella (FITC) antibodies. 4′,6-diamidino-2-phenylindole (DAPI) was used to stain the cell nuclei. Images were taken at 200x. (D) Scramble and GC-C KD cells were infected with Salmonella for four hours and then lysed to collect RNA. Realtime RT-PCR was performed on GC-C. n = 8. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Activation of GC-C by peptide agonists minimizes Salmonella invasion
We reasoned that supplementing the existing low level of GC-C activation would serve to further minimize Salmonella invasion. We used STcore which is a GC-C-binding 14 amino acid derivative of ETEC STa that is nearly identical to linaclotide, a GC-C agonist approved by the FDA for treatment of irritable bowel syndrome28. Scramble and GC-C knockdown Caco2-BBE cells were pretreated with STcore 15 minutes prior to Salmonella infection and then host-cell interaction assays were performed. These studies revealed an interesting inverse correlation between adhesion and invasion following STcore-activation of GC-C. While adhesion increased with STcore treatment in scramble shRNA control cells, invasion was substantially decreased (Fig. 2A). As expected due to the absence of GC-C, no such differences were noted in knockdown cells. Because CFTR is an important downstream target of GC-C/cGMP signaling, we next blocked CFTR in control and GC-C knockdown cells and measured Salmonella adhesion and invasion. We found that blocking CFTR mimicked loss of GC-C with regard to Salmonella attachment and cell entry (Fig. 2B). Further, while loss of both GC-C and CFTR activity did not change numbers of bacteria adhered to the cell layer (Fig. 2B, left), there was a notable and statistically significant increase in bacterial invasion in GC-C/CFTR-inhibited cells relative to all other treatment groups (Fig. 2B, right). To confirm strong activation of GC-C receptor signaling pathways, we performed immunoblot for phosphorylated vasodilator-stimulated phosphoprotein (VASP) at phosphorylation sites Ser239 and Ser157 (Fig. 2C). Phosphorylation of VASP in control cells was evident upon treatment with STcore and to a much lesser degree in GC-C knockdown cells, confirming that GC-C activation by exogenous peptides leads to cGMP signaling and crosstalk to cAMP-dependent pathways. Collectively, these results suggest that activation of the GC-C pathway plays a potent defensive role in preventing movement of invasive pathogens like Salmonella into intestinal epithelial cells.

Activation of GC-C by STcore reduces Salmonella invasion. (A,B) Adhesion and invasion assays in scramble and GC-C knockdown (GC-C KD) cells pretreated with STcore or CFTR inhibitor (CFTR(inh)-172). (C) Immunoblot of protein lysates from scramble and GC-C knockdown cell lines treated without or with STcore for 30 minutes. VASP phosphorylation (pVASP) at sites Ser239, Ser157 was measured while general transcription factor IIB (TF2B) was used as loading control. *P < 0.05, **P < 0.01, ns: not significant.
GC-C is essential for maintaining mucus layer integrity and minimizing systemic spread of Salmonella in mice
In an effort to better understand the importance of GC-C-regulated cGMP production on host-bacteria interactions, we next used an in vivo murine model of Salmonella infection that is similar to human typhoid fever. We chose this approach because it allows us to investigate systemic dissemination of an enteric pathogen without significant epithelial architecture distortion or ulceration at early time points. GC-C knockout mice were infected with 107 colony forming units (CFU) Salmonella using oral gavage. Significant weight loss was observed in knockout mice one day post-infection (Fig. 3A). Spleen and liver (Fig. 3B,C) were collected at study day 2 and showed significant increases in Salmonella CFUs in GC-C−/− mice as compared to WT. There was a significant loss in ceca weight in infected knockout mice compared to control group (Fig. 3D).

GC-C knockout mice show significant weight loss and increased systemic dissemination upon oral Salmonella infection. Mice were infected by oral gavage with 107 Salmonella. (A) Weight loss was calculated based on the initial weights. (B,C) Salmonella CFUs were measured after two days of infection in spleen and liver. (D) Ceca were collected and weighed from uninfected and infected mice (48 hrs). *P < 0.05, **P < 0.01, ***P < 0.001.
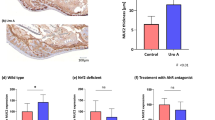
GC-C signaling is a potent regulator of chloride, bicarbonate, and water secretion and therefore has a putative role in production and/or expansion of intestinal mucus. We next determined if poor pathogen containment could be attributed, in part, to defects in the intestinal mucus layer in GC-C knockout mice. We focused on terminal ileum in these studies because small bowel is an important entry point in this model of Salmonella infection. Ileum from naïve and infected mice were processed through alcian blue and periodic acid-Schiff (PAS) stains to visualize mucins (Fig. 4A). Adherent, closely associated mucus was clearly evident as a thin pink line on the outer epithelial cell membrane. Thinning was more severe upon Salmonella infection in GC-C−/− animals and in some portions of the GC-C−/− bowel the mucus layer was completely lost. Similar results were noted in GC-C−/− colon. Measurements of mid-villus mucus thickness confirmed a significantly reduced layer in the knockout mice compared to WT in both naïve and infected states (Fig. 4A, right). Immunofluorescence and quantification of mucin 2 (Muc2), the primary secreted mucin in the intestine demonstrated a significant loss in Salmonella-infected GC-C knockout mice (Fig. 4B,C). Further, realtime RT-PCR indicated that infection significantly reduced Muc2 mRNA in the ileum of GC-C−/− mice relative to WT animals (Fig. 4D).

Mice lacking GC-C produce significantly less mucin at baseline and two days after Salmonella infection. (A) Ilea from naïve mice and those infected (107) with Salmonella for 48 hrs were stained with alcian blue and PAS. Thickness of the adherent mucus layer in both naïve and infected mice was measured using cellSens software. Images were obtained at 400X. (n = 6 mice and 2–5 images per mouse). (B) Ilea of infected mice were stained for mucin 2 (red) and cell nucleus was visualized using DAPI. Images were obtained at 200X. (C) Muc2 fluorescence was quantified and then normalized with DAPI intensity. (D) Realtime RT-PCR analysis of GC-C−/− ileum at 48 hrs post-infection showed a clear decrease in mucin 2 (Muc2) levels, n = 6–8 mice. *P < 0.05, ***P < 0.001, ****P < 0.0001, #P < 0.05 relative to naïve mice of same genotype.
Poor mucus layer homeostasis coupled with Salmonella translocation predicted pro-inflammatory gene expression in the intestine of GC-C−/− mice. Relative to naïve wildtype controls, realtime RT-PCR demonstrated elevated cytokine and chemokine expression in all mice on infection day 2. Notably, there was an increase in interleukin 1 beta (Il1b), monocyte chemoattractant protein 1 (Mcp1), C-X-C motif chemokine ligand 2 (Cxcl2), and C-X-C motif chemokine ligand 5 (Cxcl5) in infected knockout mice compared to WT infected (Fig. 5). Collectively, this series of studies suggest that the GC-C signaling pathway plays an important role in establishing or maintaining the intestinal mucus layer and in minimizing bacterial pathogen invasion and inflammatory gene expression in the intestine.

Cytokine and chemokine expression is elevated in Salmonella-infected GC-C knockout mice relative to control. Total RNA was extracted from ileum tissue of Salmonella infected mice at day 2. Cytokine and chemokine expression was performed using realtime RT-PCR and demonstrated an increase in interleukin 1 beta (IL-1β), monocyte chemoattractant protein 1 (Mcp-1), C-X-C motif chemokine ligand 2 (Cxcl2), and C-X-C motif chemokine ligand 5 (Cxcl5) in GC-C−/− mice. Gene expression was normalized to total RNA collected from ileum of uninfected WT control mice. n = 6–8mice. *P < 0.05.
Infection with high levels of Salmonella leads to poor survival of GC-C knockout mice
To investigate the role of GC-C signaling during a more robust infection and over a longer time frame, we increased the Salmonella dose to 109 CFU and analysis time points to 4 days and beyond. Significant differences in weight loss between WT and GC-C−/− mice were noted at day four (WT 6.55% ± 2.001, GC-C−/− 12.65% ± 1.177, P < 0.05). An elevated initial dose of Salmonella and a longer experimental time point prompted us to assess mucosal inflammation in terminal ileum and proximal colon tissues. Although inflammation was mild to moderate, there was increased mixed immune cell infiltrate in both GC-C knockout mice as compared to WT (Fig. 6A,B). In some studies, we allowed the infection to progress past 4 days and found the GC-C knockout animals developed significant morbidity. Survival of GC-C−/− mice began to drop off as early as day 5 and none of the knockout mice lived past day 10 (Fig. 6C). WT animals survived significantly longer, often past day 15 (P < 0.05). Importantly, western blotting revealed elevated levels of GC-C protein in wildtype animals during days 4 and 8 of the infection (quantitation on day 8 showed an approximately 50% increase; Fig. 6D).

Salmonella infection causes intestinal inflammation and decreased survival in GC-C knockout mice. Mice were dosed with 109 CFU Salmonella. (A) H&E staining of ileum and proximal colon from day 4 infected mice revealed more severe inflammation in GC-C−/− mice. Images were obtained at 200X. (B) Histopathology scoring of inflammation in ileum and proximal colon. (C) Decreased survival in mice lacking GC-C. (D) At days 4 and 8, whole colon protein extracts from wildtype mice were immunoblotted for GC-C and compared to naïve. Day 8 bands from multiple gels were quantitated and graphed. n = 10–12mice. *P < 0.05, **P < 0.01.
Disease course during intravenous Salmonella infection is not affected by GC-C deletion
While the intestine is the predominant site of GC-C expression, this receptor can be found outside the gut. To understand if extra-intestinal GC-C signaling affects the course of Salmonella infection once it translocates from the gut, mice were infected intravenously with Salmonella. We found no genotype-dependent differences in weight loss or liver and spleen CFUs (Supp. Fig. S1). These results suggest that that extra-intestinal expression of GC-C does not play a role in systemic Salmonella spread or expansion.
Exogenous GC-C agonist peptide reduces host-pathogen interactions and bacterial translocation
We next determined if therapeutic activation of GC-C in wildtype animals could reduce bacterial invasion of intestinal epithelia and translocation from the gut. We again used the GC-C agonist STcore because it can be orally delivered and stimulates GC-C throughout the length of the intestinal tract. We focused on localization of Salmonella within the intestine as well as bacterial load in mesenteric lymph nodes as an indicator of Salmonella translocation. Fluorescent in situ hybridization analysis showed Salmonella closely adhered and penetrating the epithelial cell layer of control wildtype mice that had been treated with water (Fig. 7A, left). Wildtype mice dosed with STcore, however, clearly indicated that robust GC-C activation reduced host-pathogen co-localization as Salmonella adherence and movement into the epithelia were substantially minimized (Fig. 7, right). Using high power microscopy, we quantitated the number of Salmonella that had invaded into or completely penetrated the epithelial cell layer and found this to be significantly reduced in the context of exogenous peptide activation of GC-C (Fig. 7B). Consistent with these data, we found that Salmonella bacterial load in mesenteric lymph nodes of STcore-treated wildtype mice was significantly decreased as compared to control animals (Fig. 7C). Notably, GC-C agonist did not impact overall levels of Salmonella colonization in the intestine as bacterial numbers in whole, unflushed cecum (cecum tissue and luminal contents) were similar in water and STcore-treated mice (Fig. 7D).

Activation of GC-C with an exogenous peptide minimizes host-pathogen interactions. Antibiotic-pretreated wildtype mice were infected with Salmonella (107), treated with repeated doses of STcore, and analyzed 24 hours later. (A) Fluorescent in situ hybridization was used to visualize Salmonella in colon of wildtype water- or STcore-treated mice. (400X, insets 1000X). (B) Numbers of Salmonella that had entered or moved below the epithelial cell layer were counted. N = 6–8 mice per group with 2–3 fields per mouse ***P < 0.001. (C) Mesenteric lymph nodes (MLN) were homogenized and plated in order to determine Salmonella colony forming units. *P < 0.05 n = 10–12 mice. (D) Salmonella load was determined in whole, unflushed cecum. n = 10–12 mice. ns: not significant.
Discussion
The current study suggests an important role for GC-C and its ligands in protecting the intestine from invasive bacterial pathogens. To our knowledge, this is the first study showing that GC-C-dependent signaling pathways directly affect physical interactions between invasive bacterial pathogens and the epithelial cell layer of the gut. Using a well-controlled in vitro model system, we show that signaling from the GC-C receptor controls bacterial adherence and invasion into epithelial cells. Enhanced invasion through the epithelial layer and subsequent pathogen dissemination is likely central to the rapid death of GC-C null mice following Salmonella infection. These data expand our understanding of the importance of GC-C during infancy. One aspect of childhood susceptibility to ETEC is elevated GC-C expression during the first few years of life29. We suggest that this early life susceptibility to STa-secreting ETEC is counter balanced by the critical role of GC-C in protecting the mucosa from non-STa producing, invasive bacterial pathogens, such as Salmonella. Concomitant with robust GC-C activity in the infant intestine is the colonization and expansion of commensal microflora and we suggest that additional studies will be necessary to determine if GC-C-regulated microflora complexity impacts susceptibility to invasive pathogen infection20,25,30.
Production, secretion, and extracellular expansion of intestinal mucins are an essential aspect of intestinal innate immunity31. Intestinal mucins are secreted as granulae by goblet cells and expand in the gut lumen. Critical to this process is chloride and bicarbonate secretion by CFTR32,33. Expression of CFTR, GC-C, and Ugn/Gn in enterocytes and/or mucin-secreting goblet cells provides a local environment that is rich in bicarbonate, chloride, and fluid34. There is a significant reduction of mucus release when bicarbonate and fluid transport are inhibited, suggesting the need for continuous secretion in order to maintain physiological mucus production and expansion33. This connection between electrolyte/water release and mucus hydration has important implications for susceptibility to bacterial pathogen infection. It was notable that we found fewer Muc2-positive goblet cells in Salmonella-infected GC-C knockout animals, raising the possibility that goblet cell ablation or dysfunction is an important cause of defective mucus layer in these mice. Importantly, poor mucus layer production and expansion in GC-C knockout mice may be related, in part, to elevated susceptibility to Salmonella infection.
It is likely that the defects in mucus layer deposition in mice lacking GC-C are only partially responsible for high levels of bacterial invasion during Salmonella infection. Using an in vitro model system that produces a negligible mucus layer, we observed diminished adherence but elevated invasion of Salmonella in GC-C knockdown cells as compared to control. We further demonstrated that activation of GC-C above baseline using exogenous ligand (STcore peptide) yields fewer numbers of intracellular Salmonella. While we found STcore to be an important experimental reagent in these studies, significant uncertainty remains about any therapeutic use of GC-C agonists in the context on on-going Salmonella infection. A likely mechanism for reduced bacteria internalization into epithelial cells is GC-C-dependent chloride and water secretion at the epithelial surface. Work by others has shown that Salmonella internalize and translocate at diminished levels in the context of chloride and water secretion elicited by a variety of secretagogues35. We demonstrate that blockade of CFTR also enhances bacterial invasion and that this elevates cell entry caused by GC-C knockdown alone. While highly suggestive, additional work will be needed to determine if this indicates that GC-C blocks bacterial invasion through both CFTR-dependent and –independent pathways. Relevant to this notion is the increased phosphorylation of VASP in cells treated with STcore, supporting a putative role for GC-C signaling in actin cytoskeleton reorganization and the mechanics of Salmonella uptake36. Phosphorylation of VASP at Ser239 and Ser157 is mediated by PKGII and Protein Kinase A, respectively37. This supports previous suggestions of a role for GC-C in direct activation of cGMP-dependent kinases as well as crosstalk with cAMP signaling pathways through cGMP-regulated phosphodiesterases10,11,38. Collectively, these data underscore the importance of this signaling pathway to enteric bacterial pathogenesis and identify areas needing further experimental work to fully determine the mechanistic role for this receptor in pathogen adhesion and invasion in the intestine.
Taken together, our work suggests that GC-C signaling plays a critical role in preventing uptake of invasive bacterial pathogens and we provide in vivo proof-of-concept that activation of this pathway using orally available GC-C agonist peptides is protective during Salmonella infection (Supp. Fig. S2). Although therapeutic GC-C agonists have been extensively studied in the treatment of irritable bowel syndrome and chronic constipation, additional studies focusing on the impact of GC-C agonists on the bacterial microflora of the bowel, pathogenic or otherwise, is warranted20.
Materials and Methods
Mice
Animal procedures were performed according to guidelines approved by the Institutional Animal Care and Use Committee at Cincinnati Children’s. Wildtype and GC-C−/− mice used in this study were bred and housed in microisolator cages within the same room of the Cincinnati Children’s specific pathogen-free barrier facility39,40,41. Colony and study animals, both wildtype control and GC-C−/− mice, were genotyped according to established PCR-based protocols. Mice in all study groups were age and sex-matched and had been bred into a C57BL/6 J background for >10 generations.
Salmonella Typhimurium infection
Salmonella enterica serovar Typhimurium type SL1344 was cultured as described elsewhere42. Most studies herein focused on systemic typhoid fever-like illness that allowed us to investigate bacterial translocation from non-inflamed intestine during the initial days of infection43. In some studies, we fasted mice for four hours and then gave 20 mg oral streptomycin one day prior to infection in order to facilitate greater gut pathogen load during fluorescence in situ hybridization imaging studies44. Bacterial culture concentration was estimated using optical density and confirmed by plating on the day of infection and counting colony forming units (CFUs). For all Salmonella infections, food was withdrawn for 4 hours and then mice were dosed by oral gavage (107 or 109 CFUs in 200 ul per mouse). Organ CFU analysis and inflammation scoring were performed as previously reported22,25. In some studies, wildtype mice were given 10 µg STcore, a 14 amino acid peptide based on ETEC STa, by oral gavage (−4 hrs, −1 hr, at infection, and +1 hr). For intravenous infection, mice were injected (104 CFUs in 100 µl) via tail vein and analyzed 48 hours later.
GC-C shRNA knockdown and adhesion/invasion assays
Lentivirus was produced in human 293T HEK cells using second generation lentivirus vectors and was transduced into the human intestinal Caco-2 Brush Border Epithelial (Caco2-BBE) cell line45,46. Caco2-BBE cells differentiate rapidly at confluence into a polarized monolayer with a well-developed brush border that expresses transporters and digestive enzymes found in small bowel47,48,49. We used two lentivirus short hairpin RNA (shRNA) constructs to target GC-C at different sites on the sequence (Cat#SHCLNG-NM_004963; GUCY2C MISSION® shRNA Bacterial Glycerol Stock, Sigma) (Supp. Table 1). Lentivirus expressing a control, scrambled sequence was designed and is referred to as ‘scramble’. Caco2-BBEs were transduced and selected with puromycin in order to ensure uniform expression of shRNA50. Control (scramble) and GC-C knockdown (shRNA 3280) Caco2-BBE cells were grown at least one week past confluence. Assays were performed as previously described by Gagnon et al. with minor alterations50. After 24 hours of antibiotic-free media, Salmonella was added at 100MOI and incubated at 37 C in a humidified aerobic incubator for 30 min for each adhesion assay. Adhesion was expressed as the percentage of the CFUs from adhered bacteria to CFUs from total bacteria added to each well. For invasion assays, cells were incubated at 37 C for 90 minutes with 100MOI Salmonella after which media was removed. New media supplemented with 150 µg/mL gentamicin (cat# G1397, Sigma) was then added for 60 minutes to kill bacteria that had not been taken into the cell layer. Invasion efficiency was expressed as the percentage of colonies from the cells before (cell-associated) and after (cell-invaded) gentamicin treatment. In the studies using CFTR inhibitor and GC-C agonist, cells were treated with 10 μM CFTR(inh)-172 (Sigma; cat#C2992) and with 1 µM STcore at 90 minutes and 15 minutes, respectively, prior to addition of Salmonella to the media.
Immunoblotting and immunofluorescence
Frozen ileum tissue or Caco2-BBE cells were homogenized and lysed in cold RIPA buffer to collect protein supernatant, immunoblots were performed, and band intensity quantitated as described previously22,25. Primary antibodies pVASP S239 (cat#3114) and pVASP S157 (cat#3111) were purchased from Cell Signaling. GC-C antisera has been published and validated previously40. Muc2 (SC-15334) antibody was used for immunofluorescence was performed as previously detailed (Santa Cruz Biotechnology)25. All images were taken with the same exposure times using an Olympus BX51 microscope running cellSens software (Olympus).
Mucus measurements, fluorescence in situ hybridization, realtime RT-PCR
Tissues were extracted so as not to disturb luminal contents and immediately fixed in Carnoy’s solution or frozen in OCT compound as previously described25. Mucus measurements were obtained following alcian blue and periodic acid-Schiff staining using calibrated imaging software (cellSens, Olympus). Tissue sections were incubated with 1 μM Salmonella FISH DNA Oligo (5′-/5TexRd-XN/TCT CTG GAT TCT GTG GA-3′) at 55 °C for 90 min and at 37 °C overnight. Tissues were then washed and counter-stained with DAPI to visualize nuclei. All images were taken with the same exposure times using an Olympus BX51 microscope running cellSens software (Olympus). Realtime RT-PCR was performed as previously described and primer sequences are listed in Supplemental Table S151.
Statistical analysis
Data were analyzed using t test or ANOVA using GraphPad Prism software. For normally distributed data, significance was determined using an unpaired t test. For data sets found not to be distributed normally, we used nonparametric Mann-Whitney t test. Analysis of variation was coupled with Tukey’s post-hoc test. Data in figures are expressed as mean ± SEM and P values of <0.05 were considered to be statistically significant. All data presented herein was generated from three or more independent experiments.
References
Walker, C. L. et al. Global burden of childhood pneumonia and diarrhoea. Lancet 381, 1405–1416, https://doi.org/10.1016/S0140-6736(13)60222-6 (2013).
Kotloff, K. L. The Burden and Etiology of Diarrheal Illness in Developing Countries. Pediatr Clin North Am 64, 799–814, https://doi.org/10.1016/j.pcl.2017.03.006 (2017).
Field, M., Graf, L. H. Jr., Laird, W. J. & Smith, P. L. Heat-stable enterotoxin of Escherichia coli: in vitro effects on guanylate cyclase activity, cyclic GMP concentration, and ion transport in small intestine. Proc Natl Acad Sci USA 75, 2800–2804 (1978).
Hughes, J. M., Murad, F., Chang, B. & Guerrant, R. L. Role of cyclic GMP in the action of heat-stable enterotoxin of Escherichia coli. Nature 271, 755–756 (1978).
Schulz, S., Green, C. K., Yuen, P. S. & Garbers, D. L. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell 63, 941–948, https://doi.org/10.1016/0092-8674(90)90497-3 (1990).
Currie, M. G. et al. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc Natl Acad Sci USA 89, 947–951 (1992).
Hamra, F. K. et al. Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc Natl Acad Sci USA 90, 10464–10468 (1993).
French, P. J. et al. Isotype-specific activation of cystic fibrosis transmembrane conductance regulator-chloride channels by cGMP-dependent protein kinase II. J Biol Chem 270, 26626–26631 (1995).
Vaandrager, A. B. et al. Differential role of cyclic GMP-dependent protein kinase II in ion transport in murine small intestine and colon. Gastroenterology 118, 108–114 (2000).
Chao, A. C. et al. Activation of intestinal CFTR Cl- channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J 13, 1065–1072 (1994).
De Jonge, H. R. et al. cGMP inhibition of type 3 phosphodiesterase is the major mechanism by which C-type natriuretic peptide activates CFTR in the shark rectal gland. Am J Physiol Cell Physiol 306, C343–353, https://doi.org/10.1152/ajpcell.00326.2013 (2014).
Stahl, K., Stahl, M., de Jonge, H. R. & Forrest, J. N. Jr. ANP and CNP activate CFTR expressed in Xenopus laevis oocytes by direct activation of PKA. J Recept Signal Transduct Res 35, 493–504, https://doi.org/10.3109/10799893.2015.1015738 (2015).
Borowitz, D. et al. Gastrointestinal outcomes and confounders in cystic fibrosis. J Pediatr Gastroenterol Nutr 41, 273–285 (2005).
De Lisle, R. C. Altered transit and bacterial overgrowth in the cystic fibrosis mouse small intestine. Am J Physiol Gastrointest Liver Physiol 293, G104–111, https://doi.org/10.1152/ajpgi.00548.2006 (2007).
Kiela, P. R. et al. Changes in mucosal homeostasis predispose NHE3 knockout mice to increased susceptibility to DSS-induced epithelial injury. Gastroenterology 137, 965–975, 975 e961-910, https://doi.org/10.1053/j.gastro.2009.05.043 (2009).
Laubitz, D. et al. Colonic gene expression profile in NHE3-deficient mice: evidence for spontaneous distal colitis. Am J Physiol Gastrointest Liver Physiol 295, G63–G77, https://doi.org/10.1152/ajpgi.90207.2008 (2008).
Janecke, A. R. et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum Mol Genet 24, 6614–6623, https://doi.org/10.1093/hmg/ddv367 (2015).
Romi, H. et al. Meconium ileus caused by mutations in GUCY2C, encoding the CFTR-activating guanylate cyclase 2C. Am J Hum Genet 90, 893–899, https://doi.org/10.1016/j.ajhg.2012.03.022 (2012).
Fiskerstrand, T. et al. Familial diarrhea syndrome caused by an activating GUCY2C mutation. N Engl J Med 366, 1586–1595, https://doi.org/10.1056/NEJMoa1110132 (2012).
Tronstad, R. R. et al. Guanylate Cyclase C Activation Shapes the Intestinal Microbiota in Patients with Familial Diarrhea and Increased Susceptibility for Crohn’s Disease. Inflamm Bowel Dis 23, 1752–1761, https://doi.org/10.1097/MIB.0000000000001264 (2017).
Muller, T. et al. Congenital secretory diarrhoea caused by activating germline mutations in GUCY2C. Gut 65, 1306–1313, https://doi.org/10.1136/gutjnl-2015-309441 (2016).
Harmel-Laws, E., Mann, E. A., Cohen, M. B. & Steinbrecher, K. A. Guanylate cyclase C deficiency causes severe inflammation in a murine model of spontaneous colitis. PLoS One 8, e79180, https://doi.org/10.1371/journal.pone.0079180 (2013).
Steinbrecher, K. A. et al. Murine guanylate cyclase C regulates colonic injury and inflammation. J Immunol 186, 7205–7214, https://doi.org/10.4049/jimmunol.1002469 (2011).
Garin-Laflam, M. P., Steinbrecher, K. A., Rudolph, J. A., Mao, J. & Cohen, M. B. Activation of guanylate cyclase C signaling pathway protects intestinal epithelial cells from acute radiation-induced apoptosis. Am J Physiol Gastrointest Liver Physiol 296, G740–749, https://doi.org/10.1152/ajpgi.90268.2008 (2009).
Mann, E. A., Harmel-Laws, E., Cohen, M. B. & Steinbrecher, K. A. Guanylate cyclase C limits systemic dissemination of a murine enteric pathogen. BMC Gastroenterol 13, 135, https://doi.org/10.1186/1471-230X-13-135 (2013).
Pham, O. H. & McSorley, S. J. Protective host immune responses to Salmonella infection. Future Microbiol 10, 101–110, https://doi.org/10.2217/fmb.14.98 (2015).
Gayet, R., Bioley, G., Rochereau, N., Paul, S. & Corthesy, B. Vaccination against Salmonella Infection: the Mucosal Way. Microbiol Mol Biol Rev 81, https://doi.org/10.1128/MMBR.00007-17 (2017).
Arora, K. et al. Guanylate cyclase 2C agonism corrects CFTR mutants. JCI Insight 2, https://doi.org/10.1172/jci.insight.93686 (2017).
Cohen, M. B., Guarino, A., Shukla, R. & Giannella, R. A. Age-related differences in receptors for Escherichia coli heat-stable enterotoxin in the small and large intestine of children. Gastroenterology 94, 367–373 (1988).
Steinbrecher, K. A. The multiple roles of guanylate cyclase C, a heat stable enterotoxin receptor. Curr Opin Gastroenterol 30, 1–6, https://doi.org/10.1097/MOG.0000000000000020 (2014).
Johansson, M. E. & Hansson, G. C. Immunological aspects of intestinal mucus and mucins. Nat Rev Immunol 16, 639–649, https://doi.org/10.1038/nri.2016.88 (2016).
Garcia, M. A., Yang, N. & Quinton, P. M. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J Clin Invest 119, 2613–2622, https://doi.org/10.1172/JCI38662 (2009).
Gustafsson, J. K. et al. Bicarbonate and functional CFTR channel are required for proper mucin secretion and link cystic fibrosis with its mucus phenotype. The Journal of Experimental Medicine 209, 1263–1272, https://doi.org/10.1084/jem.20120562 (2012).
Ikpa, P. T. et al. Guanylin and uroguanylin are produced by mouse intestinal epithelial cells of columnar and secretory lineage. Histochem Cell Biol, https://doi.org/10.1007/s00418-016-1453-4 (2016).
Keely, S. et al. Activated fluid transport regulates bacterial-epithelial interactions and significantly shifts the murine colonic microbiome. Gut Microbes 3, 250–260, https://doi.org/10.4161/gmic.20529 (2012).
de Souza Santos, M. & Orth, K. Subversion of the cytoskeleton by intracellular bacteria: lessons from Listeria, Salmonella and Vibrio. Cell Microbiol 17, 164–173, https://doi.org/10.1111/cmi.12399 (2015).
Smolenski, A. et al. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. J Biol Chem 273, 20029–20035 (1998).
Stangherlin, A. et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res 108, 929–939, https://doi.org/10.1161/CIRCRESAHA.110.230698 (2011).
Steinbrecher, K. A., Wowk, S. A., Rudolph, J. A., Witte, D. P. & Cohen, M. B. Targeted inactivation of the mouse guanylin gene results in altered dynamics of colonic epithelial proliferation. Am J Pathol 161, 2169–2178 (2002).
Mann, E. A., Jump, M. L., Wu, J., Yee, E. & Giannella, R. A. Mice lacking the guanylyl cyclase C receptor are resistant to STa-induced intestinal secretion. Biochem Biophys Res Commun 239, 463–466, https://doi.org/10.1006/bbrc.1997.7487 (1997).
Lorenz, J. N. et al. Uroguanylin knockout mice have increased blood pressure and impaired natriuretic response to enteral NaCl load. J Clin Invest 112, 1244–1254, https://doi.org/10.1172/JCI18743 (2003).
Grassl, G. A., Valdez, Y., Bergstrom, K. S., Vallance, B. A. & Finlay, B. B. Chronic enteric salmonella infection in mice leads to severe and persistent intestinal fibrosis. Gastroenterology 134, 768–780, https://doi.org/10.1053/j.gastro.2007.12.043 (2008).
Coburn, B., Grassl, G. A. & Finlay, B. B. Salmonella, the host and disease: a brief review. Immunol Cell Biol 85, 112–118, https://doi.org/10.1038/sj.icb.7100007 (2007).
Barthel, M. et al. Pretreatment of Mice with Streptomycin Provides a Salmonella enterica Serovar Typhimurium Colitis Model That Allows Analysis of Both Pathogen and Host. Infection and Immunity 71, 2839–2858, https://doi.org/10.1128/iai.71.5.2839-2858.2003 (2003).
Tiscornia, G., Singer, O. & Verma, I. M. Production and purification of lentiviral vectors. Nat. Protocols 1, 241–245 (2006).
Cribbs, A. P., Kennedy, A., Gregory, B. & Brennan, F. M. Simplified production and concentration of lentiviral vectors to achieve high transduction in primary human T cells. BMC Biotechnology 13, 98, https://doi.org/10.1186/1472-6750-13-98 (2013).
Tyska, M. J. & Mooseker, M. S. A role for myosin-1A in the localization of a brush border disaccharidase. J Cell Biol 165, 395–405, https://doi.org/10.1083/jcb.200310031 (2004).
Peterson, M. D., Bement, W. M. & Mooseker, M. S. An in vitro model for the analysis of intestinal brush border assembly. II. Changes in expression and localization of brush border proteins during cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci 105(Pt 2), 461–472 (1993).
Peterson, M. D. & Mooseker, M. S. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci 102(Pt 3), 581–600 (1992).
Gagnon, M., Zihler Berner, A., Chervet, N., Chassard, C. & Lacroix, C. Comparison of the Caco-2, HT-29 and the mucus-secreting HT29-MTX intestinal cell models to investigate Salmonella adhesion and invasion. J Microbiol Methods 94, 274–279, https://doi.org/10.1016/j.mimet.2013.06.027 (2013).
Steinbrecher, K. A., Harmel-Laws, E., Sitcheran, R. & Baldwin, A. S. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol 180, 2588–2599, https://doi.org/10.4049/jimmunol.180.4.2588 (2008).
Acknowledgements
We thank Dr. Sing Sing Way for providing Salmonella strain SL1344, Dr. Qishen Pang for providing second generation lentiviral plasmids, and Victoria Summey and Jeff Bailey of the CCHMC Comprehensive Mouse and Cancer Core for intravenous injection service. We also thank Ironwood Pharmaceuticals (Cambridge, MA) for providing STcore peptide. This work was supported by National Institutes of Health [AI107274 and DK047318 to K.A.S.]. This project was supported in part by NIDDK P30 DK078392 Integrative Morphology Core services of the Digestive Disease Research Core Center in Cincinnati.
Author information
Authors and Affiliations
Contributions
All authors performed experiments and analyzed data. S.A. and K.A.S. designed the studies and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Amarachintha, S., Harmel-Laws, E. & Steinbrecher, K.A. Guanylate cyclase C reduces invasion of intestinal epithelial cells by bacterial pathogens. Sci Rep 8, 1521 (2018). https://doi.org/10.1038/s41598-018-19868-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-19868-z
- Springer Nature Limited
This article is cited by
-
Trimethylamine N-oxide: a meta-organismal axis linking the gut and fibrosis
Molecular Medicine (2024)
-
Epigenetic Insights Into Necrotizing Enterocolitis: Unraveling Methylation-Regulated Biomarkers
Inflammation (2024)