
Abstract
The human intestine retains a complex microbial ecosystem, which performs crucial functions that impact on host health. Several studies have indicated that intestinal dysbiosis may impact on the establishment of life-threatening intestinal diseases such as colorectal cancer. An adenomatous polyp is the result of abnormal tissue growth, which is benign but is considered to be associated with a high risk of developing colorectal cancer, based on its grade of dysplasia. Development of diagnostic tools that are based on surveying the gut microbiota and are aimed at early detection of colorectal cancer represent highly desirable target. For this purpose, we performed a pilot study in which we applied a metataxonomic analysis based on 16S rRNA gene sequencing approach to unveil the composition of microbial communities of intestinal polyps. Moreover, we performed a meta-analysis involving the reconstructed microbiota composition of adenomatous polyps and publicly available metagenomics datasets of colorectal cancer. These analyses allowed the identification of microbial taxa such as Faecalibacterium, Bacteroides and Romboutsia, which appear to be depleted in cancerogenic mucosa as well as in adenomatous polyps, thus representing novel microbial biomarkers associated with early tumor formation. Furthermore, an absolute quantification of Fusubacterium nucleatum in polyps further compounded the important role of this microorganism as a valuable putative microbial biomarker for early diagnosis of colorectal cancer.
Similar content being viewed by others

Introduction
Adenomatous polyps or adenomas have long been recognized as precursors of colorectal cancer1,2. Colorectal cancer (CRC) is one of the most commonly diagnosed cancers worldwide and consequently one of the major causes of death in developed countries3. It is known that adenomatous polyps in some cases may evolve to colorectal cancer, even though no currently available scientific evidence unequivocally demonstrate in how many years and what are the precise causes of degeneration. So far there is a growing number of studies that highlight a direct correlation between polyp size, histology and progression of this pathology to CRC4. Furthermore, the molecular causes promoting such malignant transformation of polyps are essentially unknown5. Several factors have been investigated for polyp involvement in cancer development, such as genetics, epigenetics, diet, life style, obesity, alcohol intake and smoking6.
In recent decades, it has become evident that gut bacteria and their metabolites may participate in triggering or progression of colorectal cancer through various proposed mechanisms, including the production of reactive oxygen radicals and other genotoxins7,8,9,10, phenolic compound, and indole production11, as well as conversion of dietary factors into carcinogens and tumor promoters12, and induction of proinflammatory and procarcinogenic pathways in host epithelial cells13,14,15. These proposed mechanisms have an impact in altering the metabolic environment of the host, which may directly or indirectly influence mutagenesis rates and thus carcinogenesis16. Various studies have explored the gut microbiota of individuals with CRC, resulting in the identification of a range of different bacterial groups being associated with carcinogenesis, including Helicobacter pylori, Bacteroides fragilis, Fusobacterium nucleatum, various pathogenic Escherichia coli, Streptococcus gallolyticus and members of the genus Clostridium17,18,19,20,21,22,23,24,25,26. Although screening and lifestyle play important roles in early detection and prevention of colorectal cancer, the identification of new diagnostic tools in particular for the early detection of tumors is important. Previous studies have compared the gut microbiota composition of stool samples from healthy people versus patient with polyps27,28. However, the fecal microbiota is only partially representing the autochthonous microbiota that is in direct contact with the intestinal mucosa29,30. Thus, in order to evaluate the role of the indigenous gut microbiota of subjects undergoing colic mucosectomies of polypoid lesions, we performed a pilot study collecting gut biopsies from healthy mucosa and polyps from the same subjects. On these samples, we performed a meta-analysis involving the here reconstructed microbiota composition of adenomatous polyps and publicly available metagenomic datasets of colorectal cancer based on 16S rRNA microbial profiling data31,32,33 in order to search for the occurrence of common microorganisms between polyps and colon cancer. Altogether, these analyses revealed potential novel microbial biomarkers associated with early malignant transformation.
Materials and Methods
Patient recruitment within a pilot study
At the Gastroenterology and Digestive Endoscopy Unit, University Hospital of Parma, Italy, 12 patients were enrolled, after obtaining informed consent (Table S1). Exclusion criteria included age >of 90, absence of colic polyps in pancolonoscopy, use of antibiotics within the past 3 weeks, inability to sign informed consent. Colonic Mucosa with Polyp (CMP) and Healthy Marginal Tissue (HMT) were obtained from the same subject collected by endoscopy biopsy forceps, thus generating a total of 24 samples (mean age of subjects: 66 ± 12 years old), immediately frozen at −80 °C and stored until further processing for DNA extraction. DNA was extracted from each sample using the PowerViralTM Environmental RNA/DNA Isolation Kit Sample (Qiagen) following the manufacturer’s instructions (Qiagen Ltd, Strasse, Germany). This study protocol was approved by Ethics Committee of Parma Province and all experiments were performed in accordance with relevant guidelines and regulations. Each patient was characterized by demographic data, level of disease indicators and therapy.
16S rRNA gene amplification
Partial 16S rRNA gene sequences were amplified from extracted DNA using primer pair Probio_Uni (5′-CCTACGGGRSGCAGCAG-3′)/Probio_Rev (5′-ATTACCGCGGCTGCT-3′), which targets the V3 region of the 16S rRNA gene sequence34. Illumina adapter overhang nucleotide sequences were then added to the partial 16S rRNA gene-specific amplicons, which in turn were further processed by employing the 16S Metagenomic Sequencing Library Preparation Protocol (Part #15044223 Rev. B – Illumina; see also below). Amplifications were carried out using a Verity Thermocycler (Applied Biosystems). The integrity of the PCR amplicons was analysed by electrophoresis on a 2200 TapeStation Instrument (Agilent Technologies, USA).
MiSeq sequencing of 16S rRNA Gene-based amplicons
PCR products obtained following the amplification of a section of the 16S rRNA gene were purified by a magnetic purification step involving Agencourt AMPure XP DNA purification beads (Beckman Coulter Genomics GmbH, Bernried, Germany) in order to remove primer dimers. The DNA concentration of the amplified sequence library was estimated employing a fluorimetric Qubit quantification system (Life Technologies). Amplicons were diluted to 4 nM and 5 µl of each diluted DNA amplicon sample was mixed to prepare the pooled final library. Paired-end sequencing (250 bp × 2) was performed using an Illumina MiSeq sequencer with MiSeq Reagent Kit v3 chemicals-600 cycles (Illumina Inc., California, USA).
16S rRNA-microbial profiling analysis
The fastq files were processed using QIIME35 as previously described34. Paired-end reads were merged, and quality control implementation allowed the retention of sequences with a length between 140 and 400 bp, mean sequence quality score >25 and with truncation of a sequence at the first base if a low quality sequence within a rolling 10 bp window was found. Sequences with mismatched forward and/or reverse primers were omitted. 16S rRNA Operational Taxonomic Units (OTUs) were defined at ≥97% sequence homology using uclust36 and OTUs with less than 10 sequences were filtered. The Biological Observation Matrix (BIOM) obtained was analysed by summarize_taxa.py script in order to get the relative abundance of each taxonomic group for all samples. All reads were classified to the lowest possible taxonomic rank using QIIME35 and a reference dataset from the SILVA database37. The microbial richness of the samples (alpha-diversity) was evaluated with the Chao1 index through the alpha_rarefaction.py script included in the Qiime software suite using default parameters. Similarities between samples (beta-diversity) were calculated by unweighted uniFrac35,38. Hierarchical clusterings were performed with TMeV 4.8.1 (http://www.tm4.org) using Pearson correlation.
Statistical analyses
SPSS software (www.ibm.com/software/it/analytics/spss/) were used to complete statistical analysis. All data were presented as means ± SD. Furthermore, differential abundance of bacteria taxa and alpha-diversity were tested by one-way analysis of variance (ANOVA).
Quantitative -PCR
qPCR experiments were based on species-specific primers of Fusobacterium nucleatum targeting a previously described prostaglandin transporter-encoding gene39. The genome copy-number and the deduced cell number (since the genes targeted were in single copy per genome) was evaluated by comparing the cycle threshold (Ct) values obtained with those from a standard curve. Standard curves were calculated from serial dilutions of a culture with a known cell number (as determined by viable count assessment) for the bacterial strain versus Ct produced for each target gene. The primer sequences were as follows: forward primer 5′-CAACCATTACTTTAACTCTACCATGTTCA-3′ and reverse primer 5′-GTTGACTTTACAGAAGGAGATTATGTAAAAATC-3′. qPCR was performed using the CFX96 system (BioRad, CA, USA). Each PCR reaction mix contained the following: 12.5 μl 2x SYBR SuperMix Green (BioRad, CA, USA), 1 μl of DNA dilution, each of the forward and reverse primers at 0.5 μM and nuclease-free water was added to obtain a final volume of 20 μl. PCR products were detected with SYBR Green fluorescent dye and amplified according to the following protocol: one cycle of 95 °C for 2 minutes, followed by 42 cycles of 95 °C for 5 s and 60 °C for 30 s. Melting curve: 65 °C to 95 °C with increments of 0.5 °C/s. In each run, negative controls for each primer set were included. Cycle thresholding was calculated using the automated settings for Biorad CFX Manager 3.1 software (BioRad). The entire qPCR experiment was performed a second time using the same samples and methods as outlined above, for the purpose of replication, and very similar results were obtained.
Data Deposition
The 16S rRNA profiling data sequenced in this study were deposited in SRA database under accession number PRJNA415554.
Results and Discussion
Patients and pathological data
A total of twelve patients with polyps were included in this pilot study. From those, histological analyses revealed that four corresponded to adenomatous polyps and eight to hyperplastic polyps. All adenomatous polyps were found in males, while the remaining hyperplastic polyps were collected from three males and five females. Two biopsies were collected from each patient, one corresponding to the polyp and one to healthy mucosa. Patient characteristics and details of endoscopic treatment performed are outlined in Table S1.
Cataloguing of polyp-associated microbiota and comparison to healthy mucosa microbiota
Twelve Colonic Mucosa with Polyp (CMP) and corresponding Healthy Marginal Tissue (HMT) samples (i.e. total of 24 samples), retrieved from twelve hospitalized subjects, were used to evaluate the microbiota taxonomic profile by means of 16S rRNA gene sequencing analysis as described previously34.
Next generation sequencing of these samples produced a total of 2,059,497 sequencing reads with an average of 85,812 reads per sample (Table S2). Subsequent quality and chimera filtering produced a total of 1,479,134 filtered reads with an average of 61,631 reads, and ranging from 33,783 to 202,018 reads per sample (Table S2).
Rating of rarefaction curves based on the Chao1 biodiversity indexes calculated for the two subgroups of sequenced read pools showed that both curves tend to reach a plateau confirming the high accuracy of the performed 16S rRNA profiling analysis. Furthermore, average rarefaction curves revealed a different trend of bacterial richness of the gut microbiota of healthy samples compared to that found in the polyp sample group, i.e. higher level of HMT vs CMP samples (Fig. S1). However, these analyses displayed no statistical significance, since the ANOVA calculated at the highest rarefaction depths reached by all the samples, i.e. Chao1 average is 1895 ± 480 in HMT and 1678 ± 634 in CMP indicated a p-value > 0.05 (Fig. S1).
16S rRNA profiling of HMT and CMP samples
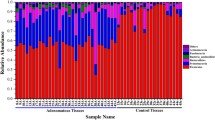
Inspection of predicted taxonomic profiles at phylum level for all samples showed that Firmicutes represented the dominant phylum (average of 57.59% ± 10.48%) of the colonic mucosa samples, out-numbering the Bacteroidetes (average of 9.24% ± 8.01%) and Proteobacteria (average of 10.26% ± 8.19%) phyla.
Nevertheless, no major shifts were observed in bacterial populations between healthy mucosa and Polyp-associated mucosa, although we did observe a moderate reduction of Actinobacteria (relative percentage difference of −13.43%) and Bacteroides (relative percentage difference of −1.02%) in CMP and a concomitant modest increase of Firmicutes (relative percentage difference of 2.84%) compared to HMT (Fig. 1a–c).

Exploration of the diversity in HMT and CMP groups. Panel a represents a bar plot of the bacterial phyla identified in the 24 analyzed samples. Panel b depicts a Venn diagram illustrating the total, unique and shared number of OTUs predicted for HMT and CMP datasets. Panel c shows the variation in terms of relative abundance of >0.4%, and showing either an increase of >10% or decrease of <−16% in CMP data sets as compared to those obtained from HMT samples.
Furthermore, at genus level, we identified 308 genera of which 225 appear to be present in at least one sample of HMT and CMP groups. In detail, 35 bacterial taxa were present in all 24 samples, thus representing the mucosa core microbiota, i.e. the permanent and common members of the microbial community40, which may include specific species important for the maintenance of an efficiently functioning gut ecosystem41 (Fig. 1b).
Moreover, HMT displayed 29 unique taxa representing the 0.015% of the total genera abundance, while CMP showed 19 unique representing 0.008% of the total genera abundance (Fig. 1b).
Taxa associated with HMT and CMP
In order to further identify differences in microbiota composition between CMP and HMT patients, we focused on 30 taxa, which were shown to be present as the absolute change of >±0.1% or that displayed a significant variation in terms of relative abundance (p-value < 0.05). In this context, these 30 taxa varied in terms of relative abundance, encompassing an increase ranging from 10% (Lachnoclostridium) to 641% (Romboutsia), or a decrease ranging from −16% (Ruminococcaceae UCG-002) to −74% (Ruminococcus I) in HMT datasets compared to those obtained from CMP samples (Fig. 1c). Interestingly, members of the Actinobacteria phylum such as the Bifidobacterium genus are present in higher abundance in HMT as compared to CMP samples. Bifidobacteria species are known to represent typical gut microorganisms with presumed positive effects on human health and recently their absence has been linked to various disease conditions42,43,44,45. In addition, the Faecalibacterium genus, which is among bacteria with purported health-promoting activities46, as well as Bacteroides taxa, which have been found at a lower abundance in IBD patients47 are present at a higher level in HMT samples. Moreover, Romboutsia taxa is the genus with the highest relative abundance in HMT vs. CMP samples. Notably, Romboutsia is a recently described bacterial genus48,49 commonly identified in the human gut50,51, and often associated with a healthy status of patients50,51. The drastic reduction of members of this particular genus (−86,51%) in mucosa associated with polyps may represent a potential microbial indicator of a disease condition50.
In contrast, the higher presence of Helicobacter and Klebsiella, which are typically associated with carcinogenesis52,53 in CMP samples reflect a typical disease condition. Interestingly, among taxa showing a significant variation in terms of relative abundance (p-value < 0.05), Unclassified member (U. m.) of Peptostreptococcaceae family occurs with an increased relative abundance of 337.30% in HMT vs. CMP samples, while Peptoclostridium is totally absent in all analyzed CMP samples.
In order to integrate and confirm the observed differences in relative abundance between HMT and CMP samples, we defined the trend (increase or decrease) for each bacterial taxa in healthy subjects versus polyp condition. Considering all patiens, we identified that only the genus Romboutsia displayed the same trend in at least 80% of HMT samples, while all taxa trend of CMP were <75%. These results reinforce the notion that Romboutsia may play a key role in maintaining the health status of the host and render this taxon a very valuable candidate as biomarker of intestinal dysbiosis.
Identification of possible cancer biomarkers
In order to reinforce our taxonomical observation and to enlarge the CMP database, we performed a survey and selection of 16S rRNA-based polyp-associated metagenomic datasets deposited in the publicly NCBI database. However, the published polyp-associated metagenomic datasets are based on fecal samples27,28. In this context, it is worth mentioning that previous studies on healthy patients or children with rectal bleeding indicated that fecal and mucosal samples belonging to the same individual displayed a very dissimilar microbial composition29,30. It has been reported that, based on different statistical analyses, fecal and mucosal samples cluster separately, at family and species taxonomic level29,30. To circumvent the issue of absence of data from polyp-associated biopsies, we decided to use human CRC datasets. In fact, as discussed above, CRC has been considered to represent a later stage of adenomas2,54. Consequently, we retrieved 293 samples from the four publically available CRC BioProjects31,32,33 (Table S3). In detail, we selected the datasets based on Illumina or 454 technologies including both healthy and CRC samples obtained from biopsies collected from the human large intestine (Table S3). Notably, to avoid biases caused by different bioinformatic analyses pipelines55, the sequence reads pools of each study were analyzed involving the same custom script56 based on the QIIME software suite35 used here for the 24 HMT/CMP samples. These datasets were enriched with the CMP and HMT 16S rRNA microbial profiling data obtained and described in our current study.
Evaluation of the difference between all Healthy Tissue (HT) and CRC-polyps Tissue (CpT) samples highlighted 38 genera with a p-value < 0.05 calculated thought Analysis of variance (ANOVA) (Fig. 2). Furthermore, we investigated the difference between each case-control, HT vs. CRC and HMT vs. CMP, in order to calculate the trend percentage.

Exploration of the diversity in HT and CpT groups. The image shows the variation in terms of relative abundance of HT and CpT samples. Only taxa with a p-vale <0.05 were shown.
Association of ANOVA and trend percentage analyses indicated that CpT samples had a higher abundance of genera belonging to Proteobacteria and Fusobacteria phyla, specifically Campylobacter (taxonomic trend prevalence of 100% in CpT) and Fusobacterium (taxonomic trend prevalence of 60% in CpT) compared to healthy samples (Fig. 3). Interestingly, these taxa are commonly found in patients with CRC57, with the Fusobacterium genus frequently being associated with tumor progression58. Moreover, CpT samples possessed microbial genera commonly involved in development of intestinal disease or pathologies, such as Streptococcus (taxonomic trend prevalence of 80.00% in CpT) and Gemella (taxonomic trend prevalence of 80% in CpT)59,60. In contrast, HT samples showed higher abundance of ‘beneficial’ bacterial genera, as well as U. m. of Lachnospiraceae family and Faecalibacterium (taxonomic trend prevalence of 100% in HT), Bacteroides (taxonomic trend prevalence of 80% in HT), and Romboutsia (taxonomic trend prevalence of 60% in HT) compared to CpT.

Evaluation of taxa trend in the gut microbiota of HT and CpT samples. Asterisks indicate statistically significant differences between each case-control for all five datasets (P-value <0.05). The heatmap shows the taxonomic trend of each 16S rRNA gene-based metagenomic dataset. Names of identified bacterial taxa are listed on the left, while names of sample groups used are listed at the top. Healthy Marginal Tissue (HMT); Colonic Mucosa with Polyps(CMP); ColoRectal Cancer (CRC); all Healthy Tissue (HT) and CRC-polyps Tissue (CpT). The green color represents high abundance in healthy cases, red color represents high abundance in disease cases and black color represents the absence of the bacterial taxa. Columns show the studies analyzed and rows indicate the bacterial taxonomy at genus level.
Furthermore, focusing on genera with a taxonomic trend with a prevalence of 100% and a significant p-value (<0.05) calculated between HT and CpT (Fig. 3), it was possible to identify five taxa characteristic of HT and one taxon of CpT subjects. In detail, healthy samples showed high abundance of U. m. of Lachnospiraceae family, Lachnoclostridium, Faecalibacterium, Ruminiclostridium and Subdoligranulum, while disease-associated CpT samples showed high abundance of Campylobacter. These results reflect the assumption that genera belonging to Lachnospiraceae and Ruminococcaceae families may protect healthy subjects from intestinal disease61, such as through the production of short-chain fatty acids (SCFAs)62. In contrast, the higher abundance of bacteria belonging to Campylobacter genus may confirm its correlation with the development of gastrointestinal diseases63,64.
Overall, we found a correspondence between the trend that we retrieved in our HMT vs CMP samples, and that of the HT vs CRC samples, revealing a small number of microbial taxa that could be considered as bacterial biomarkers for early detection of tumor or, conversely, reflecting a healthy condition.
Quantification of Fusobacterium nucleatum by qPCR
Since several studies associated the presence of Fusobacterium nucleatum with a formation and progression of colorectal cancer39,64,65,66,67 the relative abundances of this species were measured by qPCR in all 24 samples (HTM and CMP biopsies of 12 patients). Notably qPCR analyses displayed that this bacterium has been found in six patients (i.e., 2, 5, 6, 7, 9 and 13) with a genome copy number ranging from 2.24E1 to 1.25E3 for the HTM samples, and a genome copy number range of 2.08E2 to 3.26E3 for CMP biopsies (Fig. 4). These data clearly indicated that the total load of Fusobacterium nucleatum was significantly (p-value < 0.04) higher for samples 5, 6, 7 and 9 in the CMP vs. HMT mucosa for each patient. Interestingly, sample 13, showed a total load of Fusobacterium nucleatum higher respect all other samples, i.e., a mean value of 1.02E3 for the HMT as compared to a mean value of 2.37E3 for the CMP (p-value < 0.04) (Fig. 4). These preliminary data suggest that this species can, as previously indicated, not only be considered an important bacterial biomarker for the diagnosis of colorectal cancer39,66, but may also serve as a putative valuable microbial biomarker for early diagnosis of malignant displacement of polyps.

Quantification of Fusobacterium nucleatum by qPCR. Results of qPCR are represented in a grouped graph where each sample is symbolized by an individual value plot. The y-axis represents the genome copy number/gr of Fusobacterium nucleatum and x-axis shows the patient number. For each patient, HTM biopsies are schematically represented by a black circle and relative CMP portion in red square. Statistical differences were calculated by t-test (*P < 0.04).
Conclusions
Recent findings have shown that intestinal diseases are correlated with microbiota alterations such as in the case of ulcerative colitis45,68.
The contribution of gastrointestinal microbiota in the progression and development of colorectal cancer is complex and not fully understood67, but previous in vivo murine based-analyses highlight the potential role of many individual bacteria to promote carcinogenesis28,69.
In the current pilot study, we compared bacterial population in mucosa samples of patients with polyps or CRC, versus healthy mucosa, showing that the Romboutsia genus is more abundant in healthy compared to polyp-associated tissue. Remarkably, the relative abundance of Romboutsia in CRC tissue was found to be drastically reduced. Thus, we may argue that the absence of this microbial genus is a first indicator of an alteration of the mucosa. Remarkably, this microbial taxon has already been observed for other gut-associated diseases49,50,51.
Interestingly, the Peptoclostridium genus was undetectable in all CMP sample tested, while analysis of the HMT samples revealed a relatively high number of U. m. of Peptostreptococcaceae family.
Our findings were in line with the findings of a meta-analysis in which we compared the CMP samples with CRC samples from publically available metagenomic datasets31,32,33. Our metagenomic analysis revealed that CpT samples had a higher abundance of the Fusobacteria phylum compared to healthy samples. Interestingly, the Fusobacterium genus is often involved in tumor progression58,70.
At genus level, we identified five taxa characteristic of HT and one taxon typical of CpT subjects. In detail, healthy samples were shown to contain a high abundance of U. m. of Lachnospiraceae family, Lachnoclostridium, Faecalibacterium, Ruminiclostridium and Subdoligranulum, while CpT samples showed high abundance of Campylobacter. The higher abundance of bacteria belonging to the Campylobacter genus in CRC mucosa, as well as polyp-associated samples reinforces its correlation with the development of intestinal pathologies63,64. On the other hand, genera belonging to Lachnospiraceae and Ruminococcaceae families appear to be correlated to beneficial effects on the host as described previously61.
Gut microbiota include a very complex array of microorganisms and its compositional changes have been shown to impact on human health status71,72. The identification of microbial players that are distinctly altered in their abundance in CRC-associated samples may allow early diagnosis of malignant transformation as well as the identification of protective bacteria missing in polyps or CRC tissue.
In conclusion, the data obtained in this pilot study have allowed us to identify possible microbial biomarkers associated with polyps and/or CRC. However, such findings need further validation by analysis of a larger number of samples, in order to further substantiate and fully characterize such potential microbial biomarkers of CRC development, as well as beneficial bacteria, and to identify their functions and interactions with the host.
References
Fenoglio, C. M. & Lane, N. The anatomical precursor of colorectal carcinoma. Cancer 34, suppl: 819–823 (1974).
Leslie, A., Carey, F. A., Pratt, N. R. & Steele, R. J. The colorectal adenoma-carcinoma sequence. Br J Surg 89, 845–860, https://doi.org/10.1046/j.1365-2168.2002.02120.x (2002).
Boyle, P. & Ferlay, J. Cancer incidence and mortality in Europe, 2004. Ann Oncol 16, 481–488, https://doi.org/10.1093/annonc/mdi098 (2005).
Lieberman, D., Moravec, M., Holub, J., Michaels, L. & Eisen, G. Polyp size and advanced histology in patients undergoing colonoscopy screening: implications for CT colonography. Gastroenterology 135, 1100–1105, https://doi.org/10.1053/j.gastro.2008.06.083 (2008).
Grady, W. M. & Markowitz, S. D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig Dis Sci 60, 762–772, https://doi.org/10.1007/s10620-014-3444-4 (2015).
Marmol, I., Sanchez-de-Diego, C., Pradilla Dieste, A., Cerrada, E. & Rodriguez Yoldi, M. J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int J Mol Sci 18, https://doi.org/10.3390/ijms18010197 (2017).
Mandal, P. Potential biomarkers associated with oxidative stress for risk assessment of colorectal cancer. Naunyn Schmiedebergs Arch Pharmacol 390, 557–565, https://doi.org/10.1007/s00210-017-1352-9 (2017).
Huycke, M. M. & Gaskins, H. R. Commensal bacteria, redox stress, and colorectal cancer: mechanisms and models. Exp Biol Med (Maywood) 229, 586–597 (2004).
Garrett, W. S. Cancer and the microbiota. Science 348, 80–86, https://doi.org/10.1126/science.aaa4972 (2015).
Zitvogel, L., Daillere, R., Roberti, M. P., Routy, B. & Kroemer, G. Anticancer effects of the microbiome and its products. Nat Rev Microbiol 15, 465–478, https://doi.org/10.1038/nrmicro.2017.44 (2017).
Smith, E. A. & Macfarlane, G. T. Formation of Phenolic and Indolic Compounds by Anaerobic Bacteria in the Human Large Intestine. Microb Ecol 33, 180–188 (1997).
Lax, A. J. Opinion: Bacterial toxins and cancer–a case to answer? Nat Rev Microbiol 3, 343–349, https://doi.org/10.1038/nrmicro1130 (2005).
Gounaris, E. et al. Mast cells are an essential hematopoietic component for polyp development. Proc Natl Acad Sci USA 104, 19977–19982, https://doi.org/10.1073/pnas.0704620104 (2007).
Gounaris, E. et al. Live imaging of cysteine-cathepsin activity reveals dynamics of focal inflammation, angiogenesis, and polyp growth. PLoS One 3, e2916, https://doi.org/10.1371/journal.pone.0002916 (2008).
Gounaris, E. et al. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer Res 69, 5490–5497, https://doi.org/10.1158/0008-5472.CAN-09-0304 (2009).
Louis, P., Hold, G. L. & Flint, H. J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 12, 661–672, https://doi.org/10.1038/nrmicro3344 (2014).
Bonnet, M. et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res 20, 859–867, https://doi.org/10.1158/1078-0432.CCR-13-1343 (2014).
Collins, D., Hogan, A. M. & Winter, D. C. Microbial and viral pathogens in colorectal cancer. The Lancet. Oncology 12, 504–512, https://doi.org/10.1016/S1470-2045(10)70186-8 (2011).
Swidsinski, A. et al. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology 115, 281–286 (1998).
Nam, J. H. et al. Helicobacter pylori infection is an independent risk factor for colonic adenomatous neoplasms. Cancer causes & control: CCC 28, 107–115, https://doi.org/10.1007/s10552-016-0839-x (2017).
Purcell, R. V. et al. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS One 12, e0171602, https://doi.org/10.1371/journal.pone.0171602 (2017).
Scanlan, P. D. et al. Culture-independent analysis of the gut microbiota in colorectal cancer and polyposis. Environ Microbiol 10, 789–798, https://doi.org/10.1111/j.1462-2920.2007.01503.x (2008).
Doorakkers, E., Lagergren, J., Engstrand, L. & Brusselaers, N. Helicobacter pylori eradication treatment and the risk of gastric adenocarcinoma in a Western population. Gut, https://doi.org/10.1136/gutjnl-2017-315363 (2018).
Chung, L. et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe, https://doi.org/10.1016/j.chom.2018.01.007 (2018).
Eklof, V. et al. Cancer-associated fecal microbial markers in colorectal cancer detection. Int J Cancer 141, 2528–2536, https://doi.org/10.1002/ijc.31011 (2017).
Aymeric, L. et al. Colorectal cancer specific conditions promote Streptococcus gallolyticus gut colonization. Proc Natl Acad Sci USA 115, E283–E291, https://doi.org/10.1073/pnas.1715112115 (2018).
Hale, V. L. et al. Shifts in the Fecal Microbiota Associated with Adenomatous Polyps. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 26, 85–94, https://doi.org/10.1158/1055-9965.EPI-16-0337 (2017).
Peters, B. A. et al. The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome 4, 69, https://doi.org/10.1186/s40168-016-0218-6 (2016).
Durban, A. et al. Assessing gut microbial diversity from feces and rectal mucosa. Microb Ecol 61, 123–133, https://doi.org/10.1007/s00248-010-9738-y (2011).
Ouwehand, A. C., Salminen, S., Arvola, T., Ruuska, T. & Isolauri, E. Microbiota composition of the intestinal mucosa: association with fecal microbiota? Microbiol Immunol 48, 497–500 (2004).
Geng, J., Fan, H., Tang, X., Zhai, H. & Zhang, Z. Diversified pattern of the human colorectal cancer microbiome. Gut pathogens 5, 2, https://doi.org/10.1186/1757-4749-5-2 (2013).
Burns, M. B., Lynch, J., Starr, T. K., Knights, D. & Blekhman, R. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med 7, 55, https://doi.org/10.1186/s13073-015-0177-8 (2015).
Kostic, A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22, 292–298, https://doi.org/10.1101/gr.126573.111 (2012).
Milani, C. et al. Assessing the fecal microbiota: an optimized ion torrent 16S rRNA gene-based analysis protocol. PLoS One 8, e68739, https://doi.org/10.1371/journal.pone.0068739 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336, https://doi.org/10.1038/nmeth.f.303 (2010).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research 41, D590–D596, https://doi.org/10.1093/nar/gks1219 (2013).
Lozupone, C. & Knight, R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235, https://doi.org/10.1128/AEM.71.12.8228-8235.2005 (2005).
Castellarin, M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22, 299–306, https://doi.org/10.1101/gr.126516.111 (2012).
Astudillo-Garcia, C. et al. Evaluating the core microbiota in complex communities: A systematic investigation. Environ Microbiol 19, 1450–1462, https://doi.org/10.1111/1462-2920.13647 (2017).
Shetty, S. A., Hugenholtz, F., Lahti, L., Smidt, H. & de Vos, W. M. Intestinal microbiome landscaping: insight in community assemblage and implications for microbial modulation strategies. FEMS Microbiol Rev 41, 182–199, https://doi.org/10.1093/femsre/fuw045 (2017).
Ventura, M. et al. Genome-scale analyses of health-promoting bacteria: probiogenomics. Nat Rev Microbiol 7, 61–71, https://doi.org/10.1038/nrmicro2047 (2009).
Hidalgo-Cantabrana, C. et al. Bifidobacteria and Their Health-Promoting Effects. Microbiology spectrum 5, https://doi.org/10.1128/microbiolspec.BAD-0010-2016 (2017).
Arboleya, S., Watkins, C., Stanton, C. & Ross, R. P. Gut Bifidobacteria Populations in Human Health and Aging. Front Microbiol 7, 1204, https://doi.org/10.3389/fmicb.2016.01204 (2016).
Duranti, S. et al. Elucidating the gut microbiome of ulcerative colitis: bifidobacteria as novel microbial biomarkers. FEMS Microbiol Ecol 92, https://doi.org/10.1093/femsec/fiw191 (2016).
Miquel, S. et al. Faecalibacterium prausnitzii and human intestinal health. Current opinion in microbiology 16, 255–261, https://doi.org/10.1016/j.mib.2013.06.003 (2013).
Rajilic-Stojanovic, M. et al. Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 141, 1792–1801, https://doi.org/10.1053/j.gastro.2011.07.043 (2011).
Gerritsen, J. et al. Characterization of Romboutsia ilealis gen. nov., sp. nov., isolated from the gastro-intestinal tract of a rat, and proposal for the reclassification of five closely related members of the genus Clostridium into the genera Romboutsia gen. nov., Intestinibacter gen. nov., Terrisporobacter gen. nov. and Asaccharospora gen. nov. Int J Syst Evol Microbiol 64, 1600–1616, https://doi.org/10.1099/ijs.0.059543-0 (2014).
Gerritsen, J. et al. Genomic and functional analysis of Romboutsia ilealis CRIB(T) reveals adaptation to the small intestine. PeerJ 5, e3698, https://doi.org/10.7717/peerj.3698 (2017).
Ricaboni, D., Mailhe, M., Khelaifia, S., Raoult, D. & Million, M. Romboutsia timonensis, a new species isolated from human gut. New microbes and new infections 12, 6–7, https://doi.org/10.1016/j.nmni.2016.04.001 (2016).
Milani, C. et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Scientific reports 6, 25945, https://doi.org/10.1038/srep25945 (2016).
Mera, R. M. et al. Dynamics of Helicobacter pylori infection as a determinant of progression of gastric precancerous lesions: 16-year follow-up of an eradication trial. Gut. https://doi.org/10.1136/gutjnl-2016-311685 (2017).
Stecher, B., Maier, L. & Hardt, W. D. ‘Blooming’ in the gut: how dysbiosis might contribute to pathogen evolution. Nat Rev Microbiol 11, 277–284, https://doi.org/10.1038/nrmicro2989 (2013).
Dulal, S. & Keku, T. O. Gut microbiome and colorectal adenomas. Cancer journal 20, 225–231, https://doi.org/10.1097/PPO.0000000000000050 (2014).
Duvallet, C., Gibbons, S. M., Gurry, T., Irizarry, R. A. & Alm, E. J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun 8, 1784, https://doi.org/10.1038/s41467-017-01973-8 (2017).
Mancabelli, L. et al. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol Ecol 93, https://doi.org/10.1093/femsec/fix153 (2017).
Otto, C. C., Chen, L. H., He, T., Tang, Y. W. & Babady, N. E. Detection of gastrointestinal pathogens in oncology patients by highly multiplexed molecular panels. Eur J Clin Microbiol Infect Dis 36, 1665–1672, https://doi.org/10.1007/s10096-017-2981-0 (2017).
Abed, J. et al. Tumor Targeting by Fusobacterium nucleatum: A Pilot Study and FuturePerspectives. Frontiers in cellular and infection microbiology 7, 295, https://doi.org/10.3389/fcimb.2017.00295 (2017).
Allali, I. et al. Gut microbiome compositional and functional differences between tumor and non-tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes 6, 161–172, https://doi.org/10.1080/19490976.2015.1039223 (2015).
Chen, Y. et al. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Scientific reports 6, 34055, https://doi.org/10.1038/srep34055 (2016).
Lepage, P. et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 141, 227–236, https://doi.org/10.1053/j.gastro.2011.04.011 (2011).
Flint, H. J., Scott, K. P., Duncan, S. H., Louis, P. & Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306, https://doi.org/10.4161/gmic.19897 (2012).
Akutko, K. & Matusiewicz, K. Campylobacter concisus as the etiologic agent of gastrointestinal diseases. Advances in clinical and experimental medicine: official organ Wroclaw Medical University 26, 149–154, https://doi.org/10.17219/acem/61428 (2017).
Warren, R. L. et al. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 1, 16, https://doi.org/10.1186/2049-2618-1-16 (2013).
McCoy, A. N. et al. Fusobacterium is associated with colorectal adenomas. PLoS One 8, e53653, https://doi.org/10.1371/journal.pone.0053653 (2013).
Kostic, A. D. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215, https://doi.org/10.1016/j.chom.2013.07.007 (2013).
Bullman, S. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448, https://doi.org/10.1126/science.aal5240 (2017).
Chandran, P., Satthaporn, S., Robins, A. & Eremin, O. Inflammatory bowel disease: dysfunction of GALT and gut bacterial flora (I). The surgeon: journal of the Royal Colleges of Surgeons of Edinburgh and Ireland 1, 63–75 (2003).
Wu, S., Morin, P. J., Maouyo, D. & Sears, C. L. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology 124, 392–400, https://doi.org/10.1053/gast.2003.50047 (2003).
Yu, T. et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 170, 548–563 e516, https://doi.org/10.1016/j.cell.2017.07.008 (2017).
Russell, S. L. et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO reports 13, 440–447, https://doi.org/10.1038/embor.2012.32 (2012).
Arrieta, M. C., Stiemsma, L. T., Amenyogbe, N., Brown, E. M. & Finlay, B. The intestinal microbiome in early life: health and disease. Frontiers in immunology 5, 427, https://doi.org/10.3389/fimmu.2014.00427 (2014).
Acknowledgements
Part of this research is conducted using the High Performance Computing (HPC) facility of the University of Parma. The authors declare that they have no competing interests. This work was funded by the EU Joint Programming Initiative – A Healthy Diet for a Healthy Life (JPI HDHL, http://www.healthydietforhealthylife.eu/) and the MIUR to MV. We thank GenProbio srl for financial support of the Laboratory of Probiogenomics. DvS is a member of The APC Microbiome Institute funded by Science Foundation Ireland (SFI), through the Irish Government’s National Development Plan (Grant number SFI/12/RC/2273).
Author information
Authors and Affiliations
Contributions
M.M. performed laboratory experiments, L.M. and C.M. analyzed bioinformatics data, F.G. collected clinical samples and clinical data, N.de’ A., G.L.de’ A. and D.v.S. reviewed the manuscript for important intellectual content, M.V. and F.T. conceived the study, interpreted data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mangifesta, M., Mancabelli, L., Milani, C. et al. Mucosal microbiota of intestinal polyps reveals putative biomarkers of colorectal cancer. Sci Rep 8, 13974 (2018). https://doi.org/10.1038/s41598-018-32413-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32413-2
- Springer Nature Limited
Keywords
This article is cited by
-
Solobacterium moorei promotes the progression of adenomatous polyps by causing inflammation and disrupting the intestinal barrier
Journal of Translational Medicine (2024)
-
Identification of enterotype and its predictive value for patients with colorectal cancer
Gut Pathogens (2024)
-
The comparison of gut microbiota between wild and captive Asian badgers (Meles leucurus) under different seasons
Scientific Reports (2024)
-
Gut Microbiome Composition in Lynch Syndrome With and Without History of Colorectal Neoplasia and Non-Lynch Controls
Journal of Gastrointestinal Cancer (2024)
-
Investigation of the gut microbiome, bile acid composition and host immunoinflammatory response in a model of azoxymethane-induced colon cancer at discrete timepoints
British Journal of Cancer (2023)