
Abstract
The history of the domestication of rice is controversial, as it remains unknown whether domestication processes occurred once or multiple times. To date, genetic architecture and phylogenetic studies based on the rice nuclear genome have been extensively studied, but the results are quite different. Here, we found interesting results for different selections in Oryza sativa based on comprehensive studies of the rice mitochondrial (mt) genome. In detail, 412 rice germplasms were collected from around the world for variant architecture studies. A total of 10632 variants were detected in the mt genome, including 7277 SNPs and 3355 InDels. Selection signal (πw/πc) indicated that the selection sites in Oryza sativa L. ssp. japonica were different from those of Oryza sativa L. indica rice. The fixation index (FST) was higher between indica and japonica than between indica and wild rice. Moreover, haplotype and phylogenetic analyses also revealed indica clusters and japonica clusters that were well separated from wild rice. As mentioned above, our studies indicate that the selection sites of the indica type were different from those of the japonica type. This means that indica and japonica have experienced different domestication processes. We also found that japonica may have experienced a bottleneck event during domestication.
Similar content being viewed by others

Introduction
The domestication of rice is the process of transforming the natural selection process of wild characteristics into the stable desired traits from selection. For African rice, it is well established that Oryza glaberrima was independently domesticated from the wild rice Oryza barthii1. For Asian rice (Oryza sativa L.), although Oryza rufipogon is widely considered to be the ancestor of Asian rice, there is still controversy about the occurrence of single or multiple domestication processes2,3. Asian rice is mainly divided into two major varieties, namely, indica and japonica. Generally, the indica type usually shows thin and long grain and is planted in tropical Asia; japonica rice usually shows short and sticky grain and is planted at high altitudes in South Asia. Both indica and japonica are important food crops for nearly half of the global population4. Exploring the genetic information of these diverse varieties can provide deep insights into rice domestication and breeding.
One of the most basic and controversial issues regarding Asian rice is the number of times domestication occurred2. Traditionally, molecular markers (microsatellites) were used to study certain domesticated genes for domestication history5,6. Since these molecular markers represent some part of the rice genome, whole-genome sequences of rice were used to improve this situation7. However, for nuclear genomic studies of rice, the results are completely different due to introgression, bottleneck events or materials8,9. Generally, for single-domestication studies of rice, the ‘domesticated loci’ that exist in both indica and japonica provide strong evidence to support the single domestication of rice10,11. The large genomic differences and breeding barriers present in indica and japonica directly support multiple independent domestications9,12. Due to the influence of gene flow and bottleneck events, some hypotheses have emerged, such as a single domestication with multiple origins or single origin with multiple introgressions11,13,14. As mentioned above, the history of Asian rice, specifically whether Asian rice stems from a single domestication event or from multiple domestications, remains unknown. Therefore, exploring the genetic information of Oryza sativa is very important as such information may provide more evidence for the domestication of rice and many important insights into the breeding of elite varieties for sustainable agriculture.
Mitochondria are important plastids that provide energy for the growth and evolution of plants. For the mt genome, the genome size ranges from ~200 kb to 2 Mb mostly, and mitochondria have specific modes of gene expression in higher plants15. Since the first entire sequence of the rice mitochondrial genome (490,520 bp) was reported in 200216, mt analysis has been a powerful tool for us to understand the evolutionary history of rice due to the apparent lack of recombination, maternal inheritance, high copy number, and substitution rate17,18,19,20. Although there are some detailed explorations about rice mitochondrial genome such as the variations of rice mitochondrial genome and the comparison between nuclear and chloroplast genome, it is rare to associate them with the domestication of rice19,20,21. What’s more, most of these studies are limited to certain genes or to certain locations of the mitochondrial genome and do not provide evidence for comprehensive analysis. Therefore, we have used 412 rice varieties aiming to provide a comprehensive analysis of the mitochondrial genome to deepen our understanding of the rice genetic and evolutionary background.
Here, we conducted genetic variant analyses of 412 rice germplasms to investigate the evolutionary history of Oryza sativa. First, 358 Asian cultivated rice and 54 wild rice samples were collected from around the world to detect single nucleotide variants (SNPs), and insertions and deletions (InDels) based on the rice mitochondrial genome of Oryza japonica. Then, we used the selective sweep, FST, haplotype network and phylogenetic tree to comprehensively mine the genetic background of Asian rice. Our analysis focuses on the genetic architecture of the rice mt genome, which provides more insight into the evolutionary history of Oryza sativa.
Results
Variants in the mitochondrial genome
The accession information and genome sequencing of all samples are summarized in Supplementary Table S1. A total of 412 rice samples were collected from various parts of the world and sequenced with high average coverage (~16X), yielding ~3.42 TB of read data. The entire collection included 253 temperate japonica, 25 tropical japonica, 66 indica, 9 aus, 2 aromatic rice, 54 wild rice, and 3 admixture types. These germplasms were aligned to the reference mt genome of Oryza sativa japonica [NC_011033.1] for variant calling.
A total of 10,632 primary variants were identified from the rice mt genome, including 7,277 SNPs (68.4%) and 3,355 InDels (31.6%) (Table 1). Since the number of each subgroup is different, we also summarized the average of the variants for each sample (Supplementary Table S2). For all SNPs, transitions appeared most frequently, accounting for 65.3% of all SNPs, almost 2 times of transversions. The type of variant is also summarized, revealing that G/A and C/T seem to be more likely to appear in the mt genome, followed by A/T and T/C (Supplementary Fig. S1 and Supplementary Table S1). After filtering minor allele frequencies (MAFs) <0.01 and variants >20% missing calls, 2,159 high-quality (HQ) variants were obtained for subsequent statistical analysis22,23. For Asian rice, we detected a total of 755 HQ variants, with 75 HQ variants (9.9%) located in the open read frame (ORF) and 52 HQ variants (6.8%) located in the coding region. Among Oryza sativa, we found 49 common SNPs that appeared in 5 subgroups, which means that these SNPs are almost fixed in rice and may play important roles in the mitochondrial genome (Supplementary Fig. S2a). Furthermore, we detected 48 of the same SNPs in O. rufipogon and O. nivara compared with the 49 same SNPs in Oryza sativa (Supplementary Fig. S2b). This means that these common variants (48/49) of Oryza sativa may come from wild rice, and one mutation (1/49) appeared and became fixed due to the drive of selection during domestication. The variants’ distribution in the whole accession and different groups were also targeted based on the reference genome, revealing that wild rice has the highest variant, followed by indica (Fig. 1). Interestingly, these variants showed a cluster distribution in each subgroup, indicating that certain positions of the mitochondria are not allowed to change. This is consistent with a highly conserved mitochondrial genome.

The band distribution of variants (SNPs and InDels) across the mitochondrial genome. The band position is depicted as the distance of the first variant of SNPs or InDels based on the reference genome of Nipponbare. (A–F) Highlights marked on the circle map indicate the SNP and InDel positions. (A) The label name of each gene located in the mitochondrial genome based on the position of the reference genome. (B) Total variants detected among the 412 accessions. (C) Variants identified in the indica subgroup. (D) Variants identified in the temperate japonica type. (E) Variants identified in the tropical japonica type. (F) Variants in wild rice. The outside distance unit is kb. The number inside the brackets indicates the number of each accession. On account of space, not all genes are illustrated in the figure.
The evidence of different selection in Oryza sativa
In genetic analysis, different methods lead to different conclusions due to the presence of hybridization or introgression events8,9. Since mitochondria are highly conserved genomes of maternal inheritance, there is almost no genetic recombination through hybridization. Therefore, whether gene flow present between indica and japonica rice has an important impact on our subsequent analysis at the mt genome level. For recent gene flow, we analyzed the frequency of each group and their distribution based on a physical map of the reference genome7. In our results, we found that indica-specific sites (allele frequency >95% in indica) were different from japonica sites (allele frequency <5% in japonica), which means that there is no gene flow or introgression event in the rice mt genome (Supplementary Table S4). Therefore, our analysis of the genetic history of the rice mitochondrial genome is trustworthy. For rice domestication studies, we first examined the dN/dS ratio (nonsynonymous substitution rate/synonymous substitution rate) of Asian cultivated rice to calculate the evolution rate by coding region (Supplementary Fig. S3). A total of 75 genes were identified from all subgroups, and 23 genes exhibited positive selection. To identify specific positions of rice that were selected, we performed selective sweep analysis based on the diversity of rice in the mitochondrial genome. The diversity of the rice mitochondrial genome ranges from 3.7 × 10−5 to 2.0 × 10−2 (Fig. 1A) (Supplementary Table S5). Wild rice exhibited higher diversity than Asian cultivated rice (P < 0.01) (Fig. 2c). The diversity of subgroups was also analyzed based on the whole variations (SNPs and InDels), and japonica has a lower diversity compared to the other subgroups (Fig. 2b) (Supplementary Table S6). Based on the analysis of diversity, we used πwild/πcultivated of the top 5% cutoff of each Asian rice to determine selection sites (Supplementary Table S7). For 5% cutoff values, we detected a total of 8 selection sites, 4 selection sites for the indica type, and 4 sites for the japonica type. If indica and japonica were only domesticated once, they should be roughly similar in selection sites. Here, in 4000 bp cutoff areas, we only detected a 500 bp (12.5%) similar area between indica and japonica type. The selective sweep of RAiSD analysis was also conducted, which used μ statistics to detect positive selection based on multiple signatures24 (SFS, LD, and diversity) and SNPs (Supplementary Fig. S4). The results revealed that one region of japonica (100–150 kb) had experienced strong selection compared with indica.

Nucleotide diversity and selection analysis of all accessions and subgroups. (a) Nucleotide diversity of all accessions. A 500 bp window size was used in this analysis. (b) Nucleotide diversity of subgroups. The sorted values were plotted in each group. Ind: indica; Niv: O. nivara; Ruf: O. rufipogon; Te_J; temperate japonica; Tr_J; tropical japonica. (c) Nucleotide diversity of cultivated rice and wild rice. (d) The reduction in nucleotides was calculated based on previous diversity analysis. The threshold of the top 5 percentile is indicated as a red dotted circle for indica and blue circle for japonica. The regions within the 2.5 percentile are considered candidate regions under selection. The genome position unit is kb.
F ST, Tajima’s D test, PCA and MDS of populations
The fixation index (FST) was used to determine the degree of differentiation in Oryza sativa based on weighted methods25. Indica and japonica displayed higher FST values compared with wild rice in the mt genome (Fig. 3a). This finding also indicates indica and japonica rice may have reproductive barriers, although the fertility of hybrids varies from individuals26. For Tajima’s D value, temperate japonica and tropical japonica had a similar curve, and indica was shown the different curve in some part of rice mt genome compared with japonica (Fig. 3b). Principal component analysis (PCA) and multidimensional scaling (MDS) discriminated two statistically different groups of Asian cultivated rice (indica and japonica) (Fig. 3c,d). As described above, these findings indicated that mainly Asian rice indica and japonica may have far genetic distances and different genetic backgrounds.

FST, Tajima’s D test, principal component analysis and multidimensional scaling of populations. (a) The FST value between Asian rice and wild rice; the circle size displayed the diversity of each group. The FST value between each group was marked by the length of each line. R: O. rufipogon, Tr: tropical japonica, I: indica, Te: temperate japonica, N: O. nivara. (b) Tajima’s D values in subgroups based on the rice mitochondrial genome. (c) Principle component analysis of indica and japonica. (d) Multidimensional scaling plots of indica and japonica.
Haplotype network, population structure, and phylogenetic tree
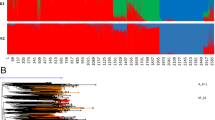
A total of 85 haplotypes were detected from 412 rice samples by DnaSP v6 based on high-quality variations27. Among these haplotypes, 38 haplotypes and 47 haplotypes were found in Asian rice and wild rice, respectively. In Asian rice, indica exhibited 31 haplotypes, whereas japonica only exhibited 4 haplotypes. If indica and japonica were domesticated once, they would have very similar haplotypes. However, we did not identify any shared haplotypes between these two subgroups at the mt level (Fig. 4a). Moreover, population structure from K = 2 to K = 7 were used to entirely distinguish the individual subgroups among the entire collection. To more accurately determine the structure, K = 5 was estimated by ChooseK.py in fastStructure (Fig. 4b). For K = 5, although japonica and wild rice are mixed together, we found a clear separation of indica and tropical japonica. We also found that the same composition exists in indica and japonica (purple color), but this composition was also found in wild rice and does not provide evidence for indica-specific or japonica-specific structure. This same structure could be obtained independently from the wild rice during a separate domestication28. To accurately assess the domestication relationship, we used all HQ SNPs to construct a phylogenetic tree using the Bayesian inference method. If Asian rice was only domesticated once, a tree with these two subpopulations as mixed or sister taxa should be most strongly supported29. However, in our results, japonica and indica types were clearly separated from wild rice (Fig. 4b). The archaeological evidence of Oryza sativa (>9,000 years) in India and China30,31 also exhibited independent domestication of Asian rice. As described above, these results demonstrate that indica and japonica may have a distinct genetic background, which supports the concept of multiple independent domestications of Asian rice.

The haplotype network, population structure and phylogenetic tree of 412 rice accessions. (a) The haplotype network of 412 rice accessions. Here, different colors represent different populations, and circle size represents the number of samples. (b) Population structure and phylogenetic tree are displayed using a rectangular cladogram. A: indica; B: tropical japonica; C: temperate japonica; D: aromatic, aus and admixture type; E: wild rice.
Discussion
The domestication history of Oryza sativa is complex. Although numerous studies on the origin of Asian rice have been conducted, results regarding whether single or multiple domestications occurred are still controversial32,33,34,35. Hybridization and gene flow in the natural state are two important factors affecting rice origin studies36,37,38. Hybridization is the critical step that brings together the high-quality features of the parents, thus disrupting the unique components of different subspecies for evolutionary studies39,40,41. Gene flow is the selection of genes from one species and the movement of such genes to the gene pool of another species13,42. Gene flow results in the genetic differentiation of local populations and plays an important role in genetic studies of specific loci in subgroups28. Civáň et al. (2018) argued that there are some potential alleles that moved to other populations by introgression events in rice, which have a critical impact on distinguishing and understanding the real history of Oryza sativa28. Wang et al. (2017) demonstrated that the different conclusions from rice genome analyses are due to extensive, continuous gene flow from cultivated rice to wild rice35. Fortunately, the mt genome is maternally inherited, and almost no genetic recombination occurs in the natural state, which provides pure and trustworthy materials for phylogenetic studies42,43. We did not detect any introgression signals between indica and japonica at the mt level based on statistical analysis of allele frequency7. Therefore, our mt genome architecture with high-quality variants is useful for solving contradictions in the domestication of Asian rice.
In the evolutionary history of rice, Huang et al. (2016) argued that japonica experienced a strong bottleneck event and that the cutoff of πw/πc should be accurate for collocated low-diversity genomic region (CLDGR) detection13. Based on this, we used the top 5 percentile of genetic diversity for a better selective sweep investigation. We detected a strong selection signal present in japonica rather than indica. In recent articles, the strong bottleneck effect was also revealed in japonica by genome-based position and the magnitude of selective sweeps13,44,45. Our selective sweep results of the rice mt genome were consistent with previous reports that performed chloroplast genome and nuclear analysis, which demonstrated that bottleneck event occurred in japonica during domestication. The comparison of the specific low diversity of a particular group may not necessarily mean independent domestication, as some selection pressures lead to areas of low diversity that may be adaptable to the local environment after the separation of Asian rice. However, if all cultivated rice came from a single domestication, the selective sweep during this event is expected to generate some of the same curves in subspecies. In our results, the selective sweep site in japonica was different from that of indica. Principal component analysis and population structure also confirmed this finding, showing distinct genetic information based on high-quality (HQ) SNPs. Furthermore, the phylogenetic analysis revealed 2 clusters of indica and japonica from wild rice. As described above, this means that japonica and indica may have been selected differently during domestication.
Methods
Samples and resequencing
A heuristic set containing 358 rice accessions with 3 types of accessions (landraces, weedy, and bred) previously generated from worldwide varieties collected from the National GeneBank of the Rural Development Administration (RDA-Genebank, Republic of Korea) using the program PowerCore46 was selected for whole-genome resequencing47. In addition, 54 wild rice accessions were obtained from the International Rice Research Institute (IRRI) in 2017.
For the 358 Asian rice and 54 wild rice accessions from our database, plants were planted in a soft field with enough water. After checking the heading date (approximately 13 days), young leaves were sampled from one plant and stored at –80 °C prior to genomic DNA extraction using the DNeasy Plant Mini Kit (Qiagen). Qualified DNA was used for whole-genome resequencing of the collected rice varieties with an average coverage of approximately 16X on the Illumina HiSeq. 2000 Sequencing Systems Platform.
Variant calling and data management
The assembly process included data preparation, filtering, mapping, sorting, and variant calling. First, the index was processed by Burrows-Wheeler Alignment v 0.7.15 (BWA)48, Samtools v1.3.149 and Picard v 2.14 (http://broadinstitute.github. io/picard/) before variant calling. Second, raw data were aligned to the Nipponbare mt genome sequence (https://www.ncbi.nlm.nih.gov/nuccore/NC_011033.1) using BWA. A sequence alignment map (SAM) file was created during mapping and converted to a binary SAM (BAM) file with sorting. Then, removal of duplicates and the addition of reading group IDs were performed using Picard Tools. Final realignment and identification of variants were performed using GATK v 3.7. Statistical analyses were applied to summarize the number and distribution of variants based on the Haplotype Map (HapMap) file generated from the VCF file. Default settings were used for most software and tools.
Statistical analysis and PCA
Statistical analyses of nucleotide diversity (π) and the fixation index (FST) were conducted using Vcftools v 0.1.1550 with a 1000-bp slide window and 500-bp steps for all collections and individuals. The FST value was used to determine the degree of population differentiation. The significance of diversity in the group was assessed using t-tests. For introgression event analysis, we followed Zhao’s method51. Generally, highly differentiated alleles of SNP loci were identified among indica, temperate japonica, and tropical japonica. SNP loci had an allele frequency greater than 0.95 in temperate indica and less than 0.05 in indica. At the SNP locus, allele information (indica-specific type, temperate-indica-specific type or tropical-japonica-specific type) of each accession was called across the mitochondrial genome. For each accession, the size of the introgression fragment in the genome was determined to estimate the proportion of potential introgression events. The selection effect of the geographic population was generated using Bottleneck v 1.2.0252,53 according to the allele frequency of each site. Regarding the reliability of the results for the detection of population bottleneck effects, minor allele frequencies <0.05 were removed from our data. To evaluate the relationship and population structure, PCA and MDS were conducted using TASSEL5 based on high quality SNPs to provide basic evidence of the population structure. Data were displayed with different groups and colors using the R package ggplot2 (https://cran.r-project.org/web/packages/ggplot2/index.html).
Haplotype network and dN/dS ratios
The TCS54 haplotype network was generated using PopART v 1.755. First, we used a python script to make FASTA data from the vcf file. Then, FASTA data alignment and transformation to nex format was performed using MEGA7. DnaSP v627 was employed for haplotype analysis (Supplementary Table S9). For dN/dS analyses, all orthologous mt genes from 23 species were aligned to the paml format using prank56. Gblocks v 0.91b57 was applied to eliminate the conservation area of the ML tree (MEGA7). The maximum likelihood method of codeml of PAML v 4.9h58 was used to estimate the ω ratio with F3X4 codon frequencies. The branch test of the null hypothesis (model = 0, NSsites = 0) was used for a single ω across branches, and the model alternative hypothesis (model = 2, NSsites = 0) was used for ω per branch site. The likelihood ratio test (LRT) was used to identify accelerated genes in the rice group. Here, indica and japonica were assigned as foreground branches, and other accessions were assigned as background branches. Genes with ω > 5 were removed because they were considered outliers59.
Population structure and evolution research
Briefly, fastStructure v 1.060 was used to investigate population clusters. InDels were removed from all high-quality (HQ) variants to obtain SNP only vcf file. Given increased K values ranging from 2 to 7, the subpopulation of an individual ancestry could be completely investigated. Bayesian inference methods were applied to construct a phylogenetic tree for the 412 accessions based on the HQ variants. After removing missing data and gaps from whole positions, the phylogenetic tree of evolutionary history was conducted by MrBayes v3.2.761 with the best nucleotide parameter (TVM + G) estimated by detection from 88 models with the software of JModelTest v 2.1.1062, with 1000 replicates and 6 categories.
Data Availability
The datasets supporting the conclusions of this article are included within the article and its additional files. In addition, the raw VCF file generated from current 412 rice accessions were also deposited in the European Variant Archive Database under Project ID: PRJEB31784.
References
Wang, M. et al. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nature genetics 46, 982 (2014).
Londo, J. P., Chiang, Y.-C., Hung, K.-H., Chiang, T.-Y. & Schaal, B. A. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proceedings of the National Academy of Sciences 103, 9578–9583 (2006).
Molina, J. et al. Molecular evidence for a single evolutionary origin of domesticated rice. Proceedings of the National Academy of Sciences 108, 8351–8356 (2011).
Khush, G. S. What it will take to feed 5.0 billion rice consumers in 2030. Plant molecular biology 59, 1–6 (2005).
Shomura, A. et al. Deletion in a gene associated with grain size increased yields during rice domestication. Nature genetics 40, 1023 (2008).
Garris, A. J., Tai, T. H., Coburn, J., Kresovich, S. & McCouch, S. Genetic structure and diversity in Oryza sativa L. Genetics 169, 1631–1638 (2005).
Wang, W. et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 557, 43 (2018).
Huang, X. et al. A map of rice genome variation reveals the origin of cultivated rice. Nature 490, 497 (2012).
Civáň, P., Craig, H., Cox, C. J. & Brown, T. A. Three geographically separate domestications of Asian rice. Nature plants 1, 15164 (2015).
Gao, L.-z & Innan, H. Nonindependent domestication of the two rice subspecies, Oryza sativa ssp. indica and ssp. japonica, demonstrated by multilocus microsatellites. Genetics 179, 965–976 (2008).
Choi, J. Y., Platts, A. E., Fuller, D. Q., Wing, R. A. & Purugganan, M. D. The rice paradox: multiple origins but single domestication in Asian rice. Molecular biology and evolution 34, 969–979 (2017).
Sang, T. & Ge, S. Genetics and phylogenetics of rice domestication. Current opinion in genetics & development 17, 533–538 (2007).
Huang, X. & Han, B. Rice domestication occurred through single origin and multiple introgressions. Nature plants 2, 15207 (2016).
Choi, J. Y. & Purugganan, M. D. Multiple origin but single domestication led to Oryza sativa. G3: Genes, Genomes, Genetics 8, 797–803 (2018).
Schuster, W. & Brennicke, A. The plant mitochondrial genome: physical structure, information content, RNA editing, and gene migration to the nucleus. Annual review of plant biology 45, 61–78 (1994).
Notsu, Y. et al. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: frequent DNA sequence acquisition and loss during the evolution of flowering plants. Molecular Genetics and Genomics 268, 434–445 (2002).
Ingman, M., Kaessmann, H., Pääbo, S. & Gyllensten, U. Mitochondrial genome variation and the origin of modern humans. Nature 408, 708 (2000).
Lang, B. F., Gray, M. W. & Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annual review of genetics 33, 351–397 (1999).
Tian, X., Zheng, J., Hu, S. & Yu, J. The rice mitochondrial genomes and their variations. Plant Physiology 140, 401–410 (2006).
Sun, C., Wang, X., Yoshimura, A. & Doi, K. Genetic differentiation for nuclear, mitochondrial and chloroplast genomes in common wild rice (Oryza rufipogon Griff.) and cultivated rice (Oryza sativa L.). Theoretical and Applied Genetics 104, 1335–1345 (2002).
Mun, J., Song, Y., Heong, K. & Roderick, G. Genetic variation among Asian populations of rice planthoppers, Nilaparvata lugens and Sogatella furcifera (Hemiptera: Delphacidae): mitochondrial DNA sequences. Bulletin of Entomological Research 89, 245–253 (1999).
Zeggini, E. et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nature genetics 40, 638 (2008).
Troyanskaya, O. et al. Missing value estimation methods for DNA microarrays. Bioinformatics 17, 520–525 (2001).
Alachiotis, N. & Pavlidis, P. RAiSD detects positive selection based on multiple signatures of a selective sweep and SNP vectors. Communications biology 1, 79 (2018).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. evolution 38, 1358–1370 (1984).
Harushima, Y., Nakagahra, M., Yano, M., Sasaki, T. & Kurata, N. Diverse variation of reproductive barriers in three intraspecific rice crosses. Genetics 160, 313–322 (2002).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular biology and evolution 34, 3299–3302 (2017).
Civáň, P. & Brown, T. A. Role of genetic introgression during the evolution of cultivated rice (Oryza sativa L.). BMC evolutionary biology 18, 57 (2018).
Yang, C.-c et al. Independent domestication of Asian rice followed by gene flow from japonica to indica. Molecular biology and evolution 29, 1471–1479 (2011).
Liu, L., Lee, G.-A., Jiang, L. & Zhang, J. Evidence for the early beginning (c. 9000 cal. BP) of rice domestication in China: a response. The Holocene 17, 1059–1068 (2007).
Fuller, D. Q., Allaby, R. G. & Stevens, C. Domestication as innovation: the entanglement of techniques, technology and chance in the domestication of cereal crops. World archaeology 42, 13–28 (2010).
Cheng, C. et al. Polyphyletic origin of cultivated rice: based on the interspersion pattern of SINEs. Molecular Biology and Evolution 20, 67–75 (2003).
Lin, Z. et al. Origin of seed shattering in rice (Oryza sativa L.). Planta 226, 11–20 (2007).
Zhu, Q. & Ge, S. Phylogenetic relationships among A‐genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytologist 167, 249–265 (2005).
Wang, H., Vieira, F. G., Crawford, J. E., Chu, C. & Nielsen, R. Asian wild rice is a hybrid swarm with extensive gene flow and feralization from domesticated rice. Genome research 27, 1029–1038 (2017).
Wilson, H. D. Artificial hybridization among species of Chenopodium sect. Chenopodium. Systematic Botany, 253–263 (1980).
Motley, T. J. & Carr, G. D. Artificial hybridization in the Hawaiian endemic genus Labordia (Loganiaceae). American Journal of Botany 85, 654–660 (1998).
Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145, 1219–1228 (1997).
Wilson, H. D. J. S. B. Artificial hybridization among species of Chenopodium sect. Chenopodium. 253–263 (1980).
Motley, T. J. & Carr, G. D. J. A. J. O. B. Artificial hybridization in the Hawaiian endemic genus Labordia (Loganiaceae). 85, 654–660 (1998).
Harrison, R. G. & Larson, E. L. Hybridization, introgression, and the nature of species boundaries. Journal of Heredity 105, 795–809 (2014).
Freeman, S. & Herron, J. C. Evolutionary analysis. (Pearson Prentice Hall Upper Saddle River, N. J., 2007).
Lonsdale, D., Brears, T., Hodge, T., Melville, S. E. & Rottmann, W. The plant mitochondrial genome: homologous recombination as a mechanism for generating heterogeneity. Philosophical Transactions of the Royal Society of London. B, Biological Sciences 319, 149–163 (1988).
Civán, P., Craig, H., Cox, C. J. & Brown, T. A. Multiple domestications of Asian rice. Nature plants 2, 16037 (2016).
Vigueira, C. C. et al. Call of the wild rice: Oryza rufipogon shapes weedy rice evolution in Southeast Asia. Evolutionary applications 12, 93–104 (2019).
Kim, K.-W. et al. PowerCore: a program applying the advanced M strategy with a heuristic search for establishing core sets. Bioinformatics 23, 2155–2162 (2007).
Kim, T.-S. et al. Genome-wide resequencing of KRICE_CORE reveals their potential for future breeding, as well as functional and evolutionary studies in the post-genomic era. BMC genomics 17, 408 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Zhao, Q. et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nature genetics 50, 278 (2018).
Cornuet, J. M. & Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144, 2001–2014 (1996).
Piry, S., Luikart, G. & Cornuet, J. BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. Journal of heredity 90, 502–503 (1999).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Molecular ecology 9, 1657–1659 (2000).
Leigh, J. W. & Bryant, D. popart: full-feature software for haplotype network construction. Methods in Ecology and Evolution 6, 1110–1116 (2015).
Löytynoja, A. In Multiple sequence alignment methods 155–170 (Springer, 2014).
Talavera, G. & Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Systematic biology 56, 564–577 (2007).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Molecular biology and evolution 24, 1586–1591 (2007).
Castillo-Davis, C. I., Hartl, D. L. & Achaz, G. cis-Regulatory and protein evolution in orthologous and duplicate genes. Genome research 14, 1530–1536 (2004).
Raj, A., Stephens, M. & Pritchard, J. K. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nature methods 9, 772–772 (2012).
Acknowledgements
This work was carried out with the support of the “Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ013405)” Rural Development Administration, Republic of Korea. This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2017R1A2B3011208).
Author information
Authors and Affiliations
Contributions
Y.P. conceived the project and oversaw writing the manuscript. Y.P., K.K. and L.C. developed the idea and edited and revised the manuscript. L.C. performed the data analysis and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cheng, L., Kim, KW. & Park, YJ. Evidence for selection events during domestication by extensive mitochondrial genome analysis between japonica and indica in cultivated rice. Sci Rep 9, 10846 (2019). https://doi.org/10.1038/s41598-019-47318-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47318-x
- Springer Nature Limited
This article is cited by
-
A large-scale population based organelle pan-genomes construction and phylogeny analysis reveal the genetic diversity and the evolutionary origins of chloroplast and mitochondrion in Brassica napus L.
BMC Genomics (2022)
-
Early Domestication History of Asian Rice Revealed by Mutations and Genome-Wide Analysis of Gene Genealogies
Rice (2022)