
Abstract
Non-small cell lung cancer (NSCLC) is featured with complex genomic alterations. Molecular profiling of large cohort of NSCLC patients is thus a prerequisite for precision medicine. We first validated the detection performance of a next-generation sequencing (NGS) cancer hotspot panel, OncoAim, on formalin-fixed paraffin-embedded (FFPE) samples. We then utilized OncoAim to delineate the genomic aberrations in Chinese NSCLC patients. Overall detection performance was powerful for mutations with allele frequency (MAF) ≥ 5% at >500 × coverage depth, with >99% sensitivity, high specificity (positive predictive value > 99%), 94% accuracy and 96% repeatability. Profiling 422 NSCLC FFPE samples revealed that patient characteristics, including gender, age, lymphatic spread, histologic grade and histologic subtype were significantly associated with the mutation incidence of EGFR and TP53. Moreover, RTK signaling pathway activation was enriched in adenocarcinoma, while PI(3)K pathway activation, oxidative stress pathway activation, and TP53 pathway inhibition were more prevalent in squamous cell carcinoma. Additionally, novel co-existence (e.g., variants in BRAF and PTEN) and mutual-exclusiveness (e.g., alterations in EGFR and NFE2L2) were found. Finally, we revealed distinct mutation spectrum in TP53, as well as a previously undervalued PTEN aberration. Our findings could aid in improving diagnosis, prognosis and personalized therapeutic decisions of Chinese NSCLC patients.
Similar content being viewed by others

Introduction
Lung cancer is the leading cause of cancer-associated mortality worldwide. Up to 90% lung cancer is non-small cell lung cancer (NSCLC)1. NSCLC is usually diagnosed at the advanced stage with limited therapeutic options (e.g., surgery, radiotherapy and chemotherapy)1.
Conventional molecular detection techniques, such as fluorescence in situ hybridization and Sanger sequencing, only detect a limited number of biomarkers2,3,4. The advent of next-generation sequencing (NGS) has broadened the landscape of genetic aberrations5,6,7, making it possible to implement targeted treatment tailored for specific mutations in individual patients1,8,9.
Lung cancer is characterized with complex genomic aberrations10. The most frequently mutated genes in NSCLC include EGFR (Epidermal growth factor receptor), TP53 (Tumor Protein p53), KRAS (Kirsten rat sarcoma viral oncogene) and PIK3CA (Phosphatidylinositol 3-kinase). In addition, Genetic mutations detected in NSCLC are complicated by patient demographic, racial, clinical and pathological characteristics1,8,9,11. For instance, the mutational frequency of EGFR can vary from 10% in Western populations to 30% in Asians, and KRAS mutation incidence in Western populations is approximately 18-26% versus 3.8-8% in Asians12, and within Asian populations, the distinct ethnicities bear EGFR aberrations at a rate ranging from 19.6% to 40.1%11. A comprehensive database of genetic aberrations in large cohorts of NSCLC patients is thus required for clinical interpretation of variants1,10. Subtyping of lung cancer based on specific molecular markers and understanding the potential association of patient genetic alterations with clinicopathological characteristics may help improve early NSCLC diagnosis, prevent NSCLC incidence, and guide more beneficial treatments6,13. For example, NSCLC patients with EGFR mutations and ALK (Anaplastic lymphoma kinase) gene rearrangement will benefit from targeted therapy of EGFR-TKIs (Tyrosine kinase inhibitors) (such as erlotinib and afatinib)14 and ALK inhibitors (crizotinib, ceratinib, etc.)15, respectively.
The tumor samples are typically preserved as formalin-fixed paraffin-embedded (FFPE), which causes DNA fragmentation and crosslinking. The reduced DNA quality in FFPE samples, along with normally low mutation frequency and tumor heterogeneity, necessitates higher sequencing coverage for reliable mutation calling16,17,18. Thus, targeted sequencing–enriching the regions of interest prior to sequencing–is more reliable and cost-effective compared with genome-wide sequencing7,19. However, stringent validation of NGS-based cancer detection in FFPE specimens for clinical testing is still very rare.
Here we developed and validated an amplification-based cancer hotspot panel, OncoAim (Singlera Genomics, Shanghai, China). The panel covers the mutational hotspots of 59 genes implicated in common cancers including lung cancer. We evaluated the detection performance of OncoAim on single nucleotide variants (SNVs) and short insertions and deletions (INDELs) in FFPE specimens using Ion Torrent PGM (Personal Genome Machine) platform. Our results established that OncoAim was suitable for clinical application in FFPE samples. This assay, along with our findings in Chinese NSCLC patients could aid in improving the diagnosis, prognosis and personalized therapeutic decisions of Chinese NSCLC patients.
Results
Performance validation of OncoAim on SNVs
For SNVs detection performance (sensitivity and specificity), we used 4 individual reference standards (i.e., HD200, HD300, HD301, and HD802) and 4 mixed reference standards (i.e., HD300-HD706, HD802-HD260, HD301-HD706, and HD200-HD706). These references together possessed 54 known SNVs with MAF ranging from 1% to 70.0% (Table S3a). The median sequencing coverage depth of these 8 libraries were between 1025× and 1469× (Table S3b). The overall SNVs detection performance was high: >99% (40/40) of expected SNVs with MAF ≥ 5% were successfully detected, and 79% (11/14) of alterations at MAF < 5% were also identified (Table S3c). In addition, high specificity was maintained with a PPV > 99% (51/51).
We then evaluated the impact of sequencing coverage depth (50-1000×) and MAF on SNV detection by randomly subsampling data (Tables S3a,c). Detection sensitivity steadily increased as coverage depth increased in all MAF sections (<5%, 5-10%, and ≥10%) (Fig. 1A), and high sensitivity was obtained up to 500× median coverage depth (Fig. 1A). At 500×, >99% (400/400) of SNVs with expected MAF ≥ 5% were successfully detected. Furthermore, PPV remained high (>99%) across the full coverage depth range (Table S4). We observed a high correlation between expected and observed MAFs (Fig. 1C), highlighting the robust quantitative characteristics of OncoAim test.

SNV and INDEL detection performance of OncoAim (panels A,C for SNV; panels B,D for INDEL). (A,B) Detection sensitivity was plotted against the median sequencing coverage depth. Error bars, standard error of the mean (SEM). (C,D) Detected allele frequencies were in concordance with those expected in reference standards.
Performance validation of OncoAim on INDELs
For INDELs detection validation, we used 3 individual reference standards (i.e., HD200, HD300, and HD802) and 3 mixed reference standards (i.e., HD200-HD706, HD300-HD706, and HD802-HD260). These references contain 7 known INDELs with a wide range of MAF and INDELs length (3-15 bp) (Table S5a). The average sequencing coverage depth across six samples was 1232× (Table S5b). We observed high overall detection performance: >99% (5/5) of expected INDELs with MAF ≥ 5% were successfully detected (Table S5c). The PPV was >99% (5/5).
The effects of sequencing coverage depth and MAF on INDELs detection performance were assessed by subsampling data (Table S5a,c). The INDELs detection sensitivity increased with increasing coverage depth at MAF ≥ 5%, and high sensitivity was observed up to 700× median coverage depth. At 700×, >99% of INDELs at MAF ≥ 5% (15/15) were successfully detected. We observed a fluctuating trend of detection sensitivity with varying coverage depth at low MAF (<5%) (Fig. 1B). Moreover, PPV achieved was 85–100% across the full coverage depth (Table S6). The correlation between measured and expected MAF for INDELs was high (Fig. 1D).
Concordance between OncoAim NGS test and orthogonal ARMS-PCR test
We tested the EGFR mutation status in 253 FFPE NSCLC specimens (a subset of 452 total cases) using both OncoAim and ARMS-PCR (Amplification refractory mutation system-Polymerase Chain Reaction) technique. In total, two methods detected 126 EGFR mutations with 94% concordance (119 aberrations were detected by both platforms) (Fig. 2). Six variants were detected by ARMS-PCR but not by NGS (Table S7), likely due to the low MAF of these variants and/or tumor heterogeneity. NGS, but not ARMS-PCR, detected one mutation that was confirmed as TP by Sanger sequencing (Table S7).

Concordance between OncoAim and ARMS-PCR approaches on FFPE specimens. Overlap of positive variant calls by NGS (OncoAim) and ARMS-PCR methods at tested sites in 253 FFPE clinical cancer specimens. Note: among the 119 common variants, 106 variants with MAF ≥ 5% was called by OncoAim when the bioinformatics analysis pipeline used 5% as cut-off for variant calling. The other 13 variants missed by OncoAim analysis pipeline (5% cut-off) had MAF < 5%, which were detected but filtered out. These 13 variants were also counted as detected by OncoAim here for better comparison of two methods.
Reproducibility of NGS test on FFPE samples
We assessed the precision (reproducibility and repeatability) of OncoAim test with 3 FFPE tumor samples that possess 5 known alterations (SNVs and INDELs including KRAS p.G12V, KRAS p.G12D, TP53 p.R248W, TP53 p.R333fs*12, EGFR p.L858R) in total. Among these 5 known alterations, mutations of KRAS and EGFR were detected previously by ARMS-PCR method, and TP53 mutations were detected previously by NGS (Illumina Miseq platform). The samples were analyzed 5 times in 3 different experiments to evaluate the inter-run and intra-run reproducibility. Concordance between replicates was 96% (Table S8), with no significant differences between inter- and intra-run replicates, demonstrating the robustness of the NGS test.
Mutational profiling of Chinese NSCLC patients
A total of 422 cases of the 452 FFPE samples was successfully sequenced for subsequent variation analysis (Fig. S1), and 30 cases were unsuccessful for several reasons, such as poor DNA quality, low library concentration, and low median read coverage and uniformity. We then utilized OncoAim to profile the mutational landscape of 422 Chinese NSCLC patients. The clinicopathological characteristics of patients were summarized in Table 1.
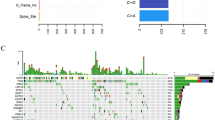
Approximately 70% (295) of 422 FFPE samples possessed at least 1 mutation. In total, 479 mutations in 21 genes were identified (Fig. 3). The gene mutation frequency, calculated by dividing the number of mutations in individual gene by the number of patients, ranged from 0.24% to 50%. The top 5 most frequently mutated genes were EGFR (211, 50%), Tumor protein p53 (TP53) (149, 35%), KRAS (24, 5%), phosphatidylinositol-4,5-bisphosphat3-kinase, catalytic subunit alpha (PIK3CA) (21, 5%), and phosphatase and tensin homolog (PTEN) (16, 4%) (Fig. 3). Some patients had more than one EGFR mutation, as 211 EGFR alterations were detected from 176 samples (Fig. 4). In addition, one patient acquired two PTEN mutations.

Mutated genes, the number of aberrations and the mutation frequency relative to the number of patients recruited in this study.

Oncoplot depicting the top 10 most frequently mutated genes. The frequency is calculated by dividing the number of variants in the specific gene by the number of patients with at least one mutation identified. Top panel shows the number of aberrations detected in each sample. The right panel displays number of variants in each gene.
Missense mutation was the most prevalent form in almost all the top 10 mutated genes, with PTEN as the exception in which in-frame-insertion was the major type (Fig. 4). In-frame-deletion and nonsense mutation ranked in 2nd place in EGFR and TP53 mutations, respectively. All mutations in KRAS, PIK3CA, nuclear factor erythroid-derived 2-like 2 (NFE2L2) and Catenin, beta-1 (CTNNB1) were missense type (Fig. 4).
Mutational hotspots in the top 5 most frequently mutated genes
The mutational hotspots of the 5 most frequently mutated genes are summarized in Fig. 5.

Mutational hotspots of the top 5 most frequently mutated genes including EGFR (A), TP53 (B), KRAS (C), PIK3CA (D), and PTEN (E). All the variants on KRAS were labelled. For other genes, only the most prevalent and critical variants were marked. The protein domains harboring these mutational hotspots were noted.
EGFR is a transmembrane tyrosine kinase belonging to HER/erbB protein family. Its activation can activate downstream PI3K-AKT-mTOR and RAS-RAF-MEK-ERK signaling pathways20. The most observed mutations in EGFR were L858R missense mutation (exon 21) and an in-frame deletion from E746 to A751 (exon 19) (Fig. 5A), consistent with previous reports21. These mutations confer increased sensitivity to both first-generation and second-generation EGFR Tyrosine kinase inhibitors (TKIs)21,22,23. Notably, the T790M variant, which is often induced by TKIs treatment and thus a resistance marker to first-generation TKIs (erlotinib and gefitinib), was detected 8 times (approximately 4% of the 211 EGFR mutations) (Fig. 5A). T790M mutation has been suggested as the primary mutation in some NSCLC patients24.
TP53 is one of the best-known tumor suppressor genes. Since TP53 protein forms a tetramer for binding to cis-elements, some of its missense mutants may have dominant-negative effects on wild-type TP53 via oligomerization25,26. We noticed 73.83% of TP53 aberrations were the missense type. TP53 mutations were clustered in the DNA binding domain (amino acid 101-300), with R273 (7.09%), G266 (5.67%), R213 (4.96%), R175 (4.26%), and R280 (4.26%) as the top 5 most frequently mutated sites (Fig. 5B). Two of these 5 residues, R273 and R175, belong to the previously reported 6 mutational hotspots of TP53 (i.e., R175, G245, R248, R249, R273 and R282)26. Among these 6 reported residues, R248 and R282 also had high frequency in the present study, both at 3.55%. However, G245 and R249 showed up at low frequency, 1.42% and 0.71%, respectively, indicating they were not prevalent in Chinese NSCLC patients. R213* nonsense mutation was the most common nonsense mutant (Fig. 5B), consistent with a previous study26. However, the high mutation frequency of R213* in our study has not previously been reported. This result, together with the observed high mutation frequency of G266 and R280, might represent the unique features of TP53 mutations in Chinese NSCLC patients.
KRAS is a member of the small GTPase superfamily whose activating mutations can constitutively activate downstream RAF-mediated signaling pathway, leading to uncontrolled cell proliferation27. NSCLC patients carrying KRAS mutations develop primary resistance to EGFR-targeted drugs such as cetuximab and gefitinib28. G12 was the most dominant mutated residue in our study (Fig. 5C), consistent with other reports1. Lung cancer patients with KRAS G12C and G12V mutations have shorter PFS (progression-free survival) compared with those patients carrying other KRAS mutations or KRAS wild type29.
PIK3CA is one of the catalytic subunits of phosphatidyl 3-kinases (PI3K), modulating various cellular processes30. We identified E542, E545 (exon 10, the helical domain) and H1047 (exon 21, the kinase domain) as the most frequently mutated sites (Fig. 5D). These PIK3CA mutations are regarded as oncogenic variants and can be targeted for drug development31.
PTEN acts as a tumor suppressor, and its best-known role is to antagonize the PI3K signaling pathway through its lipid phosphatase activity32. A333*fs10 (COSM5346961) in the C2 domain was the most frequently observed PTEN aberration in our cohort (Fig. 5E). The C2 domain is crucial for anchoring the PTEN catalytic domain onto the membrane.
Association of patient clinicopathological characteristics with mutations
Associations of mutations with the following clinicopathogical characteristics, gender, age, smoking etc were evaluated (Table 2).
Mutations in lung cancer can be associated with gender1. We observed that female patients had significantly higher frequency of EGFR mutations (P = 1.41E-11, Fisher’s exact test) but a significantly lower mutation frequency of TP53 (P = 8.912E-08) and PTEN (P = 3.2E-2) (Table 2 and Fig. S2), consistent with previous reports5,33,34. Interestingly, PTEN mutations were more prevalent in male patients.
Compared with the middle-aged group (45-65 years old), the young-aged group (18-45 years old) had a significantly lower EGFR mutation rate (P = 1.16E-03, adjusted by Bonferroni’s correction) (Table 2 and Fig. S3), which agrees with other studies35,36. Mutations in TP53 were less frequent in the young-aged group as opposed to both the middle-aged and older group (>65 years), with P values of 2.45E-06 and 2.59E-04, respectively (Table 2).
Smoking is a crucial risk factor for lung cancer1,9. The increased frequency of KRAS mutation in smokers (P = 1.7E-02) (Table 2 and Fig. S4) is consistent with other reports24,27. We observed a positive correlation of PIK3CA aberrations with smoking status (P = 1.7E-02) (Table 2), inconsistent with a previous study in Japanese lung cancer patients37. This discrepancy might result from ethnic variations.
Lymph node metastasis is a strong independent predictor of poor prognosis38. The lung tumors with lymphatic spreading were enriched with mutations in EGFR (P = 4E-03) and TP53 (P = 1.3E-02) (Table 2 and Fig. S5), indicating that tumors with EGFR or TP53 mutations were more likely to develop metastatic tumors and might have poorer prognosis. These findings were consolidated by a previous report showing that EGFR expression level is higher in lung cancer patients with lymph node metastasis39.
Lung cancer, at later developing stages, tends to spread to other body parts forming metastatic lung tumors. Compared with primary lung tumors, metastatic lung tumors had higher mutational frequency of EGFR (P = 1.6E-02, Table 2 and Fig. S6). Cautious screening and monitoring of potential metastasis should be performed for patients with primary lung tumors that harbor EGFR driver mutations.
Histologic grade of lung cancer is an independent predictor of patient survival40. We observed that the well differentiated tumor has significantly lower mutation frequency of EGFR (P = 5.25E-06, adjusted by Bonferroni’s correction) and TP53 (P = 3.54E-04, adjusted by Bonferroni’s correction) than moderately differentiated tumors (Table 2 and Fig. S7). The well-differentiated tumors also have lower EGFR and TP53 mutation frequencies compared with poorly differentiated tumors, but these differences did not reach a significant level (P = 3.99E-01 and 1.14E-01, adjusted by Bonferroni’s correction, respectively). The low mutation frequency of EGFR and TP53 in well-differentiated tumors might represent a marker for better prognosis.
Adenocarcinoma (ACA) and squamous cell carcinoma (SCC) are the two major histological subtypes of NSCLC, and they are featured with distinct gene expression profiles with different clinical implications41. While EGFR mutations were more prevalent in ACA (P = 9.12E-07), the mutations of TP53, PIK3CA, and NFE2L2 were enriched in SCC, with P values of 1.56E-08, 1.8E-02, and 2E-03, respectively (Table 2 and Fig. S8). These results indicated that RTK (Receptor Tyrosine Kinase) signaling pathway activation was enriched in ACA, whereas PI(3)K pathway activation, oxidative stress pathway activation, and TP53 pathway inhibition were more prevalent in SCC. Although such correlations for each gene have been previously reported42,43,44,45, this study is the first to reveal the histological subtype association of these 4 genes in one lung cancer cohort study. The transcription factor NFE2L2 plays central roles in modulating the expression of genes involved in antioxidant and stress-response46, and its D29Y, D29H and R34Q variants (on exon 2) detected in this study might indicate poor prognosis of lung cancer42,47.
We didn’t find differentially mutated genes in different tumor sites (left lung vs. right lung) (Table 2 and Fig. S9).
Mutually exclusive and co-occurring variants in NSCLC
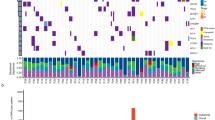
Our data demonstrated EGFR was mutated in a mutually exclusive fashion with KRAS, TP53, NFE2L2, and B-Raf proto-oncogene, serine/threonine kinase (BRAF) (Fig. 6). The mutual exclusiveness between EGFR and KRAS or BRAF has been well-known20,48,49. This is the first evidence showing NFE2L2 mutations did not coexist with EGFR mutations, consolidating the oncogenic nature of NFE2L2 mutations47. Interestingly, TP53 mutations were mutually exclusive with EGFR mutations to a significant level (Fig. 6). This new observation was, to some extent, supported by the finding that RAS and TP53 show mutual exclusiveness in acute myeloid leukemia50, suggesting that inactivating TP53 alone may be sufficient for lung cancer cells to proliferate and circumvent apoptosis.

Mutual exclusiveness and co-existance of variants. (A) Oncostip plot displayed the distribution of all the mutations across the samples containing at least one aberration in EGFR, TP53, KRAS, PTEN, NFE2L2, and BRAF. (B) Mutually exclusive gene pairs or co-occurring gene pairs were depicted.
We observed that PTEN mutations tended to coincide with BRAF mutations (Fig. 6). The PTEN mutations result in activation of the PI(3)K pathway. Studies in lung cancer have not been reported on the co-occurrence of mutations in PTEN and BRAF, but the PIK3CA mutations have been associated with BRAF mutations51. In melanoma, however, loss-of-function PTEN mutations and BRAF activation mutations coexisted52. These findings indicate that in some types of lung cancer, the PI(3)K pathway activation through loss of inhibition due to PTEN mutations can cooperate with BRAF-dependent RTK signaling pathway activation to promote cancer development.
Discussion
With deeper understanding of the underlying genetic mutations revolutionized by NGS technology, molecular testing has become an indispensable tool for lung cancer diagnosis1,10. In this study, we first performed an extensive performance evaluation of a NGS panel, OncoAim, on FFPE samples, then explored the genetic variants in Chinese NSCLC patients.
The turnaround time of the entire NGS process is about 2–3 days. We demonstrated that the targeted NGS had high sensitivity, specificity, accuracy and precision for both SNVs and INDELs. False-negative calls were predominantly low-frequency variants (MAF < 5%). Detection sensitivity rose up as sequencing coverage depth increased, up to 100% for MAF ≥ 5% variants at a certain coverage depth (500× for SNVs and 700× for INDELs) (Fig. 1A,B). High specificity was obtained across the whole sequencing coverage range (50-1000×). OncoAim test exhibited high concordance (94%) with ARMS-PCR approach for MAF ≥ 5% variants (Fig. 2), indicative of its high detection accuracy. Taken together, we concluded that the NGS-based OncoAim test had robust performance in FFPE samples. OncoAim panel covers common mutations (6000 hotspots in 59 cancer genes) in six prevalent Chinese cancer types (Oesophagus, stomach, liver, lung, breast and colon). Thus, in addition to lung cancer, it may be applied to other cancers. Further prospective studies of this panel on lung cancer and other cancer types should be performed to establish its application in clinical assay.
In five cases, EGFR exon 19 deletion was detected by ARMS-PCR, but not by NGS, which accounted for the major discordances observed between these two techniques. ARMS-PCR is super sensitive and can robustly identify alterations with 1% MAF53, whereas NGS can only reliably call variants with MAF ≥ 5%, particularly for INDELs54. Therefore, the difference in analytical sensitivity between these two methods may lead to inconsistencies in the test results. The low MAF could be caused by intratumoral heterogeneity, that is, the same tumor may possess cells that harbor different subclones with distinct mutations55,56.
We didn’t perform side-by-side comparison of OncoAim panel with previous genetic profiling tests. However, we noticed that most of our discoveries are consistent with previous reports1,10. For example, EGFR, TP53, KRAS, and PIK3CA were the top most frequently mutated genes in NSCLC. EGFR mutations were more common in female patients, and smokers tended to have higher mutation incidence of KRAS, and EGFR mutations were mutually exclusive with KRAS mutations. This validated the quality and effectiveness of our panel and bioinformatics pipeline, also reflected the robustness of these molecular signatures in NSCLC across populations with distinct demographic and racial background. In addition, our test sensitivity (>99% for variants with MAF ≥ 5% at >500X coverage depth) was comparable with that (95–99%) reported by Frampton, G.M.54.
We have also identified novel mutational patterns and novel correlations of genomic aberrations with patient characteristics in Chinese NSCLC. One intriguing finding is high mutation incidence of R213, G266 and R280 but low mutation incidence of G245 in TP53 (Fig. 5B). These mutations together may aid in examining tumorigenesis, epidemiology, and therapeutic decisions of NSCLC in Chinese population57. The high frequency of PTEN A333*fs10 represents a previously undervalued genomic variant in Chinese NSCLC, and the future functional characterization of this variant for clinical diagnosis and drug development is well justified. For the first time, we revealed the significant correlation of mutations in 4 genes, including EGFR, TP53, NFE2L2 and PIK3CA, with the specific histologic subtype of NSCLC in a single cohort study (Table 2), emphasizing the value of utilizing these molecular markers for subclassifying NSCLC patients and unearthing the distinct potential tumorigenesis mechanisms for NSCLC histologic subtypes. Moreover, OncoAim uncovered the previously unknown mutual exclusiveness of NFE2L2 mutations with EGFR mutations, which highlighted the oncogenic nature of NFE2L2.
As a retrospective study using archived FFPE specimens, one caveat of the current study is that not all the patients’ clinicopathological information was available. The small number of patients in certain characteristic groups might limit the power of statistical analysis. For example, BRAF mutations are more prevalent in nonsmokers than in smokers (P = 0.019)58. Although we detected BRAF mutations only in nonsmokers, statistical analysis failed to show difference between nonsmokers and smokers (Table 2), likely because only 13 smokers existed in our cohort.
In summary, the present findings based on NGS test could aid in subdividing NSCLC patients according to specific molecular signatures, improving diagnosis and prognosis, and implementing precision and personalized treatment for Chinese NSCLC patients.
Methods
Ethics statement
This work has been approved by the West China Hospital Sichuan University Clinical Trials and Biomedical Ethics Committee (No. 2017-114). All methods were performed in accordance with the relevant guidelines and regulations and informed consent was obtained from all participants.
Reference standards
We purchased 6 commercial reference standards (HD200, HD300, HD301, HD706, HD802, and HD260) from Horizon Diagnostics (Saint Louis, USA). These reference standards carry known mutation sites and mutation frequencies. Among them, HD200 is a Multiplex Reference Standard with known mutations in EGFR, KRAS, NRAS, PIK3CA and KIT (KIT Proto-Oncogene Receptor Tyrosine Kinase), and others are Gene-Specific Multiplex (HD260: EGFR V769_D770insASV Reference Standard; HD300: EGFR Gene-Specific Multiplex Reference Standard; HD706, EGFR V769_D770insASV Reference Standard; HD802, EGFR Gene-Specific Multiplex Reference Standard; HD301: KRAS Gene-Specific Multiplex Reference Standard).
FFPE samples
FFPE samples of 452 NSCLC patients were collected from the archives of the following hospitals from 2013 to 2016 (Table 1): Peking University Third Hospital; Tongji Hospital, Tongji Medical College of Huazhong University of Science & Technology; Xijing Hospital, Fourth Military Medical University; and The First Affiliated Hospital, Zhejiang University. The inclusion criteria for this study are: (1) clear diagnosis of non-small cell lung cancer; (2) samples within 5 years; (3) tumor cell content ≥20%. A 4-μm section of a hematoxylin and eosin-stained slide was reviewed by a pathologist to ensure a sample volume ≥1 mm3, nucleated cellularity ≥80% or ≥30000 cells and tumor cell content ≥20%. Clinicopathological information was gathered for association analysis.
DNA extraction, library preparation and sequencing
DNA of FFPE samples was extracted using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) strictly according to the manufacturer’s protocol. The extracted DNA was quantified using the Qubit dsDNA HS Assay kit and a Qubit 3.0 fluorimeter (Life Technologies). Libraries were constructed from 20 ng DNA and the OncoAim DNA Panel using the Ion AmpliSeq Library kit v2.0-96LV (Life technologies). The panel covers more than 6000 highly frequent mutation hotspots in 59 cancer genes (Table S1). Libraries were quantified with Qubit 3.0 fluorometer (Life technologies). The individual libraries were diluted and then pooled for generating an 8 pM library to amplify on Ion sphere particles (ISP) on the Ion One Touch 2 instrument (Life technologies). ISP templates were enriched, loaded on an Ion 318 chip and sequenced on the PGM sequencer (Life Technologies).
Sequencing data analysis
OncoAim panel pipeline (OncoAim version 7.2) was used for sequencing data analysis. Briefly, the quality of raw reads (fastq files) was evaluated with FastQC (version 0.9.5, Babraham Bioinformatics, Cambridge, UK). High-quality reads were aligned against the human reference genome (hg19). The Burrows-Wheeler Aligner algorithm (https://github.com/lh3/bwa) was utilized for alignment, using default parameters. Insertions and deletions in sequence alignment files were left-aligned using a custom software tool, and left-aligned reads were processed using Freebayes (https://github.com/ekg/freebayes) for variant calling. The median coverage per locus was 500–1000x to ensure confident variant calling. The minimum mutation allele frequency (MAF) for SNVs and INDELs was set to 5%. Variants were annotated for effect prediction and clinical practice guidance. All the variants were manually checked on the Integrative Genomics Viewer (https://www.broadinstitute.org/software/igv/home).
Performance validation
For SNV and indel validation, the six reference standards mentioned above were used. We mixed some of the reference standards by a 1:1 ratio to obtain more mutation sites and a wide MAF range. Then, individual standard and mixed standards were sequenced, targeting to >1000× coverage depth. Each bam file of sequencing data exported from the sequencer was randomly sampled (random selection of subsets of reads) to examine performance over a wide coverage depth range (50–1000×). We sampled 10 times for 50–500× and 3 times for 550–1000×. These sampled data were analyzed to identify variants.
For sensitivity analysis, all tested variants were assigned either a true positive (TP) if detected in the reference standards or false negative (FN) if not detected. Sensitivity at each test site was calculated as detected times/sampling times. For specificity analysis, each called variant was classified as a TP if the variant was a known mutation in the reference standards or a parental cell line, or a false positive (FP) if the variant was not a known mutation. Positive predictive value (PPV) was calculated as TP/ (TP + FP).
For accuracy analyses, we compared OncoAim panel with AmoyDx EGFR Mutations Detection Kit that uses the principle of Amplified Refractory Mutation System (ARMS) (Amoy Diagnostics, Xiamen, China). For inconsistent results, Sanger sequencing was performed using the specified primers (Table S2).
We validated test reproducibility by examining mutation calls in replicates of clinical FFPE specimens. The samples were analyzed 5 times (5 independent library preparations starting from the same extracted DNA) in three different experiments (including three replicates in a single run).
Statistical analysis
The statistical analysis was done using R (R version 3.4.1). For two nominal variables, two-sided Fisher’s exact test was performed. For analysis involving more than 2 groups, following two-sided Fisher’s exact test, pairwise comparison with Bonferroni’s correction was performed. All tests were two-sided, and P < 0.05 was considered significant. Mutual exclusivity of variants was analyzed with R package ‘maftools’ (https://github.com/PoisonAlien/maftools), which performed Fisher’s exact test for mutual exclusive events.
Data availability
All data generated or analyzed during this study are included in this article (and its Supplementary Information Files).
References
Gridelli, C. et al. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 1, 15009, https://doi.org/10.1038/nrdp.2015.9 (2015).
Yan-Fang, T. et al. Analyzing the gene expression profile of pediatric acute myeloid leukemia with real-time PCR arrays. Cancer Cell Int. 12, 40, https://doi.org/10.1186/1475-2867-12-40 (2012).
Ross, J. S. Update on HER2 testing for breast and upper gastrointestinal tract cancers. Biomark Med. 5, 307–318, https://doi.org/10.2217/bmm.11.31 (2011).
Liu, Y. et al. A comparison of ARMS and direct sequencing for EGFR mutation analysis and tyrosine kinase inhibitors treatment prediction in body fluid samples of non-small-cell lung cancer patients. J. Exp. Clin. Cancer Res. 30, 111, https://doi.org/10.1186/1756-9966-30-111 (2011).
Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550, https://doi.org/10.1038/nature13385 (2014).
Cummings, C. A., Peters, E., Lacroix, L., Andre, F. & Lackner, M. R. The Role of Next-Generation Sequencing in Enabling Personalized Oncology Therapy. Clin. Transl. Sci. 9, 283–292, https://doi.org/10.1111/cts.12429 (2016).
Kamps, R. et al. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int J Mol Sci 18, https://doi.org/10.3390/ijms18020308 (2017).
Nawaz, K. & Webster, R. M. The non-small-cell lung cancer drug market. Nat. Rev. Drug. Discov. 15, 229–230, https://doi.org/10.1038/nrd.2016.42 (2016).
Govindan, R. et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150, 1121–1134, https://doi.org/10.1016/j.cell.2012.08.024 (2012).
Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550, https://doi.org/10.1038/nature13385 (2014).
El-Telbany, A. & Ma, P. C. Cancer genes in lung cancer: racial disparities: are there any? Genes. Cancer 3, 467–480, https://doi.org/10.1177/1947601912465177 (2012).
Schabath, M. B., Cress, D. & Munoz-Antonia, T. Racial and Ethnic Differences in the Epidemiology and Genomics of Lung Cancer. Cancer Control. 23, 338–346, https://doi.org/10.1177/107327481602300405 (2016).
Davidson, M. R., Gazdar, A. F. & Clarke, B. E. The pivotal role of pathology in the management of lung cancer. J. Thorac. Dis. 5(Suppl 5), S463–478, https://doi.org/10.3978/j.issn.2072-1439.2013.08.43 (2013).
Fukuoka, M. et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J. Clin. Oncol. 29, 2866–2874, https://doi.org/10.1200/JCO.2010.33.4235 (2011).
Wang, Z. et al. Anaplastic lymphoma kinase gene rearrangement predicts better prognosis in NSCLC patients: A meta-analysis. Lung Cancer 112, 1–9, https://doi.org/10.1016/j.lungcan.2017.07.029 (2017).
Van Allen, E. M. et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 20, 682–688, https://doi.org/10.1038/nm.3559 (2014).
Kerick, M. et al. Targeted high throughput sequencing in clinical cancer settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med. Genomics 4, 68, https://doi.org/10.1186/1755-8794-4-68 (2011).
Yost, S. E. et al. Identification of high-confidence somatic mutations in whole genome sequence of formalin-fixed breast cancer specimens. Nucleic Acids Res. 40, e107, https://doi.org/10.1093/nar/gks299 (2012).
Sun, Y. et al. Next-generation diagnostics: gene panel, exome, or whole genome? Hum. Mutat. 36, 648–655, https://doi.org/10.1002/humu.22783 (2015).
Marchetti, A. et al. EGFR mutations in non-small-cell lung cancer: analysis of a large series of cases and development of a rapid and sensitive method for diagnostic screening with potential implications on pharmacologic treatment. J. Clin. Oncol. 23, 857–865, https://doi.org/10.1200/jco.2005.08.043 (2005).
Mitsudomi, T. & Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. Febs j. 277, 301–308, https://doi.org/10.1111/j.1742-4658.2009.07448.x (2010).
Rosell, R. et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 13, 239–246, https://doi.org/10.1016/S1470-2045(11)70393-X (2012).
Mok, T. S. et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 361, 947–957, https://doi.org/10.1056/NEJMoa0810699 (2009).
Li, S. et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: a comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer 110, 2812–2820, https://doi.org/10.1038/bjc.2014.210 (2014).
Olivier, M., Hollstein, M. & Hainaut, P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008, https://doi.org/10.1101/cshperspect.a001008 (2010).
Soussi, T. & Wiman, K. G. TP53: an oncogene in disguise. Cell Death Differ. 22, 1239–1249, https://doi.org/10.1038/cdd.2015.53 (2015).
Riely, G. J., Marks, J. & Pao, W. KRAS mutations in non-small cell lung cancer. Proc. Am. Thorac. Soc. 6, 201–205, https://doi.org/10.1513/pats.200809-107LC (2009).
Chong, C. R. & Janne, P. A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 19, 1389–1400, https://doi.org/10.1038/nm.3388 (2013).
Kim, E. S. et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 1, 44–53, https://doi.org/10.1158/2159-8274.CD-10-0010 (2011).
Karakas, B., Bachman, K. E. & Park, B. H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 94, 455–459, https://doi.org/10.1038/sj.bjc.6602970 (2006).
Engelman, J. A. et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 14, 1351–1356, https://doi.org/10.1038/nm.1890 (2008).
Planchon, S. M., Waite, K. A. & Eng, C. The nuclear affairs of PTEN. J. Cell Sci. 121, 249–253, https://doi.org/10.1242/jcs.022459 (2008).
Tseng, J. E. et al. Gender differences in p53 mutational status in small cell lung cancer. Cancer Res. 59, 5666–5670 (1999).
Shigematsu, H. et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J. Natl. Cancer Inst. 97, 339–346, https://doi.org/10.1093/jnci/dji055 (2005).
Wu, S. G., Chang, Y. L., Yu, C. J., Yang, P. C. & Shih, J. Y. Lung adenocarcinoma patients of young age have lower EGFR mutation rate and poorer efficacy of EGFR tyrosine kinase inhibitors. ERJ Open Res. 3, https://doi.org/10.1183/23120541.00092-2016 (2017).
Ueno, T. et al. Impact of age on epidermal growth factor receptor mutation in lung cancer. Lung Cancer 78, 207–211, https://doi.org/10.1016/j.lungcan.2012.09.006 (2012).
Kawano, O. et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer 54, 209–215, https://doi.org/10.1016/j.lungcan.2006.07.006 (2006).
Nosotti, M. et al. Lymph node micrometastases detected by carcinoembryonic antigen mRNA affect long-term survival and disease-free interval in early-stage lung cancer patients. Oncol. Lett. 4, 1140–1144, https://doi.org/10.3892/ol.2012.880 (2012).
Zhuo, Y. et al. Correlation study and significance of the EGFR expression in serum, lymph nodes and tumor tissue of NSCLC. Thorac. Cancer 5, 31–37, https://doi.org/10.1111/1759-7714.12048 (2014).
Barletta, J. A., Yeap, B. Y. & Chirieac, L. R. Prognostic significance of grading in lung adenocarcinoma. Cancer 116, 659–669, https://doi.org/10.1002/cncr.24831 (2010).
Shiran, I. et al. Non-Small-cell Lung Cancer Patients With Adenocarcinoma Morphology Have a Better Outcome Compared With Patients Diagnosed With Non-Small-cell Lung Cancer Favor Adenocarcinoma. Clin. Lung Cancer 18, 316–323 e311, https://doi.org/10.1016/j.cllc.2017.01.009 (2017).
Sasaki, H. et al. NFE2L2 gene mutation in male Japanese squamous cell carcinoma of the lung. J. Thorac. Oncol. 5, 786–789, https://doi.org/10.1097/JTO.0b013e3181db3dd3 (2010).
Yu, J. et al. PIK3CA mutation in Chinese patients with lung squamous cell carcinoma. Chin. J. Cancer Res. 25, 416–422, https://doi.org/10.3978/j.issn.1000-9604.2013.08.03 (2013).
Lindeman, N. I. et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Mol. Diagn. 15, 415–453, https://doi.org/10.1016/j.jmoldx.2013.03.001 (2013).
Gibbons, D. L., Byers, L. A. & Kurie, J. M. Smoking, p53 mutation, and lung cancer. Mol. Cancer Res. 12, 3–13, https://doi.org/10.1158/1541-7786.mcr-13-0539 (2014).
Qian, Z. et al. Nuclear factor, erythroid 2-like 2-associated molecular signature predicts lung cancer survival. Sci. Rep. 5, 16889, https://doi.org/10.1038/srep16889 (2015).
Kim, Y. R. et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. J. Pathol. 220, 446–451, https://doi.org/10.1002/path.2653 (2010).
Pao, W. et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2, e17, https://doi.org/10.1371/journal.pmed.0020017 (2005).
Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 455, 1069–1075, https://doi.org/10.1038/nature07423 (2008).
Hua, X. et al. MEGSA: A Powerful and Flexible Framework for Analyzing Mutual Exclusivity of Tumor Mutations. Am. J. Hum. Genet. 98, 442–455, https://doi.org/10.1016/j.ajhg.2015.12.021 (2016).
Wang, L. et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS One 9, e88291, https://doi.org/10.1371/journal.pone.0088291 (2014).
Tsao, H., Goel, V., Wu, H., Yang, G. & Haluska, F. G. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J. Invest. Dermatol. 122, 337–341, https://doi.org/10.1046/j.0022-202X.2004.22243.x (2004).
Ogasawara, N. et al. Feasibility and robustness of amplification refractory mutation system (ARMS)-based KRAS testing using clinically available formalin-fixed, paraffin-embedded samples of colorectal cancers. Jpn. J. Clin. Oncol. 41, 52–56, https://doi.org/10.1093/jjco/hyq151 (2011).
Frampton, G. M. et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–1031, https://doi.org/10.1038/nbt.2696 (2013).
Turajlic, S., Sottoriva, A., Graham, T. & Swanton, C. Resolving genetic heterogeneity in cancer. Nat Rev Genet, https://doi.org/10.1038/s41576-019-0114-6 (2019).
Lopez, J. I. & De Petris, G. Discovering intratumor heterogeneity: the next frontier for pathologists. Pathologica 109, 110–113 (2017).
Mogi, A. & Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 583929, https://doi.org/10.1155/2011/583929 (2011).
Ding, X. et al. Clinicopathologic characteristics and outcomes of Chinese patients with non-small-cell lung cancer and BRAF mutation. Cancer Med. 6, 555–562, https://doi.org/10.1002/cam4.1014 (2017).
Acknowledgements
We would like to thank Singlera Genomics for the precious technical help. We thank Chinese People’s Liberation Army General Hospital, Fujian Cancer Hospital and Zhongshan Hospital, Fudan University for providing their specimens and relevant information. We acknowledge grant from the National Natural Science Foundation of China (No. 81972498, 81601213); 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD18012); the Key Research and Development Project of Science and Technology Department of Sichuan Province (No: 2017SZ0005).
Author information
Authors and Affiliations
Contributions
R.J. and F.Y. conceived and designed the experiments; B.Z., X.T., P.H. and S.X., organized the work of collecting clinical cases, reviewing the slides, gathering the clinical information and performing the experiments in participating laboratories; the Singlera Genomics (Z.Z., R.L. and T.T.) developed customized pipeline for variant identifying to analyze sequencing data from reads alignment to variant calling and annotation; R.J. analyzed the NGS result; Singlera Genomics contributed reagents/materials/analysis tools; R.J. wrote the paper; F.Y. revised the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Z.Z., R.L. and T.T. are members of the Singlera Genomics Inc., Shanghai, China. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiang, R., Zhang, B., Teng, X. et al. Validating a targeted next-generation sequencing assay and profiling somatic variants in Chinese non-small cell lung cancer patients. Sci Rep 10, 2070 (2020). https://doi.org/10.1038/s41598-020-58819-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58819-5
- Springer Nature Limited