
Abstract
Drought is one of the main constraints in peanut production in West Texas and eastern New Mexico regions due to the depletion of groundwater. A multi-seasonal phenotypic analysis of 10 peanut genotypes revealed C76-16 (C-76) and Valencia-C (Val-C) as the best and poor performers under deficit irrigation (DI) in West Texas, respectively. In order to decipher transcriptome changes under DI, RNA-seq was performed in C-76 and Val-C. Approximately 369 million raw reads were generated from 12 different libraries of two genotypes subjected to fully irrigated (FI) and DI conditions, of which ~329 million (90.2%) filtered reads were mapped to the diploid ancestors of peanut. The transcriptome analysis detected 4,508 differentially expressed genes (DEGs), 1554 genes encoding transcription factors (TFs) and a total of 514 single nucleotide polymorphisms (SNPs) among the identified DEGs. The comparative analysis between the two genotypes revealed higher and integral tolerance in C-76 through activation of key genes involved in ABA and sucrose metabolic pathways. Interestingly, one SNP from the gene coding F-box protein (Araip.3WN1Q) and another SNP from gene coding for the lipid transfer protein (Aradu.03ENG) showed polymorphism in selected contrasting genotypes. These SNPs after further validation may be useful for performing early generation selection for selecting drought-responsive genotypes.
Similar content being viewed by others


Introduction
Peanut or groundnut (Arachis hypogaea L.) is the second most important legume in the world, generally grown as a rain-fed crop, ranking next to soybean in production. The United States is the third largest peanut producer, after China and India (FAO, 2018) and the highest peanut producing state is Georgia followed by Texas. In terms of acreage, among the four commercial types of peanut, the runner type is the most cultivated crop with 80% coverage, followed by Virginia (15%), Spanish (4%), and Valencia (1%) in the United States. Being a rainfed crop, peanut experiences drought stress in the cultivated regions of United States, including Texas, with an estimated annual loss of approximately $520 million1,2. The relatively low yield in the West Texas and eastern New Mexico regions may be due to the inability to provide sufficient irrigation by farmers to supplement the low rainfall received during the season. The flowering and pegging stages of the peanut life cycle are considered to be more sensitive to water deficit stress which pose adverse impact, leading to reduced yield3,4. Therefore, it is essential to breed improved varieties that can produce higher yield under water deficit conditions.
Being sessile, plants employ different pathway mechanisms to cope with different types of stressful environments. In particular, drought stress affects the plant’s architecture by triggering a wide range of physio-biochemical processes5. Plants adopt diverse strategies to combat drought stress like by reducing the stomatal conductance, decreased photosynthetic rate, accumulation of different osmoprotectants, activation of stress responsive genes and transcription factors etc.6,7. The level of stress tolerance in agricultural crops is generally evaluated based on the considerable loss of yield with respect to optimal growing conditions. The differences in the type of physiological response facilitate the plant either to be tolerant or sensitive towards the stress conditions. The yield of peanut among different genotypes under drought stress was screened to assess the variation of tolerance8,9. Different studies have reported drought-tolerant peanut genotypes, however, it is essential to check the performance of these genotypes in target locations such as West Texas and eastern New Mexico regions which faces frequent problem of limited water resources.
In West Texas and eastern New Mexico, the most widely cultivated Valencia-type peanut cultivar, Valencia-C (Val-C), is considered as standard reference, and an industrial standard variety that has been used as a control to compare yields and flavor of newly released varieties. Because of the gradual depletion of groundwater levels in these regions due to climate change, and Val-C being a low-yielding variety, it is necessary to screen different genotypes of peanuts to provide better yields for the West Texas region10. In peanut, related to drought there are very limited reports regarding the identification of candidate genes/genomic regions11,12,13 and transcriptome studies14,15. But none of these studies explored the drought tolerance mechanisms in Valencia-type peanuts; so a correct understanding of its underlying genomics will facilitate the genetic improvement of Valencia peanuts. Therefore, this study reports the identification of drought tolerance sources in the genetic background of Valencia-type peanuts. Furthermore, in recent years, the RNA-seq approach has been successfully used to understand the mechanisms behind various biotic and abiotic stresses in different crop species16,17. The availability of genome sequence information of diploid progenitors of cultivated peanut18,19 facilitates to understand the genomic complexity of the different traits in peanut. Therefore, the current study was aimed to identify the candidate genes and their molecular mechanisms involved in drought tolerance using RNA-seq approach. The results obtained from this study provide a better understanding of the tolerance mechanisms and the potential candidate genes can be utilized in breeding programs.
Results
Genotypic variation for pod yield and physiological features
In order to study the phenotypic variations and its associated physiological responses for pod yield among the 10 peanut genotypes (Supplementary Table S1), field level experiments were conducted in three consecutive years from 2013–2015 by imposing deficit irrigation (DI) stress. Adequate irrigation (100%) was based on the farmer’s well capacity, while deficit irrigation was achieved by reducing irrigation use by 50%. In the field experiments in different seasons, the observed precipitation varied greatly (Supplementary Fig. S1). The annual total precipitation and precipitation intervals also varied significantly. However, the average maximum and minimum temperatures were significantly consistent (Supplementary Figs. S2 and S3). Further, relative humidity and evapotranspiration rates were also recorded (Supplementary Fig. S4). Compared to other years, this result was consistent with the least rainfall in 2013. In addition, soil moisture content was also collected, where fully irrigated soil was very moist at 48 cm for most of the growing season and at 12, 24 cm, the soil moisture had a smaller interval of dampness and dryness due to irrigation and precipitation events (Supplementary Figs. S5 and S6).
Based on the phenotyping results from the annual analysis of variance, the irrigation treatment (T) was significant in the years 2013 and 2014 (P-value <0.0001) and in 2015 (P-value <0.01)). These results indicate that each year the yield was significantly different depending on the T, genotype (G) and T × G (Supplementary Table S2). In 2013, the fully irrigated (FI) plot produced about 2828 kg/ha−1, while the deficit irrigated plot generated about 1879 kg/ha−1, a reduction of about 34% compared to the fully irrigated plot. Further, yield reduction in the deficit-irrigated plots was about 67% and 13% in 2014 and 2015, respectively (Supplementary Table S3). Overall, in three years, the best genotypes obtained under full irrigation conditions in West Texas were Tamspan-90, ICGS 76 and C76-16 (C-76) and poor yielders were Valencia-C (Val-C), ICGV 86388 and TMV-2. On the other hand, under the conditions of water deficit, C-76 showed better response accounted by its mean yield 3278 kg ha−1, higher among the other genotypes followed by Tamspan-90, ICGS 76, COC-041, ICGV 86051 and Val-C (Table 1).
Net photosynthesis at the leaf level was measured during the water deficit treatment to monitor the impact of deficit irrigation (DI) stress on the plant metabolism. In 2013, the water deficit treatment resulted in a moderate decrease in net photosynthesis for most of the genotypes, but no significant differences were observed during stress, recovery, or complete irrigation among the genotypes. In 2014, treatment resulted in a significant decrease in net photosynthesis (ranging from 18 to 38 µmolesm−2s−1) compared to the full irrigated treatment (ranging from 20 to 40 µmolesm−2s−1) but again no statistically significant differences could be observed between the genotypes. General trends emerged in both the years for photosynthetic responses, but in this case, net photosynthesis was used to guide the collection of leaf material for transcriptome studies (Fig. 1a,b). Further, SLA and chlorophyll content were also measured in 2014 but the values obtained were not statistically significant between the treatments of the same genotypes (Fig. 1c,d).

Physiological observations from fully irrigated and deficit irrigation plots at different time intervals: (a,b) Mean gas exchange measurements in the years 2013 and 2014. (c) Mean specific leaf area (SLA) during irrigated and deficit irrigation conditions in selected genotypes. (d) Mean chlorophyll content during irrigated and deficit irrigation conditions in selected genotypes.
Sequencing and mapping statistics using reference guided assembly
Based on the observed phenotypic variations from our previous9 and present drought studies, two genotypes- C76-16 (C-76, runner type) and Valencia-C (Val-C, Valencia type) were identified as the contrasting genotypes towards drought response and thus selected for transcriptome studies. Leaf samples for the RNA-seq were collected from C-76 and Val-C genotypes in 2014 in three biological replicates for transcriptome profiling and generated ~369 million raw reads. The raw reads were filtered to obtain high-quality reads which were then further used in the downstream analysis and for mapping on A (A. duranensis) and B (A. ipaensis) diploid ancestor genomes18 of peanut separately.
A total of 329.2 million (90.2%) and 329.4 million (90.3%) filtered reads were mapped to A and B genomes, respectively. In C-76, on an average, 30 and 26 million reads per replicate were mapped under FI and DI conditions, respectively on A and B genomes. Whereas in Val-C genotype, it was observed that approximately ~25 and ~27 million reads per replicate were mapped under FI and DI conditions, respectively on A and B genomes (Table 2). About 81.2% of the filtered reads were mapped to the exonic regions of the A and B genomes, while ~6% were mapped on the intronic region, and ~12.8% were mapped on the intergenic regions (Supplementary Table S4 and Fig. 2A). Expression analysis was performed for the four samples, namely C-76_FI, C-76_DI, Val-C_FI and Val-C_DI. The level of gene expression was calculated in the four samples using fragments per kilobase of transcript per million mapped reads (FPKM) values. A total of 28,524 and 29,490 genes were observed to be expressed on A and B genomes, respectively. Further, in dendrogram constructed based on expression values, individual replicates of C-76 and Val-C were falling in two different groups, one belonging to the Val-C group and the other to the C-76 group consisting of their respective FI and DI samples. In A genome, the replicates of C-76 and Val-C genotypes were grouped separately. Similarly, for the two conditions- FI and DI, the replicates were also grouped separately under their respective genotypes (Fig. 2B). However, for the B genome, there is a little deviation from the A genome, where the irrigated samples of Val-C were falling under the C-76 but these were not grouped together (Fig. 2B).

Mapping statistics and dendrogram showing correlation among the different samples: (A) An overview of the mapped reads on (A,B) genomes of Arachis progenitors: The distribution of fragments represented as the percentage of reads that map onto exons, introns and intergenic regions on A and B genomes of peanut (B) Dendrogram constructed based on the expression values in fully irrigated (FI) and deficit irrigation (DI) samples in two different genotypes- C-76 and Val-C with biological replicates on two genome clades.
Differential gene expression analysis
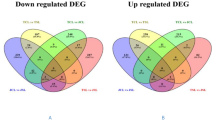
Differential gene expression analysis revealed 2,037 and 2,471 differentially expressed genes for C-76 and Val-C genotypes, respectively on comparison of their corresponding DI samples with respect to their FI samples. Genes with a P-value ≤ 0.05 and \(|lo{g}_{2}(fold\,change)|\ge 2\) were considered as differentially expressed genes (DEGs). The identified DEGs were studied for their distribution on the pseudomolecules of A and B genomes. The highest number of DEGs were present on pseudomolecules A03 (118) and B04 (119) while the minimum were observed on pseudomolecules A02 and B02 (Fig. 3a). The number of up and downregulated genes in C-76 and Val-C during DI with respect to FI on A and B genomes were identified (Fig. 3b). Differentially expressed genes in three combinations (C-76_DI vs C-76_FI; Val-C_DI vs Val-C_FI; C-76_DI vs Val-C_DI) revealed that more DEGs were upregulated in Val-C (599) than C-76 (7) during DI compared to FI and 3 DEGs were found in common on A genome. Similarly, the number of downregulated genes were 248 in the case of Val-C and 138 in C-76 during DI compared to FI, and 2 genes were common in both the genotypes on A genome and similar is the case with the B genome (Fig. 3c). In addition, the DEGs of four different combinations [C-76 vs Val-C (FI); C-76 vs Val-C(DI); C-76 (FI vs DI); Val-C(FI vs DI)] were compared to identify common and unique DEGs (Fig. 3d), where 19 (A genome) and 24 (B genome) DEGs were common across all four comparisons (Supplementary Table S5; Fig. 3d).

Differential gene expression analysis in C-76 and Val-C genotypes during deficit irrigation conditions: (a) Number of differentially expressed genes (DEGs) during deficit irrigation (DI) across 10 pseudomolecules on A and B genomes (b) Bar graph representing number of induced/upregulated and repressed/downregulated genes in C-76 and Val-C during DI with respect to fully irrigated (FI) conditions (numbers on the bars representing the number of up and downregulated genes) (c) Venn diagram representing the common and unique upregulated and down-regulated genes during deficit irrigation on A and B genomes of C-76 and Val-C (d) Venn diagram depicting the common and specific DEGs in different combinations of C-76 and Val-C genotypes during DI and FI on A and B genomes of peanut.
Differentially expressed genes during deficit irrigation (DI)
Differential expression analysis in DI revealed the expression of drought stress-responsive genes in the two selected genotypes. Interestingly, the common DEGs (19 on A genome) showed fairly contrasting expression in C-76 and Val-C (Fig. 3d; Supplementary Fig. S7). The common DEGs encode for Gcn5-related N-acetyltransferase (GNAT) gene, BON1- associated protein, the lateral organ boundary (LOB), and the late embryogenesis abundance (LEA) genes etc., (Fig. 3d and Supplementary Table S5). Especially, genes related to osmoprotectants, photosynthesis, abscisic acid, secondary metabolites and other gene families that are responsive during drought stress showed distinct expression patterns in C-76 and Val-C (Fig. 4).

An overview of differentially expressed genes in drought stress-responsive pathway mechanism: Heat maps demonstrating the expression profiles of differentially expressed drought stress-responsive genes during fully irrigated (FI) and deficit irrigation (DI). Color scale represents normalized FPKM values. Corresponding reference gene ids (with suffix XLOC) falling have been given on the right side. The color scale on the top represents normalized FPKM values.
In C-76 under DI with respect to FI, the induced expression of plant protease inhibitor genes such as trypsin inhibitors and cystatin, as well as the ATP- binding cassette (ABC) transporter and the lectin precursor genes and LOB was noted. While in Val-C, induced DEGs include genes related to cell wall and membrane-bound genes such as ureide permease 2, expansin A10, xyloglucan endotransglucosylase hydrolase, and expansin-A8 when compared DI versus FI. On the other hand, highly upregulated genes include the genes encoding cysteine endopeptidase, asparticase oryzasin-1 and ubiquitin-E3ligase in Val-C. Similarly, other drought-responsive DEGs are aquaporin PIP-2, abscisic acid (ABA) insensitive 5, ethylene-responsive transcription factor, expansin 2, L-ascorbate oxidase, LEA, peroxidase, glutathione S-transferase, thioredoxin reductase and trehalose-phosphate phosphatase, heat shock proteins (HSP) encoding genes such as Dnaj/HSP40, Class II HSPs and heat shock factor (Hsf)30 and genes involved in secondary metabolism like isoflavonoid and flavonoid synthesis, were induced under DI in Val-C with respect to DI in C-76 (Supplementary Table S6).
When FI samples from two genotypes were compared, expression of genes for drought stress response pathways such as photosynthesis-related genes-accumulation of photosystem one1 (APO1) and photosystem II family protein-coding genes (D2 protein), osmoprotectant genes like abscisic acid 8-hydroxylase, proline imino-peptidase, peroxidase genes and other genes encoding for ethylene-responsive transcription factor (ERF) and lipid transfer protein genes showed high expression levels in C-76, indicating its higher level of tolerance (Supplementary Table S7).
Gene ontology
The gene ontology of DEGs revealed their functional annotation by categorizing them into cellular, molecular, and biological components (Fig. 5A). The majority of genes have been annotated on biological processes followed by cellular component and molecular function. In total, 38.7% (A genome) and 38.0% (B genome) genes were falling under 12 categories of cellular components while 37.66% (A genome) and 38.2% (B genome) genes were falling under 21 categories of biological process, 23.7% (A genome) and 23.8% (B genome) genes were falling in molecular function under 11 categories.

Gene ontology and pathway analysis: (A) Functional categorization of drought-responsive genes under different gene ontology (GO) categories-cellular component, biological process and molecular function. (B) The top 15 different pathways involved in deficit irrigation stress of C-76 and Val-C genotypes including the representation of percent of genes involved in each pathway.
Pathways associated with deficit irrigation (DI)
Differentially expressed genes were analyzed to study their association with different metabolic pathways. The significant role was noted for 104 pathways representing biosynthesis, metabolism and degradation process. Pathways related to the biosynthesis of antibiotics (23 genes), starch and sucrose metabolism (15), biosynthesis of aromatic amino acids (11), metabolism of amino and nucleotide sugars (9), metabolism of sulfur amino acids (7), biosynthesis of flavonoids (7), etc., showed an association with DEGs (Fig. 5B and Supplementary Table S8). Different genes in the antibiotic synthesis pathway encoding for bifunctional aspartate aminotransferase, aspartate prephenate-aminotransferase, glyceraldehyde-3-phosphate dehydrogenase, pyrophosphate-fructose 6-phosphate 1-phosphotransferase, acetyl coenzyme A, 3-dehydroquinate dehydratase shikimate, methylecgonone reductase, chorismate mutase, etc. were found to be expressed (Supplementary Table S9). Similarly, genes related to starch and sucrose metabolic pathway include, endo-1,3-beta-glucosidase, alpha-amylase, glucomannan 4-beta-mannosyltransferase 2, beta lysosomal glucosidase, hexokinase, endoglucanase 6 and cellulose synthase, etc., which were found differentially expressed. Secondary metabolic pathways such as flavonoids and alkaloids were also found to be affected by drought stress. In both genotypes, the amino acid and nucleotide pathways followed by the carbohydrate and lipid metabolic pathways were found associated with water deficit stress.
Identification of transcription factors (TFs) during deficit irrigation
The differential expression of genes encoding transcription factors (TFs) were identified in the peanut under DI. A total of 715 and 839 TF genes belonging to 58 classes were differentially expressed in the peanut A and B genomes, respectively. Among the differentially expressed TF encoding genes, the highest number of genes were from MYB family for both A and B genomes followed by WRKY, and ERF during DI (Supplementary Fig. S8). The MYB, bHLH, WRKY, ERF, NAC and C2H2 transcription factor family encoding genes were abundantly expressed in both genotypes during the DI conditions (Fig. 6A,B). In addition, the analysis revealed that ABC-2 type transporter and LBD (LATERAL ORGAN BOUNDARY DOMAIN) TF were found to be upregulated during DI conditions with respect to FI in C-76. Similarly, in Val-C during DI versus FI, the ABC transporter, other TF genes, such as NAC, the bHLH family, cytochrome P-450, WRKY, and NF-Y, were found to be induced.

Heat maps representing the expression profiles of highly expressed transcription factor (TF) gene families: Expression of genes encoding (A) MYB and WRKY (B) ERF and bHLH TFs during fully irrigated (FI) and deficit irrigation (DI) conditions of C-76 and Val-C genotypes on A and B genomes of peanut. Corresponding reference gene ids (with suffix XLOC) falling under each TF family have been given on the right side. The color scale on the top represents normalized FPKM values.
Validation of DEGs from RNA-seq using quantitative real-time PCR (qRT-PCR)
The expression of RNA-seq was also validated by qRT-PCR. For this study, a total of 10 DEGs, five genes each from A genome and B genome were selected and primers were designed (Supplementary Table S10). These include- ubiquitin- ligase, NHL domain-containing, peptidyl- prolyl cis-trans isomerase, LRR receptor serine- threonine kinase, vinorine synthase, LEA, DNA mismatch repair MSH6, ATPase alpha partial, laccase-14 and plastid movement impaired 2. Although there was a small variation in the degree of expression, almost all of the 10 genes selected for validation showed the same pattern of expression as displayed in RNA-seq data (Fig. 7) confirming the results achieved through RNA-seq analysis.

Validation of RNA-seq results using quantitative real-time PCR (qRT-PCR): Expression correlation of genes using qPCR and RNA-seq data. Validation of gene expression patterns of 10 genes during deficit irrigation (DI) in comparison with their respective fully irrigated (FI) samples. The y-axis represents the relative gene expression during DI versus FI.
SNP marker identification and validation
A total of 514 SNPs were identified between C-76 and Val-C, occurring in DEGs when aligned with A (190) and B (324) genomes. The identified SNPs were present on 270 DEGs collectively, where 95 genes were present on the A genome and 175 genes on the B genome. The SNPs were identified in four different combinations and were divided into missense, 3′UTR, 5′UTR, splice region, stop gained, start lost and stop loss variants (Table 3). The distribution of SNP variants in pseudomolecules on different DEGs has been shown in Fig. 8. Several missense variants on A and B genomes in four combinations were 116 and 225, respectively. A total of 23 sets of allelic specific primers were designed for 23 genes to validate polymorphism in C-76 and Val-C (Supplementary Table S11) using qRT-PCR. Homozygous and heterozygous alleles were detected based on the variations obtained in the cycle threshold (Ct) values. If the Ct value is <30.0 for the set of allelic primers of one gene in one genotype but not in the other, this is considered to be the presence of two different alleles and vice versa. Of the 23 allelic primers designed, 21 sets of allelic primers did not show any polymorphism (Table 4), while two SNPs representing two genes exhibited polymorphism. The first SNP (Ct > 30 in C-76 and Ct <30 in Val-C) was from the gene coding the F-box protein (Araip.3WN1Q) while another SNP (Ct value of <20 in C-76 and Ct> 25.0 in Val-C) was from the gene coded for the lipid transfer protein (Aradu.03ENG). These validated SNPs from the genes Araip.3WN1Q and Aradu.03ENG can be used for performing early generation screening of the breeding material arising from crosses involving source parent for good performance under water deficit stress.

Circos representing the distribution of transcription factors (TFs), differentially expressed genes (DEGs) and SNPs across the different pseudomolecules of reference genomes of peanut: Concentric circles in the circos plot from inside to outside (a) orange color representing the expression profiles of transcription factors (b) blue color representing differentially expressed genes (DEGs) (c) green color representing the SNP density and (d) outer most track (in multicolor) representing pseudomolecules of peanut on two genomes of A and B.
Discussion
Cultivated peanut is an important legume and comprises of allotetraploid genome (type AABB; 2n = 4x = 40) with a total size of about 2.7GB. The availability of genome sequence information of the diploid progenitors (A. duranensis and A. ipaensis) of cultivated peanuts18,19 allows us to study the genome wide transcriptome variations in different peanut lines. More recently, the high-quality reference genomes for both the subspecies of cultivated peanuts have become available (was not available at time of this study), which will further enhance the precision of future genomics and transcriptomics studies in peanut20,21,22. This study is a combined approach to physiological and transcriptomic responses of peanut genotypes or breeding material under water deficit conditions. Yield and physiological attributes were measured in 10 selected peanut genotypes and further RNA-seq was performed in two different genotypes to study the transcriptome changes that occur during deficit irrigation (DI) and fully irrigated (FI) conditions. In addition, this is a unique study, where the samples from our study were collected in the field to facilitate natural environmental conditions rather than using growth chambers for controlled conditions.
Among the ten different genotypes selected for this study C-76-16 (C-76) showed better performance in terms of mean yield under DI conditions. While on the other hand, check cultivar Valencia-C (Val-C) showed lesser yield. These results are well synchronizing with our earlier study on the pod yield performance of different peanut genotypes under different soil and regional conditions9, where C-76 produced more than Val-C in rainfed conditions without irrigation but there is a considerable reduction in C-76 yield by 15–20% compared to Val-C, which was up to 50–70% reduction in yield compared to complete irrigation9. Another study in peanut23 showed a lower yield of Valencia-type pods (PI 536121) under drought conditions. Since Val-C is considered standard control for West Texas and eastern New Mexico regions, there is a need to improve the genetic potential of Val-C in terms of yield in case of water deficit conditions. This facilitates the realization of transcriptomic study on genotypes C-76 (runner type) and Val-C (Valencia type) to understand the underlying genetic mechanisms. Although the genotypes selected belong to different market classes and several factors are added, the comparison of the responses in the transcriptome of the leaf is adequate because the two genotypes represent the same species. Transcriptome analysis in C-76 and Val-C revealed the expression of 28,524 and 29,490 genes on A and B genomes of peanut, where more number of expressed genes hitting to B genome described the direct effect on the progeny of A. ipaensis from the same population that contributed to the B genome of the cultivated peanut18. Further, the analysis showed the expression of a very less number of DEGs in C-76 under DI versus FI. The number of induced genes was very less in C-76 is a rather interesting phenomenon. These results are consistent with those of the sorghum study where more DEGs were expressed in drought-sensitive than drought-tolerant subjects24.
Differential expression of functional and regulatory genes responsive to drought stress was induced in Val-C relative to C-76 on comparison of their corresponding DI samples with respect to their FI samples (Supplementary Table S6). This shows that C-76 showed a significant level of tolerance in DI and may not be experiencing water deficit stress, unlike Val-C. This can also be explained in the other direction, as the stress imposed was not sufficient for the C-76 to express more number of drought stress responsive genes during DI versus FI. However, in Val-C for the same level of imposed stress, various genes related to drought stress-responsive mechanisms/pathways were activated robustly to combat stress. Comparatively, Val-C showed the activation of a maximum number of drought-responsive pathways than C-76 during DI versus FI conditions, which could form the basis of the contrasting response of both genotypes. The adaptation to different environmental conditions varies according to the genotypes of solitary plant species, which can be explained by the phenomenon of natural variation25 and the level of tolerance to any type of stress varies from one genotype to another26.
It is interesting to note that the contrasting expression profiles of common DEGs, such as trypsin inhibitors, late embryogenesis abundant (LEA), ABC transporter and LOB containing domain, etc. in both genotypes during DI, suggest that there are significant differences between genotypes in terms of irrigation and water deficit conditions (Supplementary Fig. S7). In maize, the reciprocal expression of DEGs that responds to drought between two contrasting inbred lines, causing under water deficit stress reveals drought tolerance mechanisms27. In Arabidopsis a strong accumulation of trypsin inhibitor genes in water deficit periods was reported28. Similarly, LEA proteins are known to accumulate during water stress to prevent intracellular water loss. The increased expression of cellular LEA proteins shows tolerance to drought stress in sorghum29, and foxtail millet30.
Phytohormones, also play a crucial role in water deficit stress in plants. The accumulation of abscisic acid (ABA) can lead to stomatal closure, which reduces water loss and subsequently stops cell growth and eventually results in increased concentration of reactive oxygen species (ROS)31,32. Genes such as abscisic acid 8 hydroxylase, proline imino-peptidase and sucrose synthase, which play a crucial role in drought stress response, shown the decreased expression in Val-C during DI. On the other hand, the increased expression of these genes was noticed in C-76 compared to Val-C under FI. The increased abscisic acid can be catabolized in the cell by the abscisic acid 8-hydroxylase acid to form 8′-OH abscisic acid to maintain cellular homeostasis, has been reported in the grapevine33. The accumulation33 of ABA is negatively regulated by the plant’s mechanism through the activation of catabolic enzymes of ABA during water stress to enhance tolerance34,35. In the present study, the downregulation of genes related to abscisic acid 8-hydroxylase and cytochrome p450 (CYP) genes in Val-C implies the downregulation of abscisic acid pathway under DI, which in turn leads to an accumulation of ROS and finally cell damage. In addition, reduced expression of sucrose synthase in Val-C could play an important role in yield reduction due to the disproportion of the source to sink ratio. The sucrose synthase activity plays a crucial role in the biosynthesis of starch and storage proteins36. The increased activity of sucrose synthase leads to increased grain assimilation in wheat under water stress37.
In addition, genes related to photosynthesis showed significant discrimination between two contrasting genotypes especially during FI conditions. The genes related to light reactions of photosynthesis of photosystem I and II were found to be downregulated in Val-C versus C-76 during FI conditions (Supplementary Table S12). The downregulated genes related to the subunits of photosystems I and II under drought stress conditions, reduced plant tolerance levels to drought stress in sorghum24. Decreased expression of genes involved in light-harvesting chlorophyll a/b-binding proteins (LHCBs) and photosystems I and II results in decreased drought tolerance in plants38,39. During irrigated conditions, the genes encoding P700 apo A1 of photosystem I, and D2 of photosystem II exhibited comparatively high expression in C-76 than Val-C. The D2 protein of photosystem II plays a vital role in stress tolerance (Supplementary Table S12). Among the different DEGs identified, genes related to photosynthesis showed a distinct expression in both genotypes. Therefore, it is essential to comprehensively study the photosynthetic genes associated with drought stress in peanuts, which gives a better idea in the development of drought-tolerant genotypes. In addition, the expression of genes encoding different transcription factors (TFs) such as MYB, NF-Y, NAC, WRKY, ERF, bHLH represents the induction of the ABA-dependent signalling pathway as a stress response (Supplementary Fig. S8). The involvement of drought-responsive WRKY and NAC TFs may be useful in the detoxification of ROS by regulating downstream target genes40. In addition, the SNP validation study has provided the information on genetic markers which upon further validation in larger set of genotypes can be used in genomic-assisted breeding for enhancing drought stress tolerance.
In summary, this is a combined study of phenotypic, physiological and genome-wide transcriptome analysis in diverse peanut genotypes. The germplasm line C-76 yielded the best results among the 10 accessions tested in our study. Although demand for the high oleic trait is high, when water resources are limited, processors will contract farmers to grow a variety that can provide better performance under limited irrigation conditions. Hopefully, a variety of lineages such as C-76 can overcome the challenges where farmers can irrigate less and be able to produce a decent yield in regions of West Texas and eastern New Mexico. On the basis of yield, C-76 and Val-C were selected for transcriptome analysis. Also, this is the first attempt on the genetic improvement of Valencia-type peanut to understand the drought tolerance mechanisms. When compared to DI versus FI, the more number of DEGs in Val-C genotype than C-76 infers its steep response to water stress. While, for the C-76, the treatment of the imposed stress was not sufficient to respond and could not be under stress, which could explain in terms of the expression of less number of DEGs during DI versus FI, implies its higher tolerance. Also, the comparative transcriptome analysis of both genotypes under FI conditions, the high expression of proline imino-peptidase, peroxidase, abscisic acid 8-hydroxylase and photosystem II D2 genes in C-76 reflect its tolerance level and its ability to survive under limited water conditions. Although Val-C, is capable of activating different genes and pathways responsive to drought stress, decreased expression of key pathway genes related to ABA metabolism may result in cellular accumulation of ROS, which in turn may cause the devastating effects of the cell to resist water stress. Further, the decreased expression of the photosynthetic related genes, and genes related to sucrose metabolism may be one of the reasons for the reduced yield in Val-C. Also, identified SNPs related to F-box protein and lipid transfer genes showed polymorphism between C-76 and Val-C genotypes which can be used as linked markers for performing early generation selection in genomics-assisted breeding programs. In conclusion, this study revealed genotype information appropriate for the regions of West Texas and eastern New Mexico regions and the underlying complex mechanisms associated with contrasting peanut genotypes under water deficit conditions.
Materials and Methods
Germplasm and details of the experimental setup
Ten different peanut genotypes used in this study were obtained from four different market types, namely Spanish, Virginia, runner type and Valencia (Supplementary Table S1). Seven genotypes – ICGV 86388, ICGV 86051, TMV-2, Tamspan-90, Serenut-5R, Serenut-6T, and COC 041 belong to the Spanish market type, while ICGS 76 belongs to the Virginia group. While C-76-16 (C-76) is a breeding line which belongs to the runner type and Valencia-C (Val-C) belong to Valencia market. Detailed information on crop management, weather and meteorological conditions, soil moisture and temperature conditions have been provided in Supplementary Information). Field trials were conducted on a producer’s peanut field (Delwin Marrow Farm) in Terry County, Brownfield, Texas, USA (33° 18′N, 102° 16′W, elevation 1009 m). Two treatments, one with complete/full irrigation (FI) and the other with deficit irrigation (DI), was imposed. Complete irrigation received 100% available water content (AWC) while deficit irrigation received 50% throughout the growing season, from 2013 to 2015. Adequate irrigation (100%) was based on the farmer’s well capacity of the farmer’s wells, and the low irrigation (50%) were treatment was implemented by reducing the irrigation rate by 50%. The experiment was conducted on the basis of split-plot design using main plots with irrigation as a factor and each genotype was replicated four times. The collection of meteorological data, weather data and the total volume of water received (rain + rainfall + irrigation) as a percentage of the ETo (TWRPET) were provided in the Supplementary Information.
Crop harvest and yield measurement
At physiological maturity, the crops were mechanically harvested in September in the FI and DI fields using a specialized digging equipment to dig peanuts under the vines (Pearman Corporation, GA, USA). Maturity was determined by examining the color of the pod mesocarp along the saddle region of the dorsal seed. Black or brown colors were used as indicators of physiological maturity. To analyze the color of the pods, 25 plants were randomly selected from the middle of the plot and were extracted using a rotating nozzle pressure washer.
Leaf-level net photosynthesis
Photosynthesis at the leaf-level was measured between 60–90 days after sowing. For the fully irrigated control plants, photosynthesis was measured 48 h after an irrigation event. For the treatment of the deficit, measurements were taken during the interval of irrigation deficit interval (10 to 12 days) between the irrigation events. The recovery rates of photosynthesis for deficit treatments were measured to the plot after giving an irrigation for 48 h following 2 weeks of without irrigation. The rate of photosynthesis was measured using a portable photosynthesis system (Li-6400 XT, LI-COR, Inc.). All measurements were taken in between 10:00 and 12:00 h. For each genotype, three measurements per replicate were measured. The leaves of the Li-Cor 6400 XT cuvette were maintained and measured under the following conditions: saturated photosynthetic photon flux density (PPFD) of 2000 μmolm−2s−1, 40 Pa of CO2 and leaf temperature of 27 °C. When measuring gas exchange, carefully fill the sensor head with the leaflets to avoid errors.
Total chlorophyll content and specific leaf area (SLA)
The chlorophyll content was recorded using SPAD (502 Minolta Corp., Ramsey, NJ). Measurements were taken every 30 days after emergence (DAE). The second or third fully developed tetrafoliate leaf was used to measure the SPAD reading when attached to the plant. After recording the measurements, the leaf was detached from the plant, placed in an envelope covered with a block of ice in an ice chest for the measurement of leaf area using a leaf area meter (Li 3050 C, LI-COR, Inc.), followed by drying in a hot air oven at 60 °C for 48 h to measure the dry weight.
The dry weight was measured to determine the specific leaf area (SLA) in cm2g−1 and determined using the formula:
Statistical analysis
Data for each variable were analyzed using the PROC ANOVA procedure in Statistical Analysis System 9.3 (SAS Institute, 2013, https://www.sas.com/en_us/software/sas9.html). Before performing a separate analysis per year, the Bartlett variance, homogeneity of variance test was performed. In each year, irrigation and varieties were used as random factors, and appropriate error terms were used to calculate the mean sum of squares for each variable factor. If the F value was significant, a mean separation test was performed using the LSMEANS procedure in SAS software. In the LSMEANS procedure, additional PDIFF and PDMIX 800 options were used to evaluate the probability of significance among various main and interaction effects. Means were considered statistically different if P-value ≤0.05.
RNA isolation, quality check, and sequencing
RNA-seq leaf samples were collected during 2014 from the fully developed third leaf of the apex. Three biological replicates were collected for profiling the transcriptome. Total RNA was extracted from 12 leaf tissues (two genotypes × two treatments × three biological replicates) of genotypes C-76 (tolerant) and Val-C (susceptible) during FI and DI conditions at the active pegging and fruit development stages using the TRIzol reagent (Life Technologies, Carlsbad, CA, USA). RNA purification was performed using the RNeasy Mini Kit (Qiagen, US) and the integrity of the RNA was verified using Bioanalyzer 2100 (Agilent Technologies, US). cDNA libraries for RNA-seq were constructed using Illumina TruSeq RNA Sample Preparation Kit (Illumina Inc., San Diego, CA, USA) following the manufacturer’s instructions. Paired-end sequencing (2 × 54 bp) was performed using the Illumina HiSeq 2000 (Illumina, San Diego, CA,USA) system at National Genomic Resource Center (NCGR).
Reference-based assembly and annotation
RNA-seq data was analyzed using the Tuxedo pipeline41. The raw reads obtained from the sequencing were subjected to quality control using NGS-QCbox42 and trimmomatic v0.3543. The filtered reads of all samples were aligned to the A and B diploid progenitors of the peanut18 with Tophat v2.1.044. The aligned reads of each sample were then used to create a Reference Annotation Based Transcript (RABT) assembly using cufflinks45. The resulting assemblies were then merged into a consensus assembly using cuffmerge for downstream analysis41 (Supplementary Fig. S9). The relationship between the transcriptomes of two genotypes (C-76 and Val-C) under two conditions (FI and DI) in three biological replicates on both A and B genomes was shown by generating a dendrogram by using the cummeRbund package46.
Transcript abundance and identification of differentially expressed genes
Gene expression was estimated as fragments per kilobase of transcript per million mapped reads (FPKM) and differentially expressed genes were identified using Cuffdiff. Genes with \(|lo{g}_{2}(fold\,change)|\ge 2\) and P-value ≤ 0.05 were identified as DEGs. The identified DEGs were scanned against the National Biotechnology Information Center (NCBI) non-redundant (taxon. Viridiplantae) protein database using BLASTX with an E-value cutoff of ≤10−5 to determine their putative function.
Gene ontology (GO) and pathway analysis
Gene ontology for the expressed genes was performed using Blast2GO v 3.347. In parallel, the expressed genes were searched against Plant TFDB with an E-value cutoff of ≤10−10 to identify the genes encoding the transcription factors. The pathway analysis was then carried out using the KEGG database. Heat maps with expression profiles were generated based on the transformed FPKM values using MeV48.
SNP identification
The identification of SNPs was performed using SAMtools49, followed by their annotation using the SnpEff program50. The BatchPrimer3 tool (https://probes.pw.usda.gov/cgi-bin/batchprimer3/batchprimer3.cgi) was used to design allele-specific primers between C-76 and Val-C. The information on primers has been provided in Supplementary Tables S10 and S11. Validation of SNPs was performed by qRT-PCR using SYBR green chemistry. Homozygous and heterozygous alleles were detected based on variations in the Ct values obtained51.
Quantitative real-time PCR (qRT-PCR)
The real-time qPCR was performed on the Applied Biosystems QuantStudioTM7 Flex real-time PCR system (Life Technologies, Carlsbad, CA, USA) using RT2 SYBR Green ROX qPCR master mix chemistry (Qiagen, CA, USA). Three biological replicates per peanut genotype (C-76 and Val-C) and treatment (FI and DI), and three technical replicates per biological sample were processed by qPCR in real-time for each set of SNP primers. Each biological sample was analyzed with three technical replicates with the Actin as housekeeping gene. The conditions used for the amplification were as follows: 2 min at 50 °C, 10 min at 95 °C, followed by 45 cycles of 15 s at 95 °C, 1 min at 58 °C and analysis of the dissociation curve of 15 s at 95 °C, 1 min 58 °C, 15 s at 95 °C and 15 s at 60 °C. Expression levels were normalized at actin expression levels for each sample52. The relative fold change (log2) of the normalized data was represented by the 2−ΔΔCt method53.
Data availability
Sequencing RNA-seq data described in this article have been published in the National Biotechnology Center Archives database with BioProject IDPRJNA498570.
References
Reddy, T. Y., Reddy, V. R. & Anbumozhi, V. Physiological responses of peanut (Arachis hypogea L.) to drought stress and its amelioration: A critical review. Plant Growth Regul. 41, 75–88 (2003).
Kambiranda, D. M., Vasanthaiah, H. K. N., Ananga, R. K. A., Basha, S. M., Naik, K. Impact of drought stress on peanut (Arachis hypogaea L.) in Productivity and Food safety. Plants and Environment (eds. Vasanthaiah, H. K. N. & Kambiranda, D.) 249–272 (Croatia: InTech, 2011).
Thiyagarajan, G., Rajakumar, D., Kumaraperumal, R. & Manikandan, M. Response of yield and yield attributes of groundnut to moisture stress-a review. Agric Rev. 31(3), 210–216 (2010).
Singh, A.L. et al. Water deficit stress and its management in groundnut in Advances in Plant Physiology (eds. Hemantaranjan, A.) 371–465 (Scientific Publishers, 2013).
Anjum, S. A., Xie, X. & Wang, L. Morphological, physiological and biochemical responses of plants to drought stress. Afr J Agric Res. 6, 2026–2032 (2011).
Farooq, M., Wahid, A., Kobayashi, N., Fujita, D. B. S. M. A. & Basra, S. M. A. Plant drought stress: effects, mechanisms and management. Sust Agric 29, 185–212 (2009).
Kaur, G. & Asthir, B. Molecular responses to drought stress in plants. Biol Plantarum. 6, 201 (2017).
Pereira, J. W. L. et al. Assessment of drought tolerance of peanut cultivars based on physiological and yield traits in a semiarid environment. Agric Water Manag. 166, 70–76 (2016).
Zurweller, B. A. et al. Pod yield performance and stability of peanut genotypes under differing soil water and regional conditions. J. Crop Improv. 32, 532–551 (2018).
Steward, D. R. & Allen, A. J. Peak groundwater depletion in the high plains aquifer, projections from 1930 to 2110. Agric. Water Manag. 170, 36–48 (2016).
Ravi, K. et al. Identification of several small main-effect QTLs and a large number of epistatic QTLs for drought tolerance related traits in groundnut (Arachis hypogaea L.). Theor Appl Genet. 122, 1119–1132 (2011).
Gautami, B. et al. Quantitative trait locus analysis and construction of consensus genetic linkage map for drought tolerance traits based on three recombinant inbred line population in cultivated groundnut (Arachis hypogaea L.). Mol. Breeding. 30, 757–772 (2012).
Brasileiro, A. C. M. et al. Transcriptome profiling of wild Arachis from water-limited environments uncovers drought tolerance candidate genes. Plant Mol Biol Rep. 33, 1876–1892 (2015).
Guimarães, P. et al. Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection. BMC Genomics. 13(1), 387 (2012).
Zhao, X. et al. Transcriptomic analysis and discovery of genes in the response of Arachis hypogaea to drought stress. Mol Biol Rep. 45, 119–13 (2018).
Mashaki, K. M. et al. RNA‐Seq analysis revealed genes associated with drought stress response in kabuli chickpea (Cicer arietinum L.). PLoS ONE. 13, e0199774 (2018).
Garg, V. et al. Integrated transcriptome, small RNA and degradome sequencing approaches provide insights into Ascochyta blight resistance in chickpea. Plant Biotechnol. J. 17(5), 914–93 (2019).
Bertioli, D. J. et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. 48, 438–446.
Chen, X. et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. 113(24), 6785–6790 (2016).
Bertioli, D. J. et al. The genome sequence of peanut (Arachis hypogaea), a segmental allotetraploid. Nat Genet. 51, 877–884 (2019).
Chen, X. et al. Sequencing of cultivated peanut, Arachis hypogaea, yields insights into genome evolution and oil improvement. Mol Plant. 12(7), 920–934 (2019).
Zhuang, W. et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution, and crop domestication. Nat Genet. 51, 865–876 (2019).
Carvalho, M. J., Vorasoot, N., Puppala, N., Muitia, A. & Jogloy, S. Drought on growth, yield and yield components in Valencia peanut genotypes. Sabrao J Breed Genet. 49(3), 270–279 (2017).
Fracasso, A., Trindade, L. M. & Amaducci, S. Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. BMC Plant Biol. 16, 115 (2016).
Juenger, T. E. Natural variation and genetic constraints on drought tolerance. Curr. Opin. Plant Biol. 16(3), 274–81 (2013).
Lou, Q. et al. Quantitative trait locus mapping of deep rooting by linkage and association analysis in rice. J Exp Bot. 66, 4749–4757 (2015).
Zenda, T. et al. Key maize drought-responsive genes and pathways revealed by comparative transcriptome and physiological analyses of contrasting inbred lines. Int J Mol Sci. 20(6), 1268 (2019).
Drame, K. N., Passaquet, C., Repellin, A. & Zuily-Fodil, Y. Cloning, characterization and differential expression of a Bowman-Birk inhibitor during progressive water deficit and subsequent recovery in peanut (Arachis hypogaea) leaves. J Plant Physiol. 170, 225–229 (2013).
Nagaraju, M. et al. Genome-scale identification, classification, and tissue- specific expression analysis of late embryogenesis abundant (LEA) genes under abiotic stress conditions in Sorghum bicolor L. PLoS One. 14(1), e0209980 (2019).
Wang, M. et al. SiLEA14, a novel atypical LEA protein, confers abiotic stress resistance in foxtail millet. BMC Plant Biol. 14, 290 (2014).
Irving, H. R., Gehring, C. A. & Parish, R. W. Changes in cytosolic pH and calcium of guard cells precede stomatal movements. Proc. Natl. Acad. Sci. 89, 1790–1794 (1992).
Gonugunta, V. K., Srivastava, N. & Raghavendra, A. S. Cytosolic alkalinization is a common and early messenger preceding the production of ROS and NO during stomatal closure by variable signals, including abscisic acid, methyl jasmonate, and chitosan. Plant Signal Behav. 4, 561–564 (2009).
Rattanakon, S. et al. Abscisic acid transcriptomic signaling varies with grapevine organ. BMC Plant Biol. 16, 72 (2016).
Cutler, A. J. & Krochko, J. E. Formation and breakdown of ABA. Trends Plant Sci. 4(12), 472–478 (1999).
Ren, H. et al. Dynamic analysis of ABA accumulation in relation to the rate of ABA catabolism in maize tissues under water deficit. J Exp Bot. 58(2), 211–219 (2007).
Obata-Sasamoto, H. & Suzuki, H. Activities of enzymes relating to starch synthesis and endogenous levels of growth regulators during tuberization. Physiol. Plant. 95, 69–75 (1979).
Saeedipour, S. Effect of drought at the post-flowering stage on remobilization of carbon reserves in two wheat cultivars differing in senescence properties. Afr. J. Biotechnol. 10(18), 3549–3557 (2011).
Andersson, J. et al. Absence of the Lhcb1 and Lhcb2 proteins of the light-harvesting complex of the photosystem II-effects on photosynthesis, grana stacking and fitness. Plant J. 35, 350–361 (2003).
Xu, Y. H. et al. Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid in Arabidopsis. J Exp Bot. 63(3), 1095–1106 (2012).
Golldack, D., Li, C., Mohan, H. & Probst, N. Tolerance to drought and salt stress in plants: unraveling the signaling networks. Front Plant Sci. 5, 151 (2014).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 7, 562–578 (2012).
Katta, M. A., Khan, A. W., Doddamani, D., Thudi, M. & Varshney, R. K. NGS-QCbox and Raspberry for parallel, automated and rapid quality control analysis of large-scale next- generation sequencing (Illumina) data. PLoS ONE 10(10), e0139868 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 25, 1105–1111 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Goff L, Trapnell C, Kelley D. cummeRbund: Analysis, exploration, manipulation, and visualization of Cufflinks high-throughput sequencing data. R package version 2.26.0 (2019)
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization, and analysis in functional genomics research. Bioinformatics. 21, 3674–3676 (2005).
Howe, E. A., Sinha, R., Schlauch, D. & Quackenbush, J. RNA-Seq analysis in MeV. Bioinformatics 27(22), 3209–3210 (2011).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25(16), 2078–2079 (2009).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff. Fly. 6, 80–92 (2012).
Bosco Dhas, D. B., Ashmi, A. H., Bhat, B. V., Parija, S. C. & Banupriya, N. Modified low- cost SNP genotyping technique using cycle threshold (Ct) & melting temperature (Tm) Values in allele- specific real-time PCR. Indian J Med Res. 142(5), 555–562 (2015).
Schmittgen, T. D. & Zakrajsek, B. A. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys. Methods. 46, 69–81 (2000).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 25, 402–408 (2001).
Acknowledgements
This project was funded by in part by the NIFA-Hatch funds from the US Department of Agriculture provided to the New Mexico Agricultural Experiment Station; New Mexico Peanut Research Board; National Peanut Research Board, Office of Agriculture, Research and Policy, Beaureau of Food security, US Agency for International Development, under the terms of Award No (AID-ECG-A-00-07-0001) to the University of Georgia as management entity for the US Feed the Future Innovation lab on Peanut Productivity and Mycotoxin Control. We thank Sunil S Gangurde for helping in the preparation of the Circos figure for the manuscript. BS acknowledges the award of a research grant fellowship from the Department of Biotechnology (DBT), Government of India. AX acknowledges the Plant and Environmental Sciences, New Mexico State University for Graduate Research Assistantship during the period of study. VG acknowledges the award of a research grant fellowship from the Department of Science and Technology, Government of India. This work has been undertaken as part of the CGIAR Research Program on Grain Legumes and Dryland Cereals. ICRISAT is a member of the CGIAR consortium.
Author information
Authors and Affiliations
Contributions
S.B. analysed the data, interpreted and wrote the manuscript. A.X. collected the physiological attributes, the interpretation and contributed to the writing of the manuscript. V.G. performed analysis of transcriptome data and contributed to the writing of the manuscript. N.L. conducted a physiological analysis and contributed to the development of materials and methods. R.A. performed the SNP validation, contributed towards interpretation of the results and the revision of the manuscript. S.N.N. and M.K.P. contributed towards interpretation of the results and the revision of the manuscript. P.P. and N.P. conceptualized the research, setting up the research experiments and collection of samples in the US. R.K.V. supervised research on transcriptome analysis. S.B., M.K.P., N.P. and R.K.V. revised and finalized the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhogireddy, S., Xavier, A., Garg, V. et al. Genome-wide transcriptome and physiological analyses provide new insights into peanut drought response mechanisms. Sci Rep 10, 4071 (2020). https://doi.org/10.1038/s41598-020-60187-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-60187-z
- Springer Nature Limited