
Abstract
Inherited retinal dystrophies (IRDs), displaying pronounced genetic and clinical heterogeneity, comprise of a broad range of diseases characterized by progressive retinal cell death and gradual loss of vision. By the combined use of whole exome sequencing (WES), SNP-array and WES-based homozygosity mapping, as well as directed DNA sequencing (Sanger), we have identified nine pathogenic variants in six genes (ABCA4, RPE65, MERTK, USH2A, SPATA7, TULP1) in 10 consanguineous Iranian families. Six of the nine identified variants were novel, including a putative founder mutation in ABCA4 (c.3260A>G, p.Glu1087Gly), detected in two families from Northeastern Iran. Our findings provide additional information to the molecular pathology of IRDs in Iran, hopefully contributing to better genetic counselling and patient management in the respective families from this country.
Similar content being viewed by others
Introduction
Inherited Retinal Dystrophies (IRDs) are a genetically and clinically heterogeneous group of diseases characterized by the progressive loss of retinal photoreceptor cells. This neurodegenerative process leads in turn to increasing visual deficit, and over the years to very poor vision and blindness1. With an incidence of approximately 2.5 in 10,000, IRDs are among the most frequently observed hereditary eye diseases2,3,4. Due to their complex genetic and clinical nature, IRDs can be divided into a wide range of clinical subtypes, including: Leber congenital amaurosis (LCA; OMIM #204000), cone-rod dystrophy (CORD; #120970), Stargardt disease (STGD; #248200), retinitis pigmentosa (RP; #268000) and others. They can also manifest as individual eye disorders or involve other organs, as part of various syndromes5.
Thus far, mutations in over 270 genes have been associated with IRDs (RetNet. https://sph.uth.edu/retnet/). Recent advances in massively parallel next-generation sequencing (NGS) and genetic testing approaches have substantially improved the identification of mutations, as well as the diagnostic yield of such a genetically heterogeneous condition6. Given the intricate nature of IRDs, molecular testing in clinical settings may lead to a more accurate diagnosis of patients with ambiguous ophthalmologic evaluations7.
Consanguineous marriages have been globally practiced as a social norm for thousands of years, leading to a high degree of inbreeding in some populations, including Iranians8. As a consequence of inter-family marriages, autozygous regions are created through the inheritance of identical-by-descent haplotypes, resulting in turn in a very high prevalence of recessive conditions9.
This study aimed at integrating NGS and homozygosity mapping, a technique that identifies long stretches of homozygous haplotypes, for the genetic characterization of IRDs in 10 consanguineous Iranian families.
Results
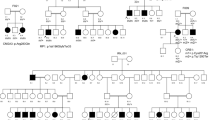
Following WES analysis in the probands from the 10 families ascertained in this study (Fig. 1), we identified nine pathogenic variants in six IRD-associated genes (ABCA4, MERTK, RPE65, SPATA7, TULP1, and USH2A). Of these variants, six were novel, and none of them was previously reported in the Iranian population (Table 1). All of the identified variants were homozygous and resided in autozygous regions of a genomic size of at least 6 Mb, hence reflecting recent endogamy in the population (Table 1, Fig. 2). Autozygome analysis using data from SNP arrays and WES revealed a relatively high number of runs of homozygosity (ROH) (Fig. 2), consistent with the high degree of consanguinity observed in these families (The ROH details for each family containing RetNet genes has been provided in Supplementary Table 1). Autozygome analysis and variant filtering on the WES data was sufficient to identify putative causative variants. SNP data confirmed that the candidate variant located to a homozygous interval in another affected family member. Additional (homozygous) candidate variants filtered for frequency, impact on protein and known to be causative of relevant diseases have been shown in Supplementary Table 2. All findings were validated by Sanger sequencing and, when DNA was available, variant segregation was confirmed within healthy and affected family members, as illustrated in Fig. 1.

Overview of the pedigrees of the 10 families presented in this study and segregation analysis of the pathogenic variants identified. Probands who underwent WES analysis are indicated with an arrow. Genotypes are also indicated, whenever available.

Homozygosity mapping for the 10 probands of this study, generated using the AutoMap tool (Quinodoz et al., manuscript under review). Autozygous regions for autosomes are depicted in blue. Intervals containing the identified pathogenic variants are highlighted in red.
In proband V.4 of family F010 (Fig. 1) we identified a novel homozygous missense variant in the RPE65 gene (NM_000329.3:c.1088C>G, p.Pro363Arg). In this same gene, another novel missense variant was found in proband IV.6 of family F031 (NM_000329.3:c.104C>T, p.Pro35Leu). Both variants are predicted to be pathogenic or likely pathogenic by different prediction tools (Table 2). In family F011, patient V.1 resulted in harboring a homozygous premature stop codon in the MERTK gene (NM_006343.3:c.392G>A, p.Trp131Ter), never published before. Family F019 was found to carry a homozygous missense variant in the USH2A gene (NM_206933.3:c.14926G>A, p.Gly4976Ser), affecting an evolutionarily conserved amino acid. Similarly, this novel variant was predicted to be pathogenic or likely pathogenic by all tested tools (Table 2).
Our NGS analyses revealed pathogenic variants in the LCA-associated genes SPATA7 and TULP1. Proband IV.5 of family F028 had a homozygous premature stop codon in SPATA7 (NM_018418.5:c.253C>T, p.Arg85Ter), previously described to cause LCA in three consanguineous families of Pakistani and Bangladeshi origins13. Segregation was confirmed in the other affected sibling and in the healthy father, who was heterozygous. Similarly, affected members of family F030 carried another homozygous premature stop codon in the same gene (NM_018418.5:c.250C>T, p.Gln84Ter). Sanger sequencing with available DNA samples confirmed that the parents and the healthy sister were both heterozygous carriers (Fig. 1). Moreover, in proband IV.3 of family F032 we identified a known pathogenic missense variant in the TULP1 gene (NM_003322.6:c.1047T>G, p.Asn349Lys). Again, segregation analysis was consistent with the recessive mode of inheritance of this disease (Fig. 1).
WES analysis of patient III.7 of family F004 identified a homozygous missense variant (NM_000350.3:c.1819G>A, p.Gly607Arg) in exon 13 of the ABCA4 gene. This same variant was previously found in patients with STGD10,15. Segregation was confirmed, as shown in Fig. 1. Lastly, in families F026 and F035 we identified a previously-unreported missense variant (NM_000350.3:c.3260A>G, p.Glu1087Gly) in exon 22 of the ABCA4 gene. Interestingly, this same codon was found to carry two missense variants in patients with this condition in previous publications10,16,17,18,19 as well as a nonsense variant in patients with cone-rod dystrophy20.
Discussion
Consanguineous marriages have been traditionally practiced in Iran as a consequence of socio-cultural factors. It is estimated that nearly 40% of Iranian marriages are between related individuals, of which ~ 21% are first cousins and ~ 19% are second cousins21,22. Endogamy, consanguinity or geographic isolation may rise the occurrence of specific mutations in selected populations, which can be isolated by homozygosity mapping. The rationale for this method is that unaffected parents who have some degree of kinship, belong to an ethnic group with high endogamy or are from a geographical isolate, could be heterozygotes for the same recessive mutation from a common ancestor. This mutation, which at the population level possibly will even be infrequent, can be brought to homozygosity since consanguinity can cause disease in these parents’ children23,24. This feature makes the Iranian gene pool one of the richest resources for genetic investigations. Several studies have so far been published concerning the genetic basis of IRDs in Iran25,26,27,28,29,30. In this study, we further refined the genetic landscape of IRDs by identifying nine pathogenic variants in genes that were previously associated with these conditions.
In particular, we identified a putative novel founder missense mutation (p.Glu1087Gly) in the ABCA4 gene, within individuals from the Northeastern region (Khorasan province) of Iran. They had a similar ethnic origin and shared an identical homozygous haplotype around ABCA4 (chr1:85′647′942–97′544′759 of size 11.9 Mb). The phenotypic features of the probands and their affected relatives were associated with those of classical STGD. According to data extracted from the database of protein families (Pfam), amino acids 946–1090 encode the ABC Transporter wherein based on the arrangement of the Pfam domains, the 1087 position is conserved and predicted to be an active site (https://pfam.xfam.org/protein/ABCA4_HUMAN). Two missenses affecting the same amino acid were already found to be pathogenic: p.Glu1087Asp and p.Glu1087Lys31,32. In addition to the increasing number of reports on pronounced clinical and genetic variability of genes that cause IRDs, mutations in three different genes, RPE65, MERTK and USH2A were ascertained in four unrelated families with members showing symptoms of RP. Fortunately, recent advances in gene therapy trials on patients with RPE65 and MERTK mutations have shown promising results, reflecting hope for a potentially revolutionary form of therapy in a subset of patients33,34,35,36. Our study stipulates the role of these variants in the pathogenesis of RP in Iranian patients, who could potentially be candidates for future therapeutic interventions. Furthermore, genetic alterations in SPATA7 and TULP1 are known to manifest with overlapping clinical symptoms of LCA and RP37,38. Evaluating the current status of detected variants and also checking them against other Iranian databases which showed nil frequencies provides supporting evidence that these variants can be classed as potential causative variants.
The advent of gene-directed interventions has enabled bridging of molecular genetics with genetic counseling and possible therapeutic interventions in ophthalmology. Specifically, since targeted gene therapy is constantly advancing, detection of genetic mutations may soon be a routine procedure to allow patients to access suitable therapies.
In this context, our study expands the current understanding of the molecular basis of IRDs in Iran, while providing at the same time elements of information for genetic and prenatal counselling as well as possible indications for future therapy.
Materials and methods
Ethics statement
This study was designed in compliance with the tenets of the Declaration of Helsinki. Written informed consent was obtained from each participant or legal guardian prior to their participation. This study was approved by the Institutional Review Boards of our respective Universities: Mashhad University of Medical Sciences (MUMS), and University of Lausanne.
Families and preparation of samples
Families were selected according to three main criteria: (i) they included two or more individuals with confirmed IRD diagnosis, (ii) showed a likely autosomal recessive (AR) pattern of inheritance, and (iii) had a history of consanguinity. Pedigrees were drawn using the Pedigree Chart Designer software (CeGaT, Tubingen, Germany). Clinical information and clinical data were extracted from patients’ medical records of Mashhad Khatamolanbia Eye Hospital. All patients were evaluated by an ophthalmologist and Optical Coherence Tomography (OCT) was performed for individuals who had no recent follow up. Six milliliters of peripheral blood from available affected and unaffected members of each family were collected and mixed with EDTA (Merck KGaA, Darmstadt, Germany). A phenol–chloroform method was used to extract genomic DNA from peripheral leukocytes. DNA quality and quantity were verified using a Nanodrop 2000 (Thermo Fisher Scientific, Wilmington, DE, USA). DNA integrity was ascertained by running DNAs on 1% agarose gel. Samples were stored at − 20 °C until used.
SNP genotyping
DNAs of studied individuals were genotyped at the iGE3 Platform of the University of Geneva, Switzerland, using an Illumina Infinium array (San Diego, CA, USA; GSAMD-24v2.0). Genotype values were obtained with GenomeStudio (Illumina). Homozygosity mapping was obtained by the use of the PLINK software. Due to the high degree of consanguinity, SNP genotyping was chosen as a complementary analysis to narrow down homozygous regions and reduce the number of candidate variants.
Whole-exome sequencing
WES was performed on one designated proband from each of the families analyzed. Exome capture and library preparation was performed using the SureSelect Human All Exon v6 kit (Agilent, Santa Clara, CA, USA) and the HiSeq Rapid PE Cluster Kit v2 (Illumina, San Diego, CA, USA), from 2 μg genomic DNA. Libraries were sequenced on a HiSeq 2500 instruments (Illumina). Raw reads were mapped to the human genome reference sequence (build hg19) with the Novoalign software (V3.08.00, Novocraft Technologies, Selangor, Malaysia). Duplicate reads were then removed using Picard (v. 2.14.0-SNAPSHOT). Base quality score recalibration was performed with HaplotypeCaller (GATK, v.4.0.3.0). Single nucleotide variants (SNVs) and small insertions and deletions were detected using the Genome Analysis Tool Kit (GATK v4.0) software package, using the Best Practice Guidelines identified by the developers39. ExomeDepth software was used to detect exonic deletions. Homozygous variants in IRD genes (according to RetNet) lying within ROHs were processed as follows. Filtration was performed based on quality (DP > 10, GQ > 30, FS < 25 and alternative read percentage > 25%) and allelic frequency [below 1% in ExAC, gnomAD, 1000 Genomes, ESP (NHLBI Exome Variant Server, https://evs.gs.washington.edu/EVS), GME (GME Variome https://igm.ucsd.edu/gme/index.php), and ABraOM]. The pathogenicity of the genetic variants was assessed after functional annotation through ANNOVAR40 and using in-house scripts41, where the predicted impact on protein sequence and messenger RNA (mRNA) splicing were also taken into account. Finally, we checked the frequency of variants in the Iranian population using the Iranome database (https://www.iranome.ir/). ROHs were identified by using the AutoMap software (Quinodoz et al., manuscript under review. https://automap.iob.ch/) on WES data. The number of filtered variants at each step is shown in Supplementary Table 3.
Sanger validation and segregation analysis
Primers were designed using the Primer3 online software42, at least 70 bp upstream and downstream of the variants, and PCR reactions were performed under standard conditions (for the list of primers see Supplementary Table 4). PCR products were purified by treatment with exonuclease and thermosensitive alkaline phosphatase (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed by Sanger sequencing using the Big Dye Terminator v3.1 Cycling Sequencing Kit (Applied Biosystem, Foster City, CA, USA) on an ABI 3730XL platform (Applied Biosystems). Sequencing data were analyzed using the Sequencher v4.8, SnapGene (https://www.snapgene.com), and Chromas Lite v2.01 software and compared with wild-type samples and reference sequences from NCBI and Ensembl databases. Furthermore, frequency of all candidate variants was verified in a database of 800 Iranian healthy individuals (https://www.iranome.ir/).
References
Berson, E. L. Retinitis pigmentosa. The Friedenwald Lecture. Investig. Ophthalmol Vis. Sci. 34, 1659–1676 (1993).
Haer-Wigman, L. et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 25, 591–599. https://doi.org/10.1038/ejhg.2017.9 (2017).
Finger, R. P., Fimmers, R., Holz, F. G. & Scholl, H. P. Incidence of blindness and severe visual impairment in Germany: projections for 2030. Invest. Ophthalmol Vis. Sci. 52, 4381–4389. https://doi.org/10.1167/iovs.10-6987 (2011).
Ayuso, C. & Millan, J. M. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2, 34. https://doi.org/10.1186/gm155 (2010).
Sahel, J. A., Marazova, K. & Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 5, a017111. https://doi.org/10.1101/cshperspect.a017111 (2014).
Jespersgaard, C. et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci. Rep. 9, 1–7 (2019).
Lee, K. & Garg, S. Navigating the current landscape of clinical genetic testing for inherited retinal dystrophies. Genet. Med. 17, 245–252. https://doi.org/10.1038/gim.2015.15 (2015).
Saadat, M. & Zarghami, M. Consanguineous marriages among Iranian Mandaeans living in South-West Iran. J. Biosoc. Sci. 50, 451–456. https://doi.org/10.1017/s0021932017000207 (2018).
Wakeling, M. N. et al. Homozygosity mapping provides supporting evidence of pathogenicity in recessive Mendelian disease. Genet Med 21, 982–986. https://doi.org/10.1038/s41436-018-0281-4 (2019).
Rivera, A. et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 67, 800–813 (2000).
Briggs, C. E. et al. Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest. Ophthalmol. Vis. Sci. 42, 2229–2236 (2001).
Riveiro-Alvarez, R. et al. Frequency of ABCA4 mutations in 278 Spanish controls: an insight into the prevalence of autosomal recessive Stargardt disease. Br. J. Ophthalmol. 93, 1359–1364 (2009).
Mackay, D. S. et al. Screening of SPATA7 in patients with Leber congenital amaurosis and severe childhood-onset retinal dystrophy reveals disease-causing mutations. Invest. Ophthalmol. Vis. Sci. 52, 3032–3038 (2011).
Watson, C. M. et al. Mutation screening of retinal dystrophy patients by targeted capture from tagged pooled DNAs and next generation sequencing. PLoS ONE 9, e104281. https://doi.org/10.1371/journal.pone.0104281 (2014).
Zhou, Y. et al. Exome sequencing analysis identifies compound heterozygous mutation in ABCA4 in a Chinese family with Stargardt disease. PLoS ONE 9, e91962 (2014).
Roberts, L. J., Nossek, C. A., Greenberg, L. J. & Ramesar, R. S. Stargardt macular dystrophy: common ABCA4 mutations in South Africa—establishment of a rapid genetic test and relating risk to patients. Mol. Vis. 18, 280 (2012).
Lewis, R. A. et al. Genotype/phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease. Am. J. Hum. Genet. 64, 422–434 (1999).
Fumagalli, A. et al. Mutational scanning of the ABCR gene with double-gradient denaturing-gradient gel electrophoresis (DG-DGGE) in Italian Stargardt disease patients. Hum. Genet. 109, 326–338 (2001).
Fujinami, K. et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 122, 326–334 (2015).
Ducroq, D. et al. The ABCA4 gene in autosomal recessive cone-rod dystrophies. Am. J. Hum. Genet. 71, 1480–1482 (2002).
Hamamy, H. Consanguineous marriages. J. Community Genet. 3, 185–192 (2012).
Hosseini-Chavoshi, M., Abbasi-Shavazi, M. J. & Bittles, A. H. Consanguineous marriage, reproductive behaviour and postnatal mortality in contemporary Iran. Hum. Hered. 77, 16–25 (2014).
Ur Rehman, A. et al. Exploring the genetic landscape of retinal diseases in North-Western Pakistan reveals a high degree of autozygosity and a prevalent founder mutation in ABCA4. Genes 11, 12 (2020).
Saqib, M. A. N. et al. Homozygosity mapping reveals novel and known mutations in Pakistani families with inherited retinal dystrophies. Sci. Rep. 5, 9965 (2015).
Ghofrani, M. et al. Homozygosity mapping and targeted sanger sequencing identifies three novel CRB1 (crumbs homologue 1) mutations in Iranian retinal degeneration families. Iran. Biomed. J. 21, 294 (2017).
Eisenberger, T. et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS ONE 8, e78496 (2013).
Beheshtian, M. et al. Impact of whole exorne sequencing among Iranian patients with autosomal recessive retinitis pigmentosa. Arch. Iran. Med. 18, 776–785 (2015).
Tayebi, N. et al. Targeted next generation sequencing reveals genetic defects underlying inherited retinal disease in Iranian families. Mol. Vis. 25, 106 (2019).
Roshandel, D. et al. Rhodopsin gene mutation analysis in Iranian patients with autosomal dominant retinitis pigmentosa. Int. Ophthalmol. 39, 2523–2531. https://doi.org/10.1007/s10792-019-01099-4 (2019).
Salmaninejad, A. et al. Next-generation sequencing and its application in diagnosis of retinitis pigmentosa. Ophthalmic Genet. 40, 393–402. https://doi.org/10.1080/13816810.2019.1675178 (2019).
Runhart, E. H. et al. The common ABCA4 variant p. Asn1868Ile shows nonpenetrance and variable expression of Stargardt disease when present in trans with severe variants. Investig. Ophthalmol. Vis. Sci. 59, 3220–3231 (2018).
Jaakson, K. et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum. Mutat. 22, 395–403 (2003).
Ghazi, N. G. et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum. Genet. 135, 327–343. https://doi.org/10.1007/s00439-016-1637-y (2016).
Russell, S. et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860. https://doi.org/10.1016/s0140-6736(17)31868-8 (2017).
Darrow, J. J. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov. Today 24, 949–954. https://doi.org/10.1016/j.drudis.2019.01.019 (2019).
Miraldi Utz, V., Coussa, R. G., Antaki, F. & Traboulsi, E. I. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. 39, 671–677. https://doi.org/10.1080/13816810.2018.1533027 (2018).
Wang, H. et al. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am. J. Hum. Genet. 84, 380–387. https://doi.org/10.1016/j.ajhg.2009.02.005 (2009).
Guo, Y. et al. Advantage of whole exome sequencing over allele-specific and targeted segment sequencing in detection of novel TULP1 mutation in Leber congenital amaurosis. Ophthalmic Genet. 36, 333–338. https://doi.org/10.3109/13816810.2014.886269 (2015).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. https://doi.org/10.1038/ng.806 (2011).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164. https://doi.org/10.1093/nar/gkq603 (2010).
van Karnebeek, C. D. et al. NANS-mediated synthesis of sialic acid is required for brain and skeletal development. Nat Genet 48, 777–784. https://doi.org/10.1038/ng.3578 (2016).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115 (2012).
Acknowledgements
This research was funded by the Swiss National Science Foundation (Grant #176097 to CR). We would like to express gratitude to the patients and all their family members that participated in this study for their valuable cooperation and participation.
Author information
Authors and Affiliations
Contributions
A.S., Z.R. and N.B. wrote the manuscript. A.S., A.P., N.S. and M.M., were responsible for recruitment of patients. A.S., N.B. and M.Q. analyzed the NGS and SNP-array data. A.S. also performed segregation and analyzed Sanger sequencing data. A.P. and C.R. contributed to conceptualization, methodology, reviewing and supervision. All the authors commented and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salmaninejad, A., Bedoni, N., Ravesh, Z. et al. Whole exome sequencing and homozygosity mapping reveals genetic defects in consanguineous Iranian families with inherited retinal dystrophies. Sci Rep 10, 19413 (2020). https://doi.org/10.1038/s41598-020-75841-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75841-9
- Springer Nature Limited
This article is cited by
-
Generation of Zebrafish Models of Human Retinitis Pigmentosa Diseases Using CRISPR/Cas9-Mediated Gene Editing System
Molecular Biotechnology (2023)