
Abstract
In dairy cattle, endometritis is a severe infectious disease that occurs following parturition. It is clear that genetic factors are involved in the etiology of endometritis, however, the molecular pathogenesis of endometritis is not entirely understood. In this study, a system biology approach was used to better understand the molecular mechanisms underlying the development of endometritis. Forty transcriptomic datasets comprising of 20 RNA-Seq (GSE66825) and 20 miRNA-Seq (GSE66826) were obtained from the GEO database. Next, the co-expressed modules were constructed based on RNA-Seq (Rb-modules) and miRNA-Seq (mb-modules) data, separately, using a weighted gene co-expression network analysis (WGCNA) approach. Preservation analysis was used to find the non-preserved Rb-modules in endometritis samples. Afterward, the non-preserved Rb-modules were assigned to the mb-modules to construct the integrated regulatory networks. Just highly connected genes (hubs) in the networks were considered and functional enrichment analysis was used to identify the biological pathways associated with the development of the disease. Furthermore, additional bioinformatic analysis including protein–protein interactions network and miRNA target prediction were applied to enhance the reliability of the results. Thirty-five Rb-modules and 10 mb-modules were identified and 19 and 10 modules were non-preserved, respectively, which were enriched in biological pathways related to endometritis like inflammation and ciliogenesis. Two non-preserved Rb-modules were significantly assigned to three mb-modules and three and two important sub-networks in the Rb-modules were identified, respectively, including important mRNAs, lncRNAs and miRNAs genes like IRAK1, CASP3, CCDC40, CCDC39, ZMYND10, FOXJ1, TLR4, IL10, STAT3, FN1, AKT1, CD68, ENSBTAG00000049936, ENSBTAG00000050527, ENSBTAG00000051242, ENSBTAG00000049287, bta-miR-449, bta-miR-484, bta-miR-149, bta-miR-30b and bta-miR-423. The potential roles of these genes have been previously demonstrated in endometritis or related pathways, which reinforced putative functions of the suggested integrated regulatory networks in the endometritis pathogenesis. These findings may help further elucidate the underlying mechanisms of bovine endometritis.
Similar content being viewed by others


Introduction
One of the main goals of reproductive management in dairy cattle is leading cows to be pregnant at a normal calving interval and in an effective manner1,2. Although several reasons currently exist for reproductive inefficiency in dairy cows, postpartum uterine disease is considered as one of the most important causes in this regard3. Infection of the postpartum uterus with bacteria, namely endometritis, affects half of all dairy cattle and consequently causes infertility or subfertility by interrupting both uterine and ovarian functions4. Postpartum endometritis (during 21 days or more after calving) is commonly observed among high-producing dairy cows, which could adversely affect their reproductive performance by increasing services per conception and calving interval, decreasing the risk of pregnancy and the rate of conception, and reducing milk yield that consequently result in economic loss5. In this context, the annual economic losses from cattle uterine disorders were estimated to be $1.4 billion and $650 million in Europe and the United States, respectively4. Undoubtedly, the development of endometritis stems from some complex interactions between bacteria and host factors, and genetic factors play important roles in the development of the disease as well6. However, the molecular genetic mechanisms by which endometritis causes infertility or subfertility are not fully clear yet.
Several genes, transcription factors (TFs), biological processes and signaling pathways have been reported in the previous studies to be involved in the development of endometritis. Investigation of gene expression profile using real time-PCR (RT-PCR) showed the higher expression of CXCL5, IL1B, IL6, IL8, PTGS2 and TNF in bovine with an inflamed endometrium in comparison to cows with a healthy endometrium7. A higher expression of PTGS2 gene, which is involved in the inflammatory response, was detected in the repeat breeder cows with subclinical endometritis compared to healthy repeat breeder cows (using RT-PCR)8. Nuclear factor (NF)-κB, as a TF, has been also reported as a major regulator of pro‐inflammatory genes involving in the pathogenesis of endometritis9,10. It was found that cows with subclinical endometritis at 45–55 days postpartum had higher expression of immune factors C3, C2, LTF, PF4 and TRAPPC13 (using microarray) in comparison to healthy cows at the same time points11. Transcriptome profile analysis of Holstein Frisian cows (with and without clinical endometritis) at 42–60 days postpartum revealed that 92 genes including PTHLH, INHBA, DAPL1 and SERPINA1 were significantly upregulated (using quantitative RT-PCR) in clinical endometritis cows compared to the healthy animal group12.
The literature review showed that most of the previously performed studies have focused on the gene expression analysis, which only considers the differentially expressed genes and does not consider the interactions among them. Therefore, to fill this gap, the co-expressed gene regulatory network constructed based on the gene expression data, in order to better explain the complex etiology of biological processes or some diseases like endometritis13. In this regard, the weighted gene co-expression network analysis (WGCNA) is a popular and effective gene co-expression network-based approach, which is performed based on the “guilt-by-association” method14. WGCNA measures the connectivity among the genes based on the gene expression patterns and clusters of the highly correlated genes into modules by considering their expression similarities15,16. Moreover, WGCNA could assess the preservation of the modules to identify those modules with topological differences between those situations (such as normal vs diseases)16. It was reported that some modules that lost their preservation in other situations may be involved in the development of that situations17, so they can be considered as potential candidates for further studies18. In our previous study for the first time, WGCNA was applied to better understand the molecular mechanisms of the endometriosis. To do this, publicly available microarray data from women with minimal/mild endometriosis, mild/severe endometriosis and without endometriosis were used. In total, 16 functional modules were found in the normal samples as reference, of which nine modules were non-preserved in the two other stages. Several important hub genes were also suggested for further investigations including NF-kB, IPO9, PTPMT1 and PTGDS19.
More recent evidence showed that non-coding RNAs (ncRNAs), especially microRNAs (miRNAs)20,21,22 and long ncRNAs (lncRNAs)23,24, play important roles in different aspects of the biological processes, including innate and adaptive immunity and inflammation25. Furthermore, they can be used as biomarkers or therapeutic targets in different diseases like endometritis26. Moreover, lncRNAs are transcripts without any known protein coding potential greater than 200 nt in length27. Many biological processes have been found to have associations with lncRNAs at three levels of transcriptional, post transcriptional, and translational28. As well, the regulatory role of lncRNA Mirt2 has been demonstrated in both inflammation and macrophage polarization29. Many other lncRNAs such as THRIL, PACER, Lethe, NKILA, and lnc13, have been also identified as crucial regulators in inflammation and immune system30. MiRNAs, as another class of ncRNAs, are ∼ 22-nt long, which could post-transcriptionally regulate the expression of targeted mRNAs31. Disrupting some miRNAs regulatory mechanisms or losing some of them can lead to disorders in immune system32. Deregulation of miR-203 was also identified as a potential biomarker of psoriasis development (as one of the most prevalent chronic inflammatory skin diseases among adults)33. Many other miRNAs such as miR-223, miR-146a, miR-146b, miR-125a, miR-125b, miR‐148a, and miR-21 have been found to be associated with inflammatory diseases or immune responses22,34. Therefore, gene co-expression network construction in the presence of both mRNAs and ncRNAs may provide more biological insights compared to when performing separate analyses to study complex diseases. This type of the integrated regulatory network can help in exploring the roles of ncRNAs in endometritis onset and development.
In the present study, we hypothesized that integrating mRNAs and ncRNAs expression profiles (as miRNAs and lncRNAs) used to construct an integrated co-expression network would provide additional insights into the endometritis-associated biological pathways. Therefore, in the present study, WGCNA was applied to identify the co-expression RNA-Seq-based modules (Rb-modules) (including mRNAs and lncRNAs) and miRNA-Seq-based modules (mb-modules) in healthy samples. Afterward, to find the non-preserved Rb-modules in the endometritis samples, preservation analysis was used. Next, the mb-modules were assigned to the non-preserved Rb-modules and then an integrated co-expression network was constructed. As well, protein–protein interaction network, functional enrichment analysis, miRNA target prediction, and hub gene detection approaches were applied to help in identifying the most important candidate genes, which may consequently affect the development of endometritis. Our findings may further aid research studies to provide novel insights into the pathogenesis of endometritis and/or identifying therapeutic targets in this regard.
Material and methods
Dataset
Raw RNA-Seq and miRNA-Seq data were obtained from the Gene Expression Omnibus (GEO) database at the National Center for Biotechnology Information (NCBI) under the accession number GSE66827. The data contained endometrial samples related to nine healthy and six endometritis cows at 7 and 21 days post-partum6. In the early postpartum period, healthy cows can be differentiated from cows at risk of developing uterine disease through change in immune profile. Therefore, all cows at two time points postpartum during uterine involution were examined for the inflammation extension and categorized as either healthy or having subclinical endometritis. The Illumina® HiSeq™ 2000 was used for sequencing. Finally, 30 RNA-Seq libraries containing 49 bp reads and 20 miRNA-Seq libraries containing 51 bp reads were generated. More details of preparing data can be found in the original paper6.
RNA-Seq and miRNA-Seq data analysis
Here, to construct an integrated regulatory network, only cows with available RNA-Seq and miRNA-Seq data were considered. Hence, 20 RNA-Seq (GSE66825) and 20 miRNA-Seq (GSE66826) samples were retained for further analysis. The quality of raw RNA-Seq and miRNA-Seq reads were checked by FastQC (version 0.11.5)35. Low quality bases/reads and adaptor sequences for both RNA-Seq and miRNA-Seq reads were removed by Trimmomatic (version 0.32)36. The trimming criteria for RNA-Seq data was TRAILING: 20, MAXINFO: 40: 0.9, MINLEN: 40 and for miRNA-Seq data was TRAILING: 20, MAXINFO: 18:0.9, MINLEN: 18. Then, FastQC was used again for monitoring the quality of the reads after trimming.
Clean reads from RNA-Seq data were aligned to bovine reference genome (version ARS-UCD1.2) using Hisat2 (version 2.1.0)37. Then, Python script HTSeq-count (version 2.7.3) was used to count the RNA-Seq read numbers mapped to annotated genes using the Ensembl bovine GTF file (release 98)38. The generated gene expression matrix included mRNAs and lncRNAs. To identify transcription factors (TFs) in the expression matrix, AnimalTFDB3.0 database was used39. For miRNA-Seq data, non-miRNA reads were filtered out using Unitas (version 1.7.0)40. Bowtie software (version 1.3.0) was then used to align miRNA-based reads to bovine pre-mature miRNA sequences downloaded from miRBase database (version 22) with mismatch ≤ 241. Finally, SAMtools (version 1.9) was used to quantify the expression of each miRNA and generate miRNA expression matrix42.
Taken together, WGCNA is expanded as an unsupervised approach to analyze microarray data sets43, both expression matrix that were included raw count data were normalized to log counts per million (CPM) based on voom approach in limma package of R software (version 3.42.0)44. On the other hand, reliability of the genes with low expression levels is not high enough to be used in co-expression analysis45. Hence, for tackling this issue, genes in both expression matrices were assessed and the genes with expression levels ≥ 1 CPM in at least four samples as well as standard deviation higher than 0.25 were kept for further analysis.
Weighted co-expression network analysis
Rb- and mb-modules were constructed separately by WGCNA R package (version 1.68)16. To ensure whether network construction was reliable, the outlier samples were excluded. For this purpose, adjacency matrices of both expression matrices matrix were calculated independently and sample network connectivity according to the distances was standardized. Outlier samples were defined as samples with a standardized connectivity less than − 2.5. Then to determine whether the sample data were complete as well as exclude the unqualified genes, goodSamplesGenes function in WGCNA package was applied.
To construct Rb-modules, an appropriate soft-thresholding power was calculated to be confident that the constructed network follows the scale-free topology. The scale-free network contains several nodes with few interactions and few nodes with high interactions, which is called hub nodes46. The approximate scale-free topology in healthy samples was achieved at a soft threshold of β = 13. Afterward, Rb-modules were constructed using blockwiseModules function in the WGCNA package, with the following major parameters: power = 13, corType = “bicor”, networkType = “signed”, TOMType = “signed”, maxBlockSize = 17,000, minModuleSize = 30, reassignThreshold = 0, mergeCutHeight = 0.25. Also, β = 22 met the soft-threshold parameters for signed miRNA co-expression network construction and the curve reached R2 = 0.80. For mb-module detection, blockwiseModules function in the WGCNA package was used with the following major parameters: power = 22, corType = “bicor”, networkType = “signed”, TOMType = “signed”, maxBlockSize = 17,000, minModuleSize = 10, reassignThreshold = 0, mergeCutHeight = 0.25. Because of the smaller size of the miRNA transcriptome in comparison to the mRNA-lncRNA transcriptome, minimum module size for the miRNAs was set as 1016,47. It is worth to note that, to identify the modules in both datasets, first, based on the soft-thresholding power, weighted adjacency matrices was constructed and was transformed into the topological matrix (TOM), which describe the association strength between the genes. Then, TOM based adjacency matrices were clustered using average linkage hierarchical clustering analysis through a dynamic hybrid tree cutting algorithm and the highly similar modules were further merged, based on the correlations between the module eigengenes.
Preservation analysis
Using modulePreservation function in WGCNA package, it was investigated whether the connectivity patterns of the genes in the detected Rb-modules in healthy samples are preserved in the infected samples. Two composite module preservation statics including Zsummary and medianRank were applied to evaluate the preservation, based on connectivity and density of genes present in a module. Since Zsummary depends on the module size, especially when modules with different sizes have to be compared, medianRank, as an index that is less sensitive to the module size, can be combined with Zsummary to accurately test the preservation level. The higher value of Zsummary and the lower value of medianRank indicate strong preservation in the infected samples48. The Zsummary < 5 and MedianRank > 8 were considered as criteria for non-preserved modules. A 5 < Zsummary < 10 and MedianRank > 8 indicated evidence of semi-preserved modules. If Zsummary > 10 and MedianRank < 8, it was concluded that there was evidence of module preservation. Permutation method (N = 200 permutations) was used to evaluate statistical significance of both module preservation statics.
Detection of hub genes
It is well known that all the genes present in a module are not important to be linked with the subject of interest (here endometritis). Hence, some genes are more interesting than others in topological and functional perspective, called hub genes. In this context, the scale-free property of the biological networks indicates to this point that a small number of highly connected genes hold the network together. The WGCNA functions moduleEigengenes and signedKME were used to identify the hub mRNAs, lncRNAs and miRNAs in the relevant modules. In both of the constructed modules, expression profile of each module was summarized by module eigengene (MEs). MEs refers to the primary component in the principal component analysis of the genes in a module49. To quantify the relationship between a gene and a given module, kME or module membership was determined as the correlation between the expression profile of a gene and the ME of a module16. kME describes the degree of internal connectivity of the module or the degree of association between a gene in a particular module and other genes in the module. Hub genes were determined as the genes with kME ≥ 0.716.
Functional enrichment analysis
To elucidate the putative biological functions of the selected genes (genes of each module, hub- or hub-hub genes), GO terms (biological process) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed using the Enrichr online analysis tool (https://maayanlab.cloud/Enrichr/). Only significant terms with adjusted p-values < 0.05 were considered.
Assign the mb-modules to the Rb-modules and target prediction
To assign the mb-modules to Rb-modules, spearman correlations were calculated between the MEs of Rb-modules and mb-modules. Negative correlations suggest that mb-modules might inversely regulate Rb-modules. Hence, mb-modules with a significant (adjusted p-value < 0.20) negative correlation larger than 0.80 were defined as regulators of the Rb-module of interest.
RNAhybrid (version 2.2)50, miRanda (version 3.3a)51 and RNA22 (version 2.0)52 were applied to predict the targets of miRNAs in the mb-modules that were negatively correlated with a Rb-module. In this respect, bovine 3′ UTR sequences (mRNAs and lncRNAs genes) were retrieved from Ensemble database (https://www.ensembl.org/). Also, bovine mature miRNA sequences were obtained from miRBase database (version 22). Minimum free energy threshold was set as − 15 and other parameters were set as default for all used software. To avoid false positive results, the predicted targets for each miRNA were considered as potential targets, if were detected by at least two out of the three applied software.
Detection of hub-hub genes in Rb-modules
In order to identify the most important hub genes (hub-hub genes) in the Rb-modules with a significant assigned mb-module, the following steps were performed:
-
(1)
Protein–protein interactions (PPIs) among the mRNA hub genes of each module were obtained using Search Tool for the Retrieval of Interacting Genes (STRING) database53.
-
(2)
All the identified interactions related to hub mRNAs (including STRING-PPIs and WGCNA-calculated co-expressed mRNAs) and hub lncRNAs (WGCNA-calculated co-expressed lncRNAs) of each module were inputted to Cytoscape software (version 3.7.2) (https://cytoscape.org/) and were evaluated by cytoHubba application (version 0.1)54. CytoHubba suggests 12 topological analysis methods including maximum click centrality (MCC), density of maximum neighborhood component (DMNC), maximum neighborhood component (MNC), degree method, edge percolated component (EPC), bottleneck, EcCentricity, closeness, radiality, betweenness, stress and clustering coefficient to screen the highly connected genes in a given network54. All the methods were applied to detect the hub-hub genes in each module.
-
(3)
Once all the results from the 12 methods were obtained, the RankAggreg package (version 0.6.5) in R software55 was used to connect the results of all methods with each other and generate a consensus ranking. RankAggreg package ordered lists based on the Cross-Entropy Monte Carlo algorithm and the Genetic Algorithm. For increasing the accuracy of the result, both methods were applied and the genes that were identified as highly connected genes in both methods were reported as hub-hub genes.
Integrated regulatory network
Cytoscape (version 3.7.2) was used to combine all the identified interactions among the genes and visualize the final integrated regulatory network. For each module, the obtained interactions from STRING-PPIs, WGCNA-calculated co-expressed mRNAs, WGCNA-calculated co-expressed lncRNAs and WGCNA-calculated co-expressed miRNAs (if an mb-module was assigned to the Rb-module) were used to establish the integrated regulatory network.
Results
RNA-Seq and miRNA-Seq data analysis
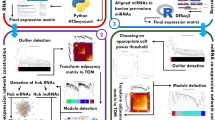
An overview of the used pipeline is provided in Fig. 1. In total, 785,810,232 RNA-Seq and 350,319,753 miRNA-Seq reads were processed in this study. After checking the quality and trimming the reads, 758,465,299 RNA-Seq and 334,769,250 miRNA-Seq reads were remained. On average, 91.58% of RNA-Seq and 79.95% of miRNA-Seq reads were aligned to the reference genome and bovine pre-mature miRNAs, respectively. The results of sequencing and mapping are presented in Supplementary File S1. Gene expression analysis and screening out the less-expressed ones led to generate a normalized gene expression matrix containing 11,744 genes (10,940 mRNAs, 775 TFs and 190 lncRNAs) and a normalized miRNA expression matrix containing 420 miRNAs.

The used pipeline for constructing the integrated regulatory network.
Detection of co-expressed genes
In this study, a network-based approach was applied to establish a better understanding on the molecular mechanisms of endometritis. No outlier samples were observed in the given datasets. After the determination of the appropriate soft threshold power beta (Supplementary File S2), 35 Rb-modules and 10 mb-modules were detected by hierarchical clustering and dynamic branch cutting, which were labelled by different colors based on the WGCNA approach (Fig. 2). The average number of genes per module in the Rb- and mb-modules were obtained as 333 and 35, respectively. Accordingly, the largest Rb-module was turquoise module (containing 3188 genes, including 152 TFs, 36 lncRNAs, and 2973 mRNAs), while the smallest module was sienna3 (containing 36 mRNAs). The greatest and smallest numbers of lncRNAs belonged to blue with 40 lncRNAs, saddlebrown, and sienna3 with no lncRNAs. Using the AnimalTFDB database, a total of 682 TFs were identified in the Rb-modules (Supplementary File S3), in which blue module with 224 TFs and sienna3, darkred, skyblue, steelblue, and yellow module with no TFs had the greatest and smallest numbers of TFs (Fig. 3). Among the mb-modules, turquoise was found as the largest one and the purple was found as the smallest one with 74 and 15 miRNAs, respectively. Additionally, 102 and 69 genes were reported as grey modules in Rb- and mb-modules, respectively, which contained some genes that were not assigned to any module. Details of all the genes of each module are represented in Supplementary File S4.

Clustering dendrogram of mRNAs and lncRNAs (a) and miRNAs (b). In WGCNA genes are clustered into modules based on their co-expression. Hierarchical clustering is a widely used method for detecting clusters. The co-expressed genes have a small degree of dissimilarity. The dissimilarity measure (1-TOM) can be used as input in hierarchical clustering. Following that, modules were defined as branches of a cluster tree and each module was labeled by a unique color using the static tree cutting method.

Percentage of mRNAs, lncRNAs and TFs in each Rb-module. Rb-modules were identified across 20 RNA-Seq datasets. A, B and C region indicate non-preserved, semi-preserved and preserved Rb-modules, respectively. As it can be seen, the percentage of mRNAs in all modules is considerably higher than percentage of TFs and lncRNAs. Despite the important regulatory role of TFs and lncRNAs, almost less than 15% of genes in each module is devoted to them.
Preservation analysis
The preservation analysis of Rb-modules was performed to dissect the connectivity patterns between the two healthy and endometritis conditions. Network properties of non- and semi-preserved modules were altered under endometritis compared to healthy conditions, so they may be related to the development of endometritis. Of 35 Rb-modules, 19, 12 and four modules were detected as non-preserved, semi-preserved and preserved modules, respectively (Fig. 4, Supplementary File S5).

The medianRank (y axis) and Zsummary (x axis) statistics of the module preservation across 20 RNA-Seq datasets. Non-preserved, semi-preserved and preserved modules are indicated by square, circle and triangle points respectively in Rb-modules. The red and black vertical lines indicate the thresholds Zsummary = 8 and Zsummary = 10 respectively. The blue horizontal line indicates a threshold MedianRank = 8. The modules with Zsummary < 5 and MedianRank > 8 were considered as non-preserved, 5 < Zsummary < 10 and MedianRank > 8 semi-preserved and Zsummary > 10 and MedianRank < 8 preserved.
Hub genes show higher connectivity inside the module and are probably more informative56. In total, 635 (including 582 mRNAs, 34 TFs and 10 lncRNAs), 5310 (including 4831 mRNAs, 366 TFs and 71 lncRNAs) and 1437 (including 1332 mRNAs, 60 TFs and 33 lncRNAs) hub genes were identified in preserved, non- and semi-preserved Rb-modules, respectively (Supplementary File S6). The highest number of hub genes was found in turquoise (1910 genes including 1,788 mRNAs, 89 TFs and 22 lncRNAs), blue (1301 genes including 1125 mRNAs, 147 TFs 19 lncRNAs) and brown (692 genes including 632 mRNAs, 49 TFs and 6 lncRNAs) Rb-modules, respectively. The total number of hub miRNAs was 212 (Supplementary File S7). Blue, turquoise and brown mb-modules showed the highest number of hub miRNAs including 37, 36 and 25, respectively. In contrast, sienna3 non-preserved Rb-module and purple mb-module had the lowest number of hubs including 30 and 13 genes, respectively.
Functional enrichment analysis
To assess the putative functions associated with the modules, all the identified Rb-modules and their hub genes were subjected to functional enrichment analysis, separately. Totally, 609 biological processes and 157 KEGG pathways were significantly enriched in six non-preserved modules including inflammatory response, response to lipopolysaccharide, regulation of MAP kinase activity, T cell chemotaxis, neutrophil migration cytokine-mediated signaling pathway, chemokine signaling pathway, IL-17 signaling pathway, TNF signaling pathway, NF-kappa B signaling and Toll-like receptor signaling pathway (Supplementary File S8, Fig. 5). The results suggesting that these genes might be related to endometritis development. Furthermore, the hub genes in the non-preserved Rb-modules found to be significantly enriched in 690 GO terms and 145 KEGG pathways. These hub genes were mostly enriched in the GO terms or KEGG pathways similar to results of the relevant modules (Supplementary File S9).

Functional enrichment analysis results (A) GO biological process (B) KEGG pathway of the non-preserved Rb-modules. Only the top 20 significant terms are displayed due to the large number of significant GO terms (biological process and KEGG pathway). The size of circles represents the enriched number of genes, and the color of circles represents the significance level of adjusted p-value. The blue color indicates the more significant terms within each module. As it can be seen, the turquoise module contains the highest number of significant terms based on GO biological process and KEGG pathway. Therefore, it can be considered one of the most important modules involved in the development of endometritis.
Enrichment analysis of the three semi-preserved Rb-modules showed that 10 biological processes and 11 KEGG pathways were significantly enriched. Sterol biosynthetic process, cholesterol metabolic process, regulation of alcohol biosynthetic process and regulation of steroid biosynthetic process were some of the GO terms. KEGG pathways were mainly associated with protein digestion and absorption, steroid biosynthesis, non-alcoholic fatty liver disease and ribosome biogenesis in eukaryotes (Supplementary File S10). Also, 29 GO terms and 12 KEGG pathways were significantly enriched in hub genes in semi-preserved modules which were according to the results of relevant modules (Supplementary File S11).
In three preserved modules, 118 biological processes and 14 KEGG pathways were significantly enriched. GO terms were related to translation, ncRNA processing, gene expression, ribosome biogenesis, mitotic cytokinesis, positive regulation of cell cycle process, regulation of cytokinesis, DNA recombination and DNA biosynthetic process (Supplementary File S12). KEGG pathways were mainly involved in cell cycle and ribosome. According to the results, preserved Rb-modules were enriched in the terms related to common activities of the cells and are less likely to be involved in endometritis development. Also, 170 GO terms and 12 KEGG pathways were significantly enriched in hub genes of the preserved Rb-modules and were similar to the results of the relevant modules (Supplementary File S13).
Assigning the mb-modules to the Rb-modules and target prediction
To explore the potential molecular mechanisms responsible for endometritis, just non-preserved Rb-modules with significant functional enrichment results were more considered. In this way, MEs of mb-modules were observed to be correlated with MEs of Rb-modules using spearman correlation. Thereafter, mb-modules that were found to be negatively correlated with Rb-modules (adjusted p < 0.20 and correlation > 0.8), were considered as the potential regulators of those Rb-modules. Totally, six significant negative correlations were found among the four mb-modules and three Rb-modules (Table 1). Finally, in order to make a stronger biological connection between mb-modules and non-preserved Rb-modules, the target prediction was performed (Supplementary File S15). The summary of the target prediction results is presented in Table 2.
Detection of hub-hub genes and integrated regulatory network construction
PPIs results of hub-mRNAs of the non-preserved Rb-modules, which were found to be negatively correlated with mb-modules, were obtained from the STRING database. Hence, two hub mRNA sets related to brown and turquoise Rb-modules were analyzed and all the obtained PPI-networks were significant (brown: 459 nodes, 1015 edges, and 1.0e−16 adjusted p-value; turquoise: 1639 nodes, 14,909 edges, and 1.0e−16 adjusted p-value). Subsequently, PPIs data were merged with the predicted interactions from WGCNA and then subjected to the hub–-hub genes’ identification. Totally, 47 hub–hub genes were identified in brown (containing 23 hub–hub genes, including 17 mRNAs and six lncRNAs) and turquoise (containing 24 hub-hub genes, including three mRNAs and 21 lncRNAs) Rb-modules. In order to construct an integrated regulatory network, the interactions obtained from STRING-PPIs, WGCNA-calculated co-expressed mRNAs, WGCNA-calculated co-expressed lncRNAs, and WGCNA-calculated co-expressed miRNAs were combined with the target prediction results. The summary of the integrated regulatory networks is presented in Table 3 (Supplementary File S16). The constructed integrated regulatory networks are displayed in Figs. 6 and 7.

Integrated regulatory network of blue and brown mb-module and brown Rb-module. Small light brown circles represent hub genes, bold large red circles represent hub-hub lncRNAs and bold large brown circles represent hub-hub mRNAs in brown Rb-module. Also, small light blue and brown diamonds represent blue and brown mb-module and large bold diamonds represent hub miRNAs in these modules. Only the targets that have been predicted by all target prediction software are shown. Cytoscape software (version 3.7.2) (https://cytoscape.org/) was used to generate this Figure.

Integrated regulatory network of purple mb-module and turquoise Rb-module. Small light turquoise circles represent hub genes, bold large red circles represent hub-hub lncRNAs and bold large turquoise circles represent hub-hub mRNAs in turquoise Rb-module. Also, small light purple diamonds represent purple mb-module and large bold diamonds represent hub miRNAs in this module. Only the targets that have been predicted by all target prediction software are shown. Cytoscape software (version 3.7.2) (https://cytoscape.org/) was used to generate this Figure.
Discussion
Bovine endometritis is known as the most common uterine disease, which occurs following parturition and affects milk yield and reproductive performance and causes serious economic burden. Most of studies in this field, just consider differentially expressed genes without identifying relationships among genes and considering other biological factors including miRNAs and lncRNAs. Whereas assessing the interactions among all regulatory factors can provide a comprehensive insight into the mechanisms involved in endometritis pathogenesis. Herein, the integrated regulatory network approach was applied to help better understanding the functional networks/pathways contributing into the endometritis development. To do this, Rb-modules (including mRNAs, TFs, and lncRNAs) as well as mb-modules (including miRNAs) were constructed based on the WGCNA approach and then assigned to each other. Next, we focused on the non-preserved Rb-modules that were found to be negatively correlated with mb-modules. Negative correlations can be indicative of inverse interactions between Rb-modules and mb-modules. It is noteworthy that the loss of connectivity among genes in the non-preserved modules can be attributed to the abnormal expression of some genes under the endometritis condition, which likely are key factors. In order to explore these potential genes that may be related to the occurrence and development of endometritis, different methods were applied, including assigning mb-modules to Rb-module, functional enrichment analysis, PPI network construction, and hub and hub-hub genes identification along with considering the relevant literature review. In addition, the target prediction analysis was used to assess the assigned genes (from Rb-modules) to miRNAs (from mb-modules) at the sequence level. Our findings provided evidence that the dysregulated genes may be associated with the development of endometritis. In this study, two most important non-preserved Rb-modules were found, namely turquoise and brown modules, which not only were negatively connected to the mb-modules, but also their hub genes were significantly connected in terms of PPI network. Therefore, genes with a high degree of connectivity within these modules, are also linked at the protein–protein interaction level. Moreover, the results of the functional enrichment analysis of these modules showed that some terms were associated with most of the features of endometritis and some of them were only related to the promotion of the establishment of the endometritis.
The functional enrichment analysis of the turquoise Rb-module revealed some enriched terms related to both inflammation and infection, including cytokine-mediated signaling pathway, Toll-like receptor signaling pathway, inflammatory response, positive regulation of NF-kappaB transcription factor activity, and MAPK cascade. Furthermore, the results of KEGG pathway analysis indicated that TNF signaling pathway, B cell receptor signaling pathway, NF-kappa B signaling pathway, Toll-like receptor signaling pathway, and MAPK signaling pathway are significantly enriched in this module. Besides, the bacterial infection of the endometrium was found to induce an inflammatory response by the secretion of chemokines and cytokines57. Of note, Toll-like receptors (TLRs) are the crucial receptors on endometrial cells and macrophages, which are used for recognizing pathogen-associated molecular patterns (PAMPs) present on bacterial cell walls like lipopolysaccharide (LPS)58. After recognizing PAMPs by TLRs, Myeloid differentiation factor 88 (MyD88) interacted with IL-1 receptor-associated kinase-4 (IRAK-4) and formed the MyD88-IRAK-4 complex. Subsequently, IRAK-1 and IRAK-2 were recruited, which can lead to the phosphorylation of IRAKs. Correspondingly, the phosphorylation of these kinases was also found to have the ability of leading to the recruitment of other proteins. Finally, the nuclear factor-kB (NF-kB) signaling and mitogen-activated protein kinase (MAPK) signaling pathways were activated. The activation of these downstream signaling pathways led to a cascade of inflammatory response as well as the secretion of chemokines and cytokines58. Interestingly, in our study, IRAK1 was detected as the hub gene in turquoise module, which is known as one of the most important genes in TLRs signaling pathway. In the current study, intersection of the results of the three target prediction software revealed that IRAK1 is a potential target of hub miRNAs, including bta-miR-449a and bta-miR-449b. Accordingly, these two miRNAs could also target hub-hub lncRNA ENSBTAG00000049936 in turquoise module. It was confirmed that miR-449a is up-regulated in children’s with inflammatory bowel disease (IBD) and plays some roles in inflammatory response. The inhibition of miR-449a has been previously shown to inhibit inflammatory response59. Jianga et al. in their study have reported that miR-449a plays a crucial role in patients with atherosclerosis via regulating TNF-a expression60. Moreover, the results of the study by CAI et al. demonstrated that IL-6 is targeted by miR-449b-3p, which consequently affects the JAK2/STAT3 signaling pathway61. The constructed integrated regulatory sub-network related to IRAK1 in turquoise Rb-module is shown in Fig. 8.

The constructed integrated regulatory sub-network related to IRAK1 gene in turquoise Rb-module. The green and yellow circles indicate hub genes and hub TFs related to IRAK1, respectively. The purple diamonds represent the hub miRNAs that be predicted to regulate IRAK1 and hub-hub lncRNA ENSBTAG00000049936. Cytoscape software (version 3.7.2) (https://cytoscape.org/) was used to generate this Figure.
Among the genes in turquoise module, CASP3 (which is a hub gene) was previously found to be associated with inflammatory response and predicted to be targeted by hub bta-miR-484 (based on both the miRanda and RNAhybrid results). Of note, several hub-hub lncRNAs in turquoise module, including ENSBTAG00000050527, ENSBTAG00000052755, and ENSBTAG00000054306, are also targeted by bta-miR-484. In mammals, caspase gene family consists of 15 members that can be grouping into inflammatory caspases and apoptotic caspases62. In this regard, apoptotic caspases have an association with cellular dismantling, while inflammatory caspases only mediate the activation of inflammatory cytokines62. Caspase-3 (CASP3) is known as a key mediator of apoptosis62. In addition, it has been demonstrated that CASP3 plays a crucial role in the differentiation of various cells such as monocytes, osteoblasts, and platelet63. The caspase-3 family, including caspases-3 and -7, plays an important role in activating pro-inflammatory cytokines64. On the other hand, myocardial cells can prevent ischemia–reperfusion injury via the role of mir-484 in suppressing both caspase-3 and caspase-9 expressions during cardiomyocyte apoptosis. Besides, mir-484 reduces the expressions of IL-6, TNF-α, and IL-1β65. The constructed integrated regulatory sub-network regarding CASP3 in turquoise Rb-module is shown in Fig. 9.

The constructed integrated regulatory sub-network related to CASP3 in turquoise Rb-module. The green and yellow circles indicate hub genes and hub TFs related to casp3, respectively. The purple diamond represents hub miRNA that be predicted to regulate CASP3 and hub-hub lncRNAs including ENSBTAG00000050527, ENSBTAG00000052755 and ENSBTAG00000054306. Cytoscape software (version 3.7.2) (https://cytoscape.org/) was used to generate this Figure.
Some of the highly connected genes in turquoise module, including AKT1 (hub-hub), STAT3 (hub), IL10 (hub), TLR4 (hub), CD68 (hub), and FN1 (hub-hub), have been reported to be related to immune response and inflammation. The potential regulatory networks of these genes are available in Supplementary File S17. AKT1 is a primary effector molecule of PI3K-AKT signaling pathway, acting as a suppressor for the NF-κB activation66. It was previously found that miR-29a could regulate the NF-κB pathway via targeting AKT1, thereby promoting inflammatory responses67. In contrast, Kane et al. in their study have reported that AKT1 plays roles in the IκB proteins degradation and subsequently in the NF-κB activation68. In the patients with endometrial cancer, some hub genes, including PBK, BIRC5, AURKA, GTSE1, KNSTRN, and PSMB10, were detected to be associated with AKT1. Hence, the higher expression of AKT1 was observed to be significantly associated with developing the endometrial cancer69. STAT3, as one of the hub TFs in turquoise Rb-module, is known as a regulator of expression of many genes, inducing some important cytokines. Furthermore, it is considered as a mediator of both innate and adaptive immunities. Notably, lack of STAT3 in immune cells induces severe inflammation in response to pathogens70. In the bovine endometrium, IL-6 acts as an activator of STAT3, the activation of which more increases in the secretion of both IL-6 and IL-871. It has been shown that phosphorylation and activation of STAT3 in the endometrium are essential for a successful implantation72. Pro-inflammatory cytokines could initiate and increase the inflammatory response, while anti-inflammatory cytokines only modulate the pro-inflammatory cytokines73. IL-10 is known as one of the anti-inflammatory cytokines, deficiencies in which can lead to tissue damage74. As mentioned earlier, TLRs, which are involved in inflammation and immune system, play some crucial roles in initiating inflammatory response. TLRs could also activate immune cells via two separate signaling pathways, namely MYD88 and TRIF. Furthermore, TLR4 uses both signaling pathways for the activation of immune cells. Several investigations have reported TLR4 as the main molecule in the development of the pro-inflammatory-based diseases75. Several studies have previously shown the upregulation of CD68 expression in macrophages in response to inflammatory stimuli. Accordingly, it is also commonly used as a histochemical/cytochemical marker of inflammation76. Fibronectin (FN1), which is an extracellular matrix molecule, has been identified as an activator of TLRs77. The interaction between FN and integrin β1 in macrophages results in reinforcing Toll-like receptor 2/4 (TLR2/TLR4) signaling pathways as well as phagocytosis by macrophages78. Generally, due to the functional roles of the above-mentioned genes in inflammation and immune system, these hub genes as well as the suggested integrated regulatory network may be involved in both the pathogenesis and progression of endometritis. As well, their possible cellular roles and the biological mechanisms related to this infection can be further assessed.
In the current study, cilium organization and cilium movement GO terms were significantly enriched in brown Rb-module. In this regard, Samatha et al. in their study investigated endometrial biopsies samples obtained from infertile buffaloes in terms of the histopathological and immunohistochemical properties and as a result, they reported that surface epithelial cells contain few ciliated and non-ciliated cells. Moreover, they observed the loss of both cilia and microvilli of the surface epithelium in acute endometritis cases79. Cilia are microtubule-based, conserved organelles growing from basal bodies80. It has been hypothesized that cilia play the role of antennae by the signal detection80. In this context, the role of primary cilia in NF-κB signaling pathway through the regulation of IKK activity has been demonstrated, as well81,82. Additionally, Beak et al. in their study reported a link between the expression of pro-inflammatory cytokines and ciliary function83. The disruption of ciliary intraflagellar transport (IFT) could alter the cell response to IL-1β, which supports the existence of a putative link between cilia and inflammation84. IFT includes motor proteins, kinesins, and dyneins and plays a crucial role in transporting ciliary proteins within the cilium85. Some of the highly connected genes related to cilium organization in brown Rb-module are CCDC39, CCDC40, ZMYND10, DNAAF11 and FOXJ1.
CCDC40 is one of the identified hub–hub genes in the brown Rb-module, which is required for axonemal assembly and the proper formation and/or maintenance of cilia. In addition, it is essential for both cytoplasmic preassembly and transportation of the axonemal components, including CCDC39, GAS11, and DNALI186. The key roles of CCDC39 and CCDC40, which are involved in axonemal disorganization and inner dynein arm loss, in patients with primary ciliary dyskinesia (PCD) have been demonstrated previously. These genes are known as major candidates for genetic testing in families affected by this ciliary phenotype87. According to the target prediction results of miRanda, RNAhybrid, and RNA22, CCDC39 was found as a potential target of bta-miR-149 hub miRNA. Moreover, hub-hub lncRNAs, including ENSBTAG00000051242 and ENSBTAG00000054677, were observed to be targeted by bta-miR-149. It is worth noting that the regulatory role of miR-149 in inflammation has been reported in several previous studies88,89. The constructed integrated regulatory sub-network regarding both CCDC40 and CCDC39 in brown Rb-module is displayed in Fig. 10.

The constructed integrated regulatory sub-network related to CCDC40 and CCDC39 in brown Rb-module. The pink circles indicate hub and hub-hub lncRNAs. The brown diamond represents hub miRNA that be predicted to regulate CCDC39 and hub-hub target lncRNAs including ENSBTAG00000051242 and ENSBTAG00000054677. Cytoscape software (version 3.7.2) (https://cytoscape.org/) was used to generate this Figure.
In the present study, ZMYND10 was identified as a hub-hub gene potentially predicted to be targeted by hub miRNA bta-miR-30b. In this regard, ZMYND10 plays an important role in the dynein-arm synthesis, assembly or transport. In flies, ZMYND10 is primarily considered as a cytoplasmic component of cells containing motile cilia90. Moreover, both ZMYND10 and DNAAF11 are assembled into a protein complex, which plays a role in the transcriptional regulation of some dynein components91. Moreover, the regulatory role of miR-30b has been reported in inflammation92. The constructed integrated regulatory sub-network regarding both ZMYND10 and DNAAF11 in brown Rb-module is provided in Fig. 11.

The constructed integrated regulatory sub-network related to ZMYND10 and DNAAF11 in brown Rb-module. The pink circles indicate hub-hub lncRNAs related to the mentioned genes. The brown diamond represents hub miRNA that be predicted to regulate ZMYND10. The yellow circle indicates hub TF. Cytoscape software (version 3.7.2) (https://cytoscape.org/) was used to generate this Figure.
One of the hub TFs in brown Rb-module is FOXJ1, which is involved in the transcriptional regulation of those genes that encode essential components for the synthesis and function of motile cilia93. According to the results of the target prediction analysis, FOXJ1 is targeted by bta-miR-423 in blue mb-module and bta-miR-149 (identified as a hub miRNA) in brown mb-module. Accordingly, these miRNAs could also target hub-hub lncRNAs, including ENSBTAG00000049287, ENSBTAG00000051242, and ENSBTAG00000054677 in the brown Rb-module. Interestingly, the roles of both miRNAs (miR-149 and miR-423) in inflammatory response have been reported, reinforcing the potential regulatory roles of these genes in endometritis89,94. The constructed integrated regulatory sub-network regarding FOXJ1 in brown Rb-module is provided in Fig. 12.

The constructed integrated regulatory sub-network related to FOXJ1 in brown Rb-module. The pink circles indicate hub-hub lncRNAs related to the mentioned gene. The brown and blue diamond represents miRNAs that be predicted to regulate FOXJ1. Cytoscape software (version 3.7.2) (https://cytoscape.org/) was used to generate this Figure.
In summary, our results suggest, in accordance with literature, the potential role of the brown Rb-module in ciliogenesis and inflammation, which may provide a foundation for obtaining further understanding on the underlying molecular mechanisms of endometritis development in the bovine. In addition, our findings showed that the integration of mRNAs, lncRNAs, and miRNAs based on the WGCNA networks along with different data such as PPI and miRNA target prediction results can be considered as a robust approach to provide greater insights on the disease‐related biological process.
Conclusion
In the present study, integrated regulatory networks related to bovine endometritis were constructed by assigning the mb-modules to the non-preserved Rb-modules. These modules were constructed based on the gene co-expression approach (WGCNA) and were also assessed by PPI data, functional enrichment analysis and target prediction analysis. Moreover, only highly connected genes (hubs and hub-hubs) were considered, which are the key components of the networks and more likely play an important role in endometritis. Two important non-preserved Rb-modules (brown and turquoise) were found that were assigned to three mb-modules (brown, blue and purple). In addition, several sub-networks in brown and turquoise modules were identified, including mRNAs (such as IRAK1, AKT1, STAT3, CCDC39 and ZMYND10), lncRNAs (such as ENSBTAG00000049936, ENSBTAG00000050527, ENSBTAG00000054306, ENSBTAG00000051242 and ENSBTAG00000049287) as well as miRNAs (such as bta-miR-449a, bta-miR-484, bta-miR-30b, bta-miR-149 and bta-miR-423), as potential pathways/genes that may contribute to the progression of bovine endometritis and could be regarded as biomarkers and therapeutic targets of this infection. The proposed integrated regulatory networks can provide a basis for additional inspection of the regulatory mechanisms of endometritis; however, further experimental works are needed to validate our findings and elucidate the importance of these networks in bovine endometritis.
References
Dijkhuizen, A., Stelwagen, J. & Renkema, J. Economic aspects of reproductive failure in dairy cattle. I. Financial loss at farm level. Prev. Vet. Med. 3, 251–263 (1985).
Plaizier, J., King, G., Dekkers, J. & Lissemore, K. Estimation of economic values of indices for reproductive performance in dairy herds using computer simulation. J. Dairy Sci. 80, 2775–2783 (1997).
Radostits, O. M., Leslie, K. & Fetrow, J. Herd Health: Food Animal Production Medicine (WB Saunders Company, 1994).
Sheldon, I. M., Cronin, J., Goetze, L., Donofrio, G. & Schuberth, H.-J. Defining postpartum uterine disease and the mechanisms of infection and immunity in the female reproductive tract in cattle. Biol. Reprod. 81, 1025–1032 (2009).
Kasimanickam, R. et al. Endometrial cytology and ultrasonography for the detection of subclinical endometritis in postpartum dairy cows. Theriogenology 62, 9–23 (2004).
Foley, C. et al. Integrated analysis of the local and systemic changes preceding the development of post-partum cytological endometritis. BMC Genomics 16, 811 (2015).
Gabler, C., Fischer, C., Drillich, M., Einspanier, R. & Heuwieser, W. Time-dependent mRNA expression of selected pro-inflammatory factors in the endometrium of primiparous cows postpartum. Reprod. Biol. Endocrinol. 8, 152 (2010).
Janowski, T. et al. Endometrial mRNA expression of prostaglandin synthase enzymes PTGS 2, PTGFS and mPTGES 1 in repeat-breeding cows with cytologically determined endometritis. Acta Vet. Hung. 65, 96–104 (2017).
González-Ramos, R. et al. Involvement of the nuclear factor-κB pathway in the pathogenesis of endometriosis. Fertil. Steril. 94, 1985–1994 (2010).
Jiang, K. et al. Matrine alleviates Staphylococcus aureus lipoteichoic acid-induced endometritis via suppression of TLR2-mediated NF-κB activation. Int. Immunopharmacol. 70, 201–207 (2019).
Raliou, M. et al. Subclinical endometritis in dairy cattle is associated with distinct mRNA expression patterns in blood and endometrium. PLoS ONE 14, e0220244 (2019).
Salilew-Wondim, D. et al. Clinical and subclinical endometritis induced alterations in bovine endometrial transcriptome and miRNome profile. BMC Genomics 17, 1–21 (2016).
Ma, T. & Zhang, A. Reconstructing context-specific gene regulatory network and identifying modules and network rewiring through data integration. Methods 124, 36–45 (2017).
Mamdani, M. et al. Integrating mRNA and miRNA weighted gene co-expression networks with eQTLs in the nucleus accumbens of subjects with alcohol dependence. PLoS ONE 10, e0137671 (2015).
Zhang, B. & Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. https://doi.org/10.2202/1544-6115.1128 (2005).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Bakhtiarizadeh, M. R., Mirzaei, S., Norouzi, M., Sheybani, N. & Vafaei Sadi, M. S. Identification of gene modules and hub genes involved in mastitis development using a systems biology approach. Front. Genet. 11, 722 (2020).
Botía, J. A. et al. An additional k-means clustering step improves the biological features of WGCNA gene co-expression networks. BMC Syst. Biol. 11, 47 (2017).
Bakhtiarizadeh, M. R., Hosseinpour, B., Shahhoseini, M., Korte, A. & Gifani, P. Weighted gene co-expression network analysis of endometriosis and identification of functional modules associated with its main hallmarks. Front. Genet. 9, 453 (2018).
Zhao, G. et al. The potential therapeutic role of miR-223 in bovine endometritis by targeting the NLRP3 inflammasome. Front. Immunol. 9, 1916 (2018).
Hailemariam, D. et al. MicroRNA-regulated molecular mechanism underlying bovine subclinical endometritis. Reprod. Fertil. Dev. 26, 898–913 (2014).
Jiang, K. et al. miR-148a suppresses inflammation in lipopolysaccharide-induced endometritis. J. Cell Mol. Med. 24, 405–417 (2020).
Walther, K. & Schulte, L. N. The role of lncRNAs in innate immunity and inflammation. RNA Biol. 18, 1–17 (2020).
Rapicavoli, N. A. et al. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife 2, e00762 (2013).
Chen, W., Liu, D., Li, Q.-Z. & Zhu, H. The function of ncRNAs in rheumatic diseases. Epigenomics 11, 821–833 (2019).
La Ferlita, A. et al. Non-coding RNAs in endometrial physiopathology. Int. J. Mol. Sci. 19, 2120 (2018).
Mercer, T. R., Dinger, M. E. & Mattick, J. S. Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159 (2009).
Castro-Oropeza, R., Melendez-Zajgla, J., Maldonado, V. & Vazquez-Santillan, K. The emerging role of lncRNAs in the regulation of cancer stem cells. Cell. Oncol. 41, 585–603 (2018).
Du, M. et al. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat. Commun. 8, 1–18 (2017).
Mathy, N. W. & Chen, X.-M. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J. Biol. Chem. 292, 12375–12382 (2017).
Cai, Y., Yu, X., Hu, S. & Yu, J. A brief review on the mechanisms of miRNA regulation. Genomics Proteomics Bioinform. 7, 147–154 (2009).
Xiao, C. & Rajewsky, K. MicroRNA control in the immune system: Basic principles. Cell 136, 26–36 (2009).
Sonkoly, E. et al. MicroRNAs: Novel regulators involved in the pathogenesis of psoriasis? PLoS ONE 2, e610 (2007).
Lee, H.-M., Kim, T. S. & Jo, E.-K. MiR-146 and miR-125 in the regulation of innate immunity and inflammation. BMB Rep. 49, 311 (2016).
Andrews, S. Babraham Bioinformatics-FastQC a Quality Control Tool for High Throughput Sequence Data (Babraham Institite, 2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Langmead, B. & Salzberg, S. hisat2. Nat. Methods 12, 357 (2015).
Anders, S., Pyl, P. T. & Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Hu, H. et al. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 47, D33–D38 (2019).
Gebert, D., Hewel, C. & Rosenkranz, D. Unitas: The universal tool for annotation of small RNAs. BMC Genomics 18, 644 (2017).
Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 32, 11–14 (2010).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Malki, K. et al. Integrative mouse and human mRNA studies using WGCNA nominates novel candidate genes involved in the pathogenesis of major depressive disorder. Pharmacogenomics 14, 1979–1990 (2013).
Smyth, G. K., Ritchie, M., Thorne, N. & Wettenhall, J. LIMMA: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Statistics for Biology and Health (eds Gentleman, R. et al.) (Springer, 2005).
Łabaj, P. P. et al. Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics 27, i383–i391 (2011).
Amrine, K. C., Blanco-Ulate, B. & Cantu, D. Discovery of core biotic stress responsive genes in Arabidopsis by weighted gene co-expression network analysis. PLoS ONE 10, e0118731 (2015).
Betel, D., Koppal, A., Agius, P., Sander, C. & Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 11, R90 (2010).
Langfelder, P., Luo, R., Oldham, M. C. & Horvath, S. Is my network module preserved and reproducible? PLoS Comput. Biol. 7, e1001057 (2011).
Xia, W.-X. et al. Identification of four hub genes associated with adrenocortical carcinoma progression by WGCNA. PeerJ 7, e6555 (2019).
Krüger, J. & Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–W454 (2006).
Enright, A. et al. MicroRNA targets in drosophila. Genome Biol. 4, 1–27 (2003).
Rigoutsos, I., Miranda, K. & Huynh, T. rna22: A Unified Computational Framework for Discovering miRNA Precursors, Localizing Mature miRNAs, Identifying 3’UTR Target-Islands, and Determining the Targets of Mature-miRNAs (Ibm Corporation, 2007).
Szklarczyk, D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Chin, C.-H. et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8, S11 (2014).
Pihur, V., Datta, S. & Datta, S. RankAggreg, an R package for weighted rank aggregation. BMC Bioinform. 10, 62 (2009).
Filteau, M., Pavey, S. A., St-Cyr, J. & Bernatchez, L. Gene coexpression networks reveal key drivers of phenotypic divergence in lake whitefish. Mol. Biol. Evol. 30, 1384–1396 (2013).
Johnson, H., Torres, C. G., Carvallo, F., Duchens, M. & Peralta, O. A. Endometrial expression of selected transcripts in postpartum of primiparous Holstein cows with clinical and subclinical endometritis. Anim. Reprod. Sci. 156, 34–39 (2015).
Zheng, C., Chen, J., Chu, F., Zhu, J. & Jin, T. Inflammatory role of TLR-MyD88 signaling in multiple sclerosis. Front. Mol. Neurosci. 12, 314 (2020).
Zhao, D. et al. Role of microRNA-449a in the progress of inflammatory bowel disease in children. Biotechnol. Biotechnol. Equip. 34, 144–153 (2020).
Jiang, L. et al. miR-449a induces EndMT, promotes the development of atherosclerosis by targeting the interaction between AdipoR2 and E-cadherin in Lipid Rafts. Biomed. Pharmacother. 109, 2293–2304 (2019).
Cai, K., Li, H. X., Li, P. P., Guo, Z. J. & Yang, Y. MicroRNA-449b-3p inhibits epithelial-mesenchymal transition by targeting IL-6 and through the JAK2/STAT3 signaling pathway in non-small cell lung cancer. Exp. Ther. Med. 19, 2527–2534 (2020).
Chowdhury, I., Tharakan, B. & Bhat, G. K. Caspases—An update. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 151, 10–27 (2008).
Dassé, E. et al. Tissue inhibitor of metalloproteinase-1 promotes hematopoietic differentiation via caspase-3 upstream the MEKK1/MEK6/p38 α pathway. Leukemia 21, 595–603 (2007).
Canisso, I. F., Segabinazzi, L. G. & Fedorka, C. E. Persistent breeding-induced endometritis in mares-a multifaceted challenge: From clinical aspects to immunopathogenesis and pathobiology. Int. J. Mol. Sci. 21, 1432 (2020).
Liu, H., Li, S., Jiang, W. & Li, Y. MiR-484 protects rat myocardial cells from ischemia-reperfusion injury by inhibiting caspase-3 and caspase-9 during apoptosis. Korean Circ. J. 50, 250–263 (2020).
Zenke, K., Muroi, M. & Tanamoto, K.-I. AKT1 distinctively suppresses MyD88-depenedent and TRIF-dependent toll-like receptor signaling in a kinase activity-independent manner. Cell. Signal. 43, 32–39 (2018).
Tang, B. et al. MicroRNA-29a regulates lipopolysaccharide (LPS)-induced inflammatory responses in murine macrophages through the Akt1/NF-κB pathway. Exp. Cell Res. 360, 74–80 (2017).
Kane, L. P., Shapiro, V. S., Stokoe, D. & Weiss, A. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 9, 601 (1999).
Huo, X. et al. Clinical and expression significance of AKT1 by co-expression network analysis in endometrial cancer. Front. Oncol. 9, 1147 (2019).
Fu, X. Y. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 16, 214–219 (2006).
Cronin, J., Kanamarlapudi, V., Thornton, C. & Sheldon, I. Signal transducer and activator of transcription-3 licenses Toll-like receptor 4-dependent interleukin (IL)-6 and IL-8 production via IL-6 receptor-positive feedback in endometrial cells. Mucosal Immunol. 9, 1125–1136 (2016).
Catalano, R. D. et al. Inhibition of Stat3 activation in the endometrium prevents implantation: A nonsteroidal approach to contraception. Proc. Natl. Acad. Sci. 102, 8585–8590 (2005).
Parham, P. The Immune System 414 (Garland, 2005).
Cyktor, J. C. & Turner, J. Interleukin-10 and immunity against prokaryotic and eukaryotic intracellular pathogens. Infect. Immun. 79, 2964–2973 (2011).
Manjili, F. A., Yousefi-Ahmadipour, A. & Arababadi, M. K. The roles played by TLR4 in the pathogenesis of multiple sclerosis. A systematic review article. Immunol. Lett. 220, 63–70 (2020).
Chistiakov, D. A., Killingsworth, M. C., Myasoedova, V. A., Orekhov, A. N. & Bobryshev, Y. V. CD68/macrosialin: Not just a histochemical marker. Lab. Investig. 97, 4–13 (2017).
Kelsh, R., You, R., Horzempa, C., Zheng, M. & McKeown-Longo, P. J. Regulation of the innate immune response by fibronectin: Synergism between the III-1 and EDA domains. PLoS ONE 9, e102974 (2014).
Fei, D. et al. Fibronectin (FN) cooperated with TLR2/TLR4 receptor to promote innate immune responses of macrophages via binding to integrin β1. Virulence 9, 1588–1600 (2018).
Samatha, V. Histopathological, Immunohistochemical and Ultrastructural Studies on Endometrial Biopsies in Infertile Buffaloes (Sri Venkateswara Veterinary University, 2018).
Eggenschwiler, J. T. & Anderson, K. V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 23, 345–373 (2007).
Wann, A., Chapple, J. & Knight, M. The primary cilium influences interleukin-1β-induced NFκB signalling by regulating IKK activity. Cell. Signal. 26, 1735–1742 (2014).
Wann, A. & Knight, M. Primary cilia elongation in response to interleukin-1 mediates the inflammatory response. Cell. Mol. Life Sci. 69, 2967–2977 (2012).
Baek, H. et al. Primary cilia modulate TLR4-mediated inflammatory responses in hippocampal neurons. J. Neuroinflamm. 14, 1–10 (2017).
McFie, M. et al. Ciliary proteins specify the cell inflammatory response by tuning NFκB signaling, independently of primary cilia. J. Cell Sci. https://doi.org/10.1242/jcs.239871 (2020).
Gerdes, J. M., Davis, E. E. & Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32–45 (2009).
Becker-Heck, A. et al. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 43, 79 (2011).
Antony, D. et al. Mutations in CCDC 39 and CCDC 40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum. Mutat. 34, 462–472 (2013).
Qin, C., Lv, Y., Zhao, H., Yang, B. & Zhang, P. MicroRNA-149 suppresses inflammation in nucleus Pulposus cells of intervertebral discs by regulating MyD88. Med. Sci. Monit. 25, 4892 (2019).
Chen, Q., Wu, S., Wu, Y., Chen, L. & Pang, Q. miR-149 suppresses the inflammatory response of chondrocytes in osteoarthritis by down-regulating the activation of TAK1/NF-κB. Biomed. Pharmacother. 101, 763–768 (2018).
Moore, D. J. et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 93, 346–356 (2013).
Zariwala, M. A. et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6. Am. J. Hum. Genet. 93, 336–345 (2013).
Zhou, T. & Chen, Y.-L. The functional mechanisms of miR-30b-5p in acute lung injury in children. Med. Sci. Monit. 25, 40 (2019).
Yu, X., Ng, C. P., Habacher, H. & Roy, S. Foxj1 transcription factors are master regulators of the motile ciliogenic program. Nat. Genet. 40, 1445–1453 (2008).
Wang, W., Gao, J. & Wang, F. MiR-663a/MiR-423-5p are involved in the pathogenesis of lupus nephritis via modulating the activation of NF-κB by targeting TNIP2. Am. J. Transl. Res. 9, 3796 (2017).
Author information
Authors and Affiliations
Contributions
M.R.B. conceived the ideas. M.R.B. and N.S. designed study and analyzed the data. M.R.B., N.S. and A.S. interpreted the data and wrote the main manuscript text. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sheybani, N., Bakhtiarizadeh, M.R. & Salehi, A. An integrated analysis of mRNAs, lncRNAs, and miRNAs based on weighted gene co-expression network analysis involved in bovine endometritis. Sci Rep 11, 18050 (2021). https://doi.org/10.1038/s41598-021-97319-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97319-y
- Springer Nature Limited