
Abstract
Pulmonary arterial hypertension (PAH) is a chronic cardiopulmonary syndrome with high pulmonary vascular load and eventually causing RV heart failure even death. However, the mechanism of pulmonary hypertension remains unclear. The purpose of this research is to detect the underlying key genes and potential mechanism of PAH using several bioinformatic methods. The microarrays GSE22356, GSE131793 and GSE168905 were acquired from the GEO. Subsequently, a host of bioinformatics techniques such as DAVID, STRING, R language and Cytoscape were utilized to investigate DEGs between PAH and healthy controls and conduct GO annotation, KEGG enrichment analysis and PPI network construction etc. Additionally, we predicted the transcription factors regulating DEGs through iRegulon plugin of Cytoscape and CIBERSORT was used to conduct immune infiltration analysis. One thousand two hundred and seventy-seven DEGs (403 up-regulated and 874 down-regulated) were identified from peripheral blood samples of 32 PAH patients and 29 controls, among which SLC4A1, AHSP, ALAS2, CA1, HBD, SNCA, HBM, SELENBP1, SERPINE1 and ITGA2B were detected as hub genes. The functional enrichment changes of DEGs were mainly enriched in protein binding, extracellular exosome, extracellular space, extracellular region and integral component of plasma membrane. The hub genes are chiefly enriched at extracellular exosome, hemoglobin complex, blood microparticle, oxygen transporter activity. Among TF-DEGs network, 42 target DEGs and 6 TFs were predicted with an NES > 4 (TEAD4, TGIF2LY, GATA5, GATA1, GATA2, FOS). Immune infiltration analysis showed that monocytes occupied the largest proportion of immune cells. The trend analysis results of infiltration immune cells illustrated that PAH patients had higher infiltration of NK cell activation, monocyte, T cell CD4 memory activation, and mast cell than healthy controls and lower infiltration of T cell CD4 naive. We detected SLC4A1, AHSP, ALAS2, CA1, HBD, SNCA, HBM, SELENBP1, SERPINE1 and ITGA2B as the most significant markers of PAH. The PAH patients had higher infiltration of NK cell activation, monocyte, T cell CD4 memory activation, and mast cell than healthy controls and lower infiltration of T cell CD4 naive. These identified genes and these immune cells probably have precise regulatory relationships in the development of PAH.
Similar content being viewed by others

Introduction
Pulmonary arterial hypertension (PAH) is a chronic cardiopulmonary syndrome characterized by salient pulmonary vascular remodeling and accelerating increase in pulmonary vascular load, bringing about right ventricular (RV) enlargement and pulmonary blood vessels remodeling1 and ultimately leading to heart failure or even death if untreated2. Plentiful widespread cardiopulmonary diseases complicated with PAH, which badly increases morbidity and mortality1. PAH is a rare disease with reported prevalence of 15–50 cases in a million people and incidence of 5–10 cases in a million people every year3. Debate on the best strategy for PHA management continues despite great progress in treating PAH in recent decades. Extensive researched have shown current therapeutic approaches in PAH included prostacyclin receptor agonists, phosphodiesterase-5 (PDE-5) inhibitors, endothelin receptor antagonists (ERAs) and a soluble guanylate cyclase activator targeting several crucial signaling pathways that predominantly regulate pulmonary vasculature4. However, PAH may also can be caused by many unknown causes, such as pulmonary vascular remodeling and inflammation, which cannot be well solved by current drug treatment and PAH is still a complicated incurable cardiopulmonary disease5. Thus, it is necessary for us to utilize bioinformatics technology to explore the pathogenesis or potential treatments of PAH.
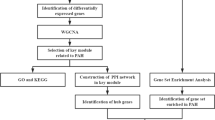
At present, bioinformatics methods have been proverbially performed to analyze gene sequencing data of various diseases to ascertain differentially expressed genes (DEGs) and implement various analyses. And more and more robust databases and powerful online tools have been established to help our repositioning of known intricate mechanism of diseases6. Increasing researches applied microarray technology for the purpose of searching DEGs and their molecular functions (MFs), biological processes (BPs), or cellular components (CCs) as well as relatedregulatory pathways in specific disease state7. For example, Habib Rahman and his colleagues used bioinformatics and machine learning methods to determine new factors that improve the identification and characterization of glioblastoma tumors and their progression8. Based on a neighborhood-based benchmarking and multilayer network topology techniques, they also identified novel putative biomarkers which manifest how type 2 diabetes (T2D) interact9. In our study, as the flow chart shown in Fig. 1, the expression datasets GSE22356, GSE131793 and GSE168905 were analyzed to identify key genes employing comprehensive bioinformatics analysis technologies, consisting of GO and KEGG analysis, protein–protein interaction (PPI) network construction, immune infiltration analysis and hub gene identification. Relying on the GEO database and R language, the distinguishing infiltration of 22 immune cells in peripheral blood of PAH patients were compared with healthy cases. The study probably revealed the pathogenic mechanism and potential therapeutic target of PAH.

Flow chart of the analysis program in this study.
Results
Identification of DEGs in PAH
There are 580 DEGs screened from GSE22356 between PAH group with healthy group which consists of 162 up-regulated and 418 down-regulated genes (Fig. 2A,D). Simultaneously, heatmaps and volcano plots vividly showed that 156 up-regulated and 13 down-regulated DEGs were identified from GSE131793, 102 up-regulated and 456 down-regulated DEGs were identified from GSE168905 (Fig. 2B,C,E,F) (p value < 0.05 and |log2FC|≥ 0.5). Then, we merged up- regulated and down-regulated DEGs in the three datasets, which revealed a total of 403 up-regulated DEGs and 874 down-regulated DEGs in the microarrays.

The identification of EDGs. (A–C) The heat-maps of differential expression genes of GSE22356, GSE131793 and GSE168905 (P < 0.05, |log FC|> 1). Up-regulated genes were in red and down-regulated genes were in blue. (D–F) Volcano plot of genes identified in PAH of GSE22356, GSE131793 and GSE168905 (P < 0.05, |log FC|> 0.5). Red dots represent up-regulated and green dots represent down-regulated.
Functional enrichment analysis of DEGs
For demonstrating PAH‐related functional annotation and pathway enrichment, these DEGs with an absolute value of log2 fold change greater than 1 were selected for GO and KEGG analysis and detailed results are presented in Tables 1 and 2. In GO analysis, the richest BP participated hydrogen peroxide catabolic process, positive regulation of leukocyte migration and homeostasis of number of cells (Fig. 3A). In the CC category, platelet alpha granule membrane, haptoglobin-hemoglobin complex and platelet alpha granule were mostly interrelated with PAH (Fig. 3B). Organic acid binding, chemokine binding and haptoglobin binding were most obviously enriched for DEGs in the MF category (Fig. 3C). As we all know, KEGG mapping is a predictive method via reestablishing molecular network systems from molecular constructing blocks grounded on functional orthologs10,11,12. In KEGG analysis, the result indicated that genes were mainly associated with hematopoietic cell lineage, TGF-beta signaling pathway and ECM-receptor interaction (Fig. 3D).

GO and KEGG analysis of DEGs between PAH and healthy groups. (A–C) The top 10 GO annotation results of DEGs. (D) KEGG pathway analyses of DEGs (top 10 according to enrichment score).
PPI network construction and hub genes identification
Uploading 47 DEGs into the STRING database, a PPI network with the desired interaction score > 0.48 was constructed and the network containing 33 nodes and 57 edges was visualized via Cytoscape software (Fig. 4A). Then the most remarkable module was recognized by cytoHubba plug-in according to the maximal clique centrality topology analysis methods (Fig. 4B). The top 10 significant nodes with the highest connectivity among the network were appraised as hub genes: SLC4A1, AHSP, ALAS2, CA1, HBD, SNCA, HBM, SELENBP1, SERPINE1, ITGA2B (Table 3). Additionally, the most significantly enriched BPs showed that hub genes were related to oxygen transport, erythrocyte development, hydrogen peroxide catabolic process bicarbonate transport. The changes of CCs containing hemoglobin complex, blood microparticle, haptoglobin-hemoglobin complex and platelet alpha granule membrane. MFs were mainly enriched in hemoglobin binding, haptoglobin binding, oxygen transporter activity and organic acid binding (Fig. 4C).

(A) The PPI network of DEGs was established using string and Cytoscape, containing 33 nodes and 57 edges. The upregulated genes are red and downregulated are green. (B) The top 10 hub genes were detected from the PPI network. (C) GO terms of the top 10 key genes.
Analysis of TF-target regulating networks
To gain further insight into the regulatory TFs and selected DEGs, we used the iRegulon plugin to predict the TFs of DEGs. There were 48 nodes were predicted consisting of 42 DEGs and 6 TFs with an NES > 4 in this TF-DEGs network (TEAD4, TGIF2LY, GATA5, GATA1, GATA2, FOS) (Fig. 5). Specifically, the results show that 36 DEGs were forecasted as targets of TGIF2LY, 15 DEGs were forecasted as targets of TEAD4, GATA2 is predicted to have 17 DEG targets, GATA5 has 25 DEG targets, GATA1 has19 DEGs, and FOS has 23 DEGs13. Three predicted TFs belong to the family of GATA zinc finger TFs that bind to various motifs in the promoters of DEGs. It can be speculated that the TFs regulates the development and progression of PAH by activating or repressing the transcription of DEGs (details of transcription factors are shown in Table 4).

The TF-DEGs network including 42 DEGs and 6 TFs with an NES > 4.
Immune cells infiltration analysis
Using CIBERSORT, the relative proportions of 22 infiltration immunocytes in PAH and control samples were determined (Fig. 6A) and details were presented in Table 3. Furthermore, the heatmap showed that the 22 subpopulations of immunocytes were significantly different between PAH and controls and it was found that monocytes occupied the largest proportion of immune cells (Fig. 6B). Correlation analysis discovered that macrophages M had the most obvious positive relationship with both T cells follicular helper and T cells regulatory (Tregs) (r = 0.9, r = 0.9), dendritic cells resting and mast cells activated were also highly positively correlated with r = 0.83, while the strongest negative correlation was between dendritic cell activation and NK cell quiescence with r = − 0.6 (Fig. 6C). And the trend analysis results of infiltration immune cells illustrated that PAH patients had higher infiltration of NK cell activation, monocyte, T cell CD4 memory activation, and mast cell than healthy controls and lower infiltration of T cell CD4 naive (Fig. 6D; p < 0.05).

Summary of immune cell subpopulations between normal controls and PAH. (A) Composition of 22 kinds of immune cells subsets from GSE22356, GSE131793 and GSE168905 datasets. (B) Heatmap shows differences between the 22 subpopulations cell types and samples. (C) Correlation matrix of the 22 immunocyte proportions in healthy control and PAH samples. Red colors represent positive and blue colors represent negative correlations. (D) The difference of immune cells infiltration between 9 normal controls and 12 PAH (blue color represent normal controls group and red colors represent PAH group. P values < 0.05).
Discussion
PAH is a rare chronic refractory syndrome accompanied with pathological changes of distal pulmonary arteries such as vasculature remodeling, lumen stenosis, laminar intimal proliferation and fibrosis, plexiform lesions with excessive poorly formed capillaries14. The current clinical definition of PAH is that the average pulmonary arterial pressure (m PAP) ≥ 25 mm Hg measured during right heart catheterization (1 mmHg = 0.133 kPa)15. PAH is divided into many subgroups due to different etiologies worldwide. According to the latest Chinese PAH registry, coronary heart disease-related PAH was reported as the most prevalent subtype, accounting for about 43% of all cases3. In addition to anticoagulant, calcium channel blockers16, diuretics, exercise rehabilitation17, oxygen therapy18 and other basic measures and conventional therapies to reduce right ventricular preload and improve left ventricular filling, the treatment of PAH also includes drugs targeting the prostacyclin pathway, endothelin pathway, nitric oxide-cGMP pathway like epoprostenol, iloprost, treprostinil, bosentan, ambrisentan, cediline, nafil, sildenafil, riociguat etc.5,19. Previous study has reported that tyrosine kinase inhibition (TKI) has an inhibitory effect on cell proliferation against pulmonary vascular remodeling20. Despite substantial efforts were made in PAH field in recent years, the pathogenesis of PAH remains to be further elucidated. Nearly decades, rapid advances in gene sequencing technologies and bioinformatics related science have made it possible to utilize abundant sequencing data for further analyzing. In this research, we identified 403 up-regulated and 874 down-regulated DEGs from peripheral blood samples belong to 29 controls and 32 PAH patients whose expression profiles downloaded via GEO database. Besides, PPI network construction and functional enrichment analyses were performed to filter key genes and important pathways. GO annotation results manifested that DEGs were enriched in extracellular exosome, integral component of plasma membrane, platelet degranulation, blood microparticle, extracellular space, extracellular region, immune response, cell surface and protein binding. KEGG pathway results declared that DEGs were mainly mapped to hematopoietic cell lineage, cytokine-cytokine receptor interaction, transcription growth factor beta (TGF-β) signaling pathway, extracellular mechanism (ECM) receptor interaction and focal adhesion.
Establishing PPI network is friendly for researchers to investigate the underlying molecular mechanism of PAH for the reason that the DEGs would be grouped and ordered in the network judging by their interactions21. Of which, 10 hub genes (SLC4A1, AHSP, ALAS2, CA1, HBD, SNCA, HBM, SELENBP1, SERPINE1, ITGA2B) related to PAH were detected owing to Cytoscape. It is speculated that these significant DEGs theoretically lead to the occurrence and progression of PAH. SLC4A1, solute carrier family 4 member 1, a member of the anion exchanger (AE) family, expressed mainly in the erythrocyte plasma membrane, functions as a chloride/bicarbonate exchanger in the erythrocyte plasma membrane associated with the transport of carbon dioxide from tissues to the lungs22. Recently a genome-wide association study revealed that candidate genes including SLC4A1 known to regulate structure of erythrocyte, metabolic process and ion channels23. Kaneda et al. used a lung cancer (LC) model mice to examine the proteome of cancer cell-secreted extracellular vesicles (cEVs) and found that SLC4A1 was co-expressed in CHL1-expressing EVs24. AHSP, Alpha-hemoglobin-stabilizing protein, is reported as a molecular chaperone, which peculiarly attaches to free α-globin and participates in hemoglobin assembly. This protein links to monomeric alpha-globin while it is transferred to beta-globin to compose a heterodimer, which then conversely unites to another heterodimer to shape a sturdy tetrameric hemoglobin25. During normal erythroid development, this protein functions as a chaperone to guard against deleterious aggregation of alpha-hemoglobin, particularly, to defend free alpha-hemoglobin from precipitation. AHSP regulates pathological conditions with excess alpha-hemoglobin like beta-thalassemia26. Protein 5′-Aminolevulinate Synthase 2 of ALAS2 is an erythrocyte-specific mitochondrial localization enzyme and that step one of the heme biosynthetic pathway is catalyzed by this product27. Blemishes on ALAS2 will lead to the development of X-linked pyridoxine-responsive sideroblastic anemia28.
The evidence presented thus far supports the idea that carbonic anhydrases (CAs) are zinc metalloenzymes mainly involved in catalyzing the reversible hydration of carbon dioxide29. CAs are involved in diverse biological processes, consisting of respiration, calcification, acid–base balance, bone resorption and so on. Studies have confirmed that CA1 gene is tightly related to the CA2 and CA3 genes on chromosome 8 and encrypts the highest levels of cytosolic proteins found in red blood cells30. Previous studies said the HBD gene was normally expressed in adults and encodes a delta chain that two delta chains add two alpha chains make up HbA-2, then together with HbF makes up adult hemoglobin. The research to date has tended to display that mutation in the HBD gene give rise to hemoglobin leprosy-beta-thalassemia syndrome and fetal hemoglobin quantitative trait loci31. The HBM gene has an open reading frame (ORF) that enciphers a 141 amino acid polypeptide resemble the delta globin unearthed in reptiles and birds. HBD-related pathways include factors involved in megakaryocyte development and thrombopoiesis and responses to elevated platelet cytoplasmic Ca2+. Alpha-synuclein, encoded by SNCA, belongs to the synuclein family including beta- and gamma-synuclein as well. Synuclein is plentifully expressed in the cerebrum, α- and β-synuclein selectively inhibits phospholipase D2. Protein SNCA can be used to analyze and integrate presynaptic semaphore and membrane transportation. It has been reported that SNCA peptides are the governing elements of amyloid patches in the brains of victims suffering from Alzheimer32. SERPINE1 gene a serine protease that is classified to the serine protease inhibitor (serpin) superfamily and a major inhibitor for tissue plasminogen activator (tPA) and urokinase (uPA). In addition, it is capable of congenial antiviral immunity. Studies have shown that SERPINE1 deficiency is responsible for plasminogen activator inhibitor 1 deficiency (PAI-1), and the high consistence proteins linked to thrombophilia33. Selenium Binding Protein 1, encoded by SELENBP1, pertain to the selenium-binding protein family. As we all known selenium is an indispensable trace element that possesses formidable anticarcinogenic characters. Researchers have proved that lack of selenium probably results in some certain neurological diseases. The role of selenium in the precaution of carcinoma and neurological diseases perchance is regulated through selenium-united proteins, and alleviated expression of SELENBP1 presumably is relevant to a few kinds of cancer34. This protein might selenium-dependently function in degrading ubiquitination/deubiquitination-regulated protein. SELENBP1 relational disorders include Extraoral Halitosis due to methanethiol oxidase deficiency and methionine adenosyltransferase deficiency35. Integrin Subunit Alpha 2b (ITGA2B) is geared to integrin alpha chain proteins family. The preproprotein undergoes proteolytic processing to produce light and heavy chains that binding via disulfide bonds to constitute subunits of the alpha-IIb/beta-3 integrin cell adhesion receptor. This receptor plays a vital character in coagulation system via attracting platelet gathering. Platelet-type bleeding disorders such as Glanzmann thrombasthenia are caused by mutations in ITGA2B manifesting the inability of platelets to aggregate36.
Furthermore, our current analysis also identified some TFs (TEAD4, TGIF2LY, GATA5, GATA1, GATA2, FOS) related to PAH, insinuating that these DEGs serve an important function in PAH. Next, based on current relevant studies we take a closer look at the association between PAHs and identified TFs. TEAD4, TEA domain transcription factor 4, is important component in Hippo signaling pathway which is involved in controlling organ growth and inhibiting tumor through limiting cell proliferation and facilitating apoptosis. TEAD4 mediates cell proliferation, migration, and epithelial-mesenchymal transition (EMT) by mediating the gene expression of YAP1 and WWTR1/TAZ. Previous study showed that Luteolin ameliorates reconstitution of pulmonary blood vessels and RV hypertrophy on rats PAH model and inhibited smooth muscle cells proliferation and migration at a dose-dependent manner through inhibition mediated by HIPPO-YAP/PI3K/AKT pathway37. TGIF2LY, belongs to the TALE/TGIF homeobox family of transcription factors, is located in the specific region of chromosome Y, in a large stretch of sequence which is considered as a X-to-Y transposition. It is possible to exert transcriptional regulation in testis and become a competitor or regulator of gene TGIF2LX. There are no studies to date that TGIF2LY is associated with PAH yet. The GATA family of transcription factors members GATA1, GATA2and GATA5 are associated with the extent of amino acid sequence identity in DNA zinc finger binding domains and they all have the capability to link the GATA sequence. Considering reprogramming function of GATA1/2/5, the three GATA family members are inducers of pluripotency reprogramming. Specifically, as a transcriptional activator or repressor, GATA1 possibly play a general switch role during erythroid development. GATA1 combines to the DNA sites with the common sequence 5′-GATA-3′ inside modulatory districts of globin genes and others genes of red blood cells and motivates genes transcription concerned with erythroid differentiation of K562 erythroleukemia cells, containing HBB, HBG1/2, ALAS2 and HMBS. GATA2 is a transcription factor participating in stem cell sustain, hematopoiesis and mediating endothelin-1 gene expression of endothelial cells. GATA5 is an indispensable TF during cardiovascular development38 and transcriptional program, which is the basis of diversity of smooth muscle cell. It’s reported that GATA5 united in the CEF-1 nucleoprotein binding domain of cardiac-specific slow/cardiac troponin C transcriptional enhance39. FOS acts as a nuclear phosphoprotein that composes a close but non-covalently connected complex with JUN/AP-1 transcription factor. Both FOS and the JUN/AP-1 basic region appear to interact with symmetrical DNA half-sites in heterodimers. Multimeric SMAD3/SMAD4/JUN/FOS complexes are formed at the AP1/SMAD binding site during TGF-β activation to mediate TGF-β signaling. It has been documented that FOS critically functions in modifying the formation and maintenance of skeletal cell40 and it is believed FOS plays an significant role in signal switching, cellular proliferation and differentiation.
For further investigating functions of immune cells in PAH, CIBERSORT was performed for immune infiltration assays. Results displayed that NK cell activation, monocyte, T cell CD4 memory activation and mast cell infiltration were increased and T cell CD4 naive infiltration was decreased in peripheral blood of PAH patients contrasted with the control group, which may be related to the development and exacerbation of PAH. It is noteworthy that that it was monocyte that was the highest proportion in PAH peripheral blood. Previous researches showed that for coping with hypoxia, pulmonary monocytes could perceive hypoxia, permeate pulmonary arterioles, and accelerate vascular reforming, which suggested that the monocyte lineage functions directly in the morbidity of PAH41. NK cells activation is able to identify and eliminate cells infected with viruses or tumor lesions and degranulate perforin-containing granules and granzymes with the participation of FasL and TRAIL molecules42. Ormiston et al. demonstrated that NK cells in PAH patients generated a greater amount of matrix metalloproteinase 9, leading to deleterious effects on pulmonary vascular remodeling and functional impairment43. At most concomitant states, PAH is bound up with CD4 T cells defects and related to HIV or HHV-8 infection and some experimental studies have displayed that the application of NK cells and T cells suppressed the development of PAH44. In a clinical study, mast cells were abundantly found in PAH by quantitatively measuring the number and activity of mast cells in blood and urine among 44 PAH patients and 29 healthy individuals45. Besides, mast cells were discerned as connective tissue types expressing tryptase and chymotrypsin46. The above findings indicated that mast cells were conducive to the vascular pathophysiology of PAH, which is consistent with the findings of our study.
Conclusion
In summary, results in present study manifested that the development of pulmonary hypertension probably is the result of imbalance between pulmonary vascular remodeling and immune micro environment. We discovered that genes SLC4A1, AHSP, ALAS2, CA1, HBD, SNCA, HBM, SELENBP1, SERPINE1, ITGA2B are the most notable markers of PAH. In addition, (TEAD4, TGIF2LY, GATA5, GATA1, GATA2, FOS) are predicted TFs regulating DEGs and may be regarded as potential targets for the prevention of pulmonary vascular restructure. Regulatory NK cell activation, monocyte, T cell CD4 memory activation and mast cell were detected as the most obvious immune cells infiltrating the peripheral blood of PAH. Since a large number of immune cells are clearly changed in PAH, although it is not clear how the immune system affects vascular remodeling, it can be conjectured that immune system goes a long way in this pathological process. The inadequacies of this study are that the insufficient amount of data may lead to results bias and yet we did not carry out trials to verify the results obtained. Further experiments on immune cells can identify targets to perfect immuno-modulatory therapy for PAH patients. To a certain extent, it could be interpreted like this, key genes and TFs are tightly related to the occurrence of PAH and regarded as underlying targets for therapeutic makers as well, however, it is necessary to explore the specific mechanism using some animal and cell experiments.
Materials and methods
Raw data acquisition and preprocessing
In the study, three gene expression profiles GSE22356, GSE131793 and GSE168905 were obtained from GEO (Home–GEO–NCBI (nih.gov)) database. GSE22356 was based on the GPL570 platform including 10 normal samples and 10 PAH samples. GSE131793 was based on the GPL6244 platform involving 10 healthy samples and 10 PAH samples. GSE168905 was based on the GPL16791 platform containing 9 control samples and 12 PAH samples. Raw data of these profiles were annotated according to their respective platform files following by probes switched into gene symbols, respectively47. The software Perl and R (version 4.0.2) were employed for data preprocessing.
Identification of DEGs
For the purpose of identifying the significant DEGs between the PAH and the healthy group, Limma (linear models for microarray data) package was adopted processing three datasets with |log2 fold change (FC)|≥ 1 and adjusted p value < 0.05 as the thresholds21. We obtained the DEGs of the three datasets respectively. Additionally, volcano plots and heatmaps were generated to evaluate these selected DEGs.
Functional and pathway enrichment analysis
To assess the biological functions of the total DEGs, we combined the DEGs for gene ontology (GO) annotation and Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis applying online website DAVID (https://david.ncifcrf.gov/tools.jsp), which classifies gene functions into BP, CC and MF48. Then use R to generate bubble and bar charts to visualize the obtained results. P value < 0.05 was defined statistically significant.
Protein–protein interaction (PPI) network and hub gene identification
PPI network among all DEGs was established based on an online tool STRING (https://cn.string-db.org/) and then software Cytoscape (version 3.7.2) was employed to adjust and visualize PPI networks49. Subsequently, we utilized a plug-in Cytohubba of Cytoscape to determine top 10 hub genes according to the MCC algorithms. All key genes’ scores were over 15 degrees.
Construction of TF-DEG regulation network
For searching potential interactions between DEGs and transcription factors (TFs), another plugin iRegulon in Cytoscape used for the identification of DEG-targeted TFs. The enriched motifs in iRegulon were ranked depending on the direct targets by means of position weight matrix50. The TF-DEG crosstalk pairs were gained from databases such as TRANSFAC, TRED and so on. Then the TF-DEG regulatory network was visualized by Cytoscape.
Immune cell infiltration analysis
Numerous studies have shown that PAH has a certain relationship with the immune micro environment, so we performed this analysis. The corrected gene matrix was uploaded to CIBERSORT website (CIBERSORT (stanford.edu)), a powerful online immune assay calculation tool, via a deconvolution algorithm to calculate the percentages of 22 kinds of infiltrating immune cells with P < 0.05 as standard51. Hereafter, the results of deduced immunocytes infiltration analysis were visualized by R language in different manners (Spplemetary Table 1).
Data availability
The gene expression profiles of GSE22356, GSE131793 and GSE168905 were downloaded from Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22356, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131793 and https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168905).
Abbreviations
- PAH:
-
Pulmonary arterial hypertension
- RV:
-
Right ventricular
- DEG:
-
Differentially expressed gene
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- TFs:
-
Transcription factors
- PPI:
-
Protein–protein interaction
- BP:
-
Biological process
- CC:
-
Cell component
- MF:
-
Molecular function
- PDE-5:
-
Phosphodiesterase-5
- ERAs:
-
Endothelin receptor antagonists
- TKI:
-
Tyrosine kinase inhibition
- ECM:
-
Extracellular mechanism
- TGF-β:
-
Transcription growth factor beta
- AE:
-
Anion exchanger
- GO:
-
Gene ontology
- GWAS:
-
Enome-wide association study
- NHLBI:
-
American national heart, lung, and blood institute
- cEVs:
-
Cell-secreted extracellular vesicles
- LC:
-
Lung cancer
- CAs:
-
Carbonic anhydrases
- ORF:
-
Open reading frame
- tPA:
-
Tissue plasminogen activator
- uPA:
-
Urokinase
- PAI-1:
-
Plasminogen activator inhibitor 1 deficiency
- EMT:
-
Epithelial mesenchymal transition
References
Hassoun, P. M. & Taichman, D. B. Pulmonary arterial hypertension. N. Engl. J. Med. 385, 2361–2376 (2021).
Thenappan, T., Ormiston, M. L., Ryan, J. J. & Archer, S. L. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ 360, j5492 (2018).
Lau, E. M. T., David, E. G., Celermajer, S. & Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 20, 14 (2017).
Maron, B. A. et al. Pulmonary arterial hypertension: Diagnosis, treatment, and novel advances. Am. J. Respir. Crit. Care Med. 203, 1472–1487 (2021).
Burki, T. K. Pharmacotherapy for pulmonary arterial hypertension. Lancet Respir. Med. 8, 25 (2020).
Stearman, R. S. et al. Systems analysis of the human pulmonary arterial hypertension lung transcriptome. Am. J. Respir. Cell Mol. Biol. 60, 637–649 (2019).
Saygin, D. et al. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulmonary Circ. 10, 1–15 (2020).
Rahman, M. H. et al. Bioinformatics and machine learning methodologies to identify the effects of central nervous system disorders on glioblastoma progression. Brief Bioinform. 22, 25 (2021).
Rahman, M. H. et al. A Network-based bioinformatics approach to identify molecular biomarkers for type 2 diabetes that are linked to the progression of neurological diseases. Int. J. Environ. Res. Public Health 17, 25 (2020).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021).
Goto, M. K. A. S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Xiaomei, Y. C. W., Yicheng, L. & Peng, Z. Identification of crucial hub genes and differential T cell infiltration in idiopathic pulmonary arterial hypertension using bioinformatics strategies. Front. Mol. Biosci. 9, 25 (2022).
Rosenzweig, E. B. et al. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 53, 25 (2019).
Hoeper, M. M. et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 62, D42-50 (2013).
Galie, N. et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur. Respir. J. 53, 25 (2019).
Mainguy, V. et al. Peripheral muscle dysfunction in idiopathic pulmonary arterial hypertension. Thorax 65, 113–117 (2010).
Ulrich, S. et al. Effect of nocturnal oxygen and acetazolamide on exercise performance in patients with pre-capillary pulmonary hypertension and sleep-disturbed breathing: Randomized, double-blind, cross-over trial. Eur. Heart J. 36, 615–623 (2015).
Olsson, K. M. et al. Anticoagulation and survival in pulmonary arterial hypertension: Results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 129, 57–65 (2014).
Rieg, A. D. et al. Tyrosine kinase inhibitors relax pulmonary arteries in human and murine precision-cut lung slices. Respir. Res. 20, 111 (2019).
Cai, W., Li, H., Zhang, Y. & Han, G. Identification of key biomarkers and immune infiltration in the synovial tissue of osteoarthritis by bioinformatics analysis. PeerJ 8, e8390 (2020).
Gallagher, P. G. Disorders of erythrocyte hydration. Blood 130, 2699–2708 (2017).
Page, G. P. et al. Multiple-ancestry genome-wide association study identifies 27 loci associated with measures of hemolysis following blood storage. J. Clin. Invest. 131, 25 (2021).
Kaneda, H. et al. Proximity proteomics has potential for extracellular vesicle identification. J. Proteome Res. 20, 3519–3531 (2021).
Han, G. et al. Nrf2 expands the intracellular pool of the chaperone AHSP in a cellular model of β-thalassemia. Redox Biol. 50, 102239 (2022).
Kihm, A. J. et al. An abundant erythroid protein that stabilizes free alpha-haemoglobin. Nature 417, 758–763 (2002).
Phillips, J. D. Heme biosynthesis and the porphyrias. Mol. Genet. Metab. 128, 164–177 (2019).
Balwani, M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: Pathophysiology, genetics, clinical manifestations, and management. Mol. Genet. Metab. 128, 298–303 (2019).
Alkadhi, K. A. Cellular and molecular differences between area ca1 and the dentate gyrus of the hippocampus. Mol. Neurobiol. 56, 6566–6580 (2019).
Dong, C., Madar, A. D. & Sheffield, M. E. J. Distinct place cell dynamics in CA1 and CA3 encode experience in new environments. Nat. Commun. 12, 2977 (2021).
Wienert, B., Martyn, G. E., Funnell, A. P. W., Quinlan, K. G. R. & Crossley, M. Wake-up sleepy gene: Reactivating fetal globin for β-hemoglobinopathies. Trends Genet. 34, 927–940 (2018).
Yao, R. Q., Ren, C., Xia, Z. F. & Yao, Y. M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 17, 385–401 (2021).
Udumudi, A. & Lava, C. Genetic markers for inherited thrombophilia related pregnancy loss and implantation failure in Indian population—implications for diagnosis and clinical management. J. Maternal Fetal Neonatal Med. 20, 1–9 (2022).
Caswell, D. R. et al. Tumor suppressor activity of selenbp1, a direct Nkx2-1 target, in lung adenocarcinoma. Mol. Cancer Res. 16, 1737–1749 (2018).
Pol, A. et al. Mutations in SELENBP1, encoding a novel human methanethiol oxidase, cause extraoral halitosis. Nat. Genet. 50, 120–129 (2018).
Botero, J. P. et al. Glanzmann thrombasthenia: Genetic basis and clinical correlates. Haematologica 105, 888–894 (2020).
Zuo, W. et al. Luteolin ameliorates experimental pulmonary arterial hypertension via suppressing Hippo-YAP/PI3K/AKT signaling pathway. Front. Pharmacol. 12, 663551 (2021).
Wei, D. et al. GATA5 loss-of-function mutations underlie tetralogy of fallot. Int. J. Med. Sci. 10, 34–42 (2013).
Zhang, X. L. et al. GATA5 loss-of-function mutation in familial dilated cardiomyopathy. Int J Mol Med 35, 763–770 (2015).
Almada, A. E. et al. FOS licenses early events in stem cell activation driving skeletal muscle regeneration. Cell Rep. 34, 108656 (2021).
Yu, Y. A. et al. Nonclassical monocytes sense hypoxia, regulate pulmonary vascular remodeling, and promote pulmonary hypertension. J. Immunol. 204, 1474–1485 (2020).
Newman, K. C. & Riley, E. M. Whatever turns you on: Accessory-cell-dependent activation of NK cells by pathogens. Nat. Rev. Immunol. 7, 279–291 (2007).
Ormiston, M. L. et al. Impaired natural killer cell phenotype and function in idiopathic and heritable pulmonary arterial hypertension. Circulation 126, 1099–1109 (2012).
Hassoun, P. M. et al. Inflammation, growth factors, and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 54, S10-s19 (2009).
Farha, S. et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm. Circ. 2, 220–228 (2012).
Liu, J. et al. The role of pulmonary mast cells activation and degranulation in the process of increased pulmonary artery pressure. Gen. Physiol. Biophys. 40, 183–195 (2021).
Wang, H., Liu, D. & Zhang, H. Investigation of the underlying genes and mechanism of macrophage-enriched ruptured atherosclerotic plaques using bioinformatics method. J. Atheroscler. Thromb. 26, 636–658 (2019).
Teng, P. et al. Identification of key genes in calcific aortic valve disease by integrated bioinformatics analysis. Medicine (Baltimore) 99, e21286 (2020).
Yin, Y. et al. Identification of potential hub genes of atherosclerosis through bioinformatic analysis. J. Comput. Biol. 20, 20 (2020).
Janky, R. et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 10, e1003731 (2014).
Wang, D. et al. Predicting the key genes involved in aortic valve calcification through integrated bioinformatics analysis. Front. Genet. 12, 650213 (2021).
Author information
Authors and Affiliations
Contributions
S.X.T. and Y.L. were agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved and drafted the manuscript. S.X.T. and Y.L. made substantial contributions to conception and design and revised the manuscript critically for important intellectual content; B.L. made substantial contributions to acquisition of data; Y.L. made substantial contributions to analysis and interpretation of data. B.L. given final approval of the version to be published. All authors read and approved the final manuscript. S.X.T. and Y.L. contributed equally to the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, S., Liu, Y. & Liu, B. Integrated bioinformatics analysis reveals marker genes and immune infiltration for pulmonary arterial hypertension. Sci Rep 12, 10154 (2022). https://doi.org/10.1038/s41598-022-14307-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14307-6
- Springer Nature Limited
This article is cited by
-
An Analysis Regarding the Association Between DAZ Interacting Zinc Finger Protein 1 (DZIP1) and Colorectal Cancer (CRC)
Molecular Biotechnology (2024)
-
Molecular Mechanism of Yangshen Maidong Decoction in the Treatment of Chronic Heart Failure based on Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulations
Cell Biochemistry and Biophysics (2024)
-
GSDMD deficiency ameliorates hyperoxia-induced BPD and ROP in neonatal mice
Scientific Reports (2023)