
Abstract
Microbial colonization plays a direct role in host health. Understanding the ecology of the resident microbial community for a given host species is thus an important step for detecting population vulnerabilities like disease. However, the idea of integrating microbiome research into conservation is still relatively new, and wild birds have received less attention in this field than mammals or domesticated animals. Here we examine the composition and function of the gut microbiome of the endangered Galapagos penguin (Spheniscus mendiculus) with the goals of characterizing the normal microbial community and resistome, identifying likely pathogens, and testing hypotheses of structuring forces for this community based on demographics, location, and infection status. We collected fecal samples from wild penguins in 2018 and performed 16S rRNA gene sequencing and whole genome sequencing (WGS) on extracted DNA. 16S sequencing revealed that the bacterial phyla Fusobacteria, Epsilonbacteraeota, Firmicutes, and Proteobacteria dominate the community. Functional pathways were computed from WGS data, showing genetic functional potential primarily focused on metabolism—amino acid metabolism, carbohydrate metabolism, and energy metabolism are the most well-represented functional groups. WGS samples were each screened for antimicrobial resistance, characterizing a resistome made up of nine antibiotic resistance genes. Samples were screened for potential enteric pathogens using virulence factors as indicators; Clostridium perfringens was revealed as a likely pathogen. Overall, three factors appear to be shaping the alpha and beta diversity of the microbial community: penguin developmental stage, sampling location, and C. perfringens. We found that juvenile penguins have significantly lower alpha diversity than adults based on three metrics, as well as significantly different beta diversity. Location effects are minimal, but one site has significantly lower Shannon diversity than the other primary sites. Finally, when samples were grouped by C. perfringens virulence factors, we found dramatic changes in beta diversity based on operational taxonomic units, protein families, and functional pathways. This study provides a baseline microbiome for an endangered species, implicates both penguin age and the presence of a potential bacterial pathogen as primary factors associated with microbial community variance, and reveals widespread antibiotic resistance genes across the population.
Similar content being viewed by others


Introduction
Resident microbes diversely affect animal health. Some long-term members of the microbiome may facilitate digestion or provide immune system training for the host1. Colonization resistance is an important benefit, as mutualistic microbes may out-compete a pathogenic invader for space and nutrients or even release toxins as a deterrent2. Some community members may also be opportunistically pathogenic, occupying an inconspicuous position in the microbiome until a disruption occurs, then infecting the host3,4.
Characterizing the normal microbiome for species of concern provides an essential baseline from which to measure changes. This has the potential to help zoos improve their level of care for collection animals as well as to increase reintroduction success by mimicking a species’ normal microbiome5,6. However, microbial community assessments can also improve management of wild populations in a number of ways. Dysbiosis in the microbiome (i.e. disruption of the normal bacterial community) can be an indicator of health problems and is associated with a number of diseases1,7. Metagenomic assessments can reveal potential pathogens that may pose a threat to the population8. Sequencing data can additionally be used to assess the level of antimicrobial resistance in the community, known as the “resistome,” which at high levels can indicate a potentially dangerous level of connectivity between humans and wildlife9. Thus, microbiome research may provide a critical perspective for conservation efforts.
Factors structuring these resident microbial communities are complex and can vary substantially between taxa. Diet is known to be a primary driver of microbiome composition and taxa with atypical diets tend to have distinct microbiomes10,11,12. For instance, the vampire finch (Geospiza septentrionalis) supplements its diet in an unusual way by eating eggs, guano, and the blood of larger birds, and the species also exhibits a unique microbiome profile compared to the other Darwin’s finches13. These dietary changes can be rapid—one study of human diet found that switching from a plant- to animal-based diet resulted in reduced carbohydrate fermentation and increased protein fermentation by gut microbes in a matter of days, with corresponding abundance changes in bacteria associated with those activities14. Environmental factors such as season and habitat are known to affect gut microbiomes as well, though these differences are largely attributed to changes in food availability between seasons15,16. However, diet is a better predictor for microbiome composition and function in mammals than it is for birds, and the effect of diet is weakest in the microbiomes of both bats and flying birds, possibly due to the shorter intestines associated with flight adaptation17.
Demographic factors such as the host’s sex or developmental stage can also play a role in shaping the microbiome18,19. One study found that cloacal microbiomes differed between male and female rufous-collared sparrows (Zonotrichia capensis) during breeding seasons, with the male microbiome becoming more diverse at the onset of the breeding season19. Hormonal differences or immune variation between sexes may be responsible to some degree19,20,21. Many taxa also undergo microbiome changes throughout development; for example, little penguins (Eudyptula minor) exhibit increased abundances of Firmicutes and Bacteroidetes as they mature22. Microbial community differences based on sex and age are not consistent across taxa, and in some cases the effect size is quite small compared to other factors, indicating a continued need to assess community drivers on a case-by-case basis in wild populations23,24.
Once the normal microbiome is understood for a given species, monitoring the microbiome using fecal samples may be a valuable non-invasive assay for wild populations of concern25,26. Scat microbiome assays are being developed to provide insights into population demographics and host health. For example, one study in Rocky Mountain elk (Cervus canadensis) determined microbial predictors for host age, sex, body fat, and biogeography, reliably classifying individuals based on fecal microbiome samples25. Microbiome diversity (i.e. the number of species in a microbial community) can also indicate vulnerability to disease—for example, juvenile ostriches with initially low bacterial diversity can be more likely to develop pathogen-associated dysbiosis and later succumb to enterocolitis mortality7. Testing the fecal microbiome can also reveal anthropogenic influence on a wild population, often through dysbiosis in the microbial composition and abundance26,27. Human influence may also be detected through the increased presence of antibiotic resistance genes in the microbiome; human-associated factors such as wastewater or livestock can introduce resistance genes to new environments, where the resistance genes can be rapidly disseminated through microbial communities via horizontal gene transfer9,28. However, many wild microbiome studies rely solely on 16S rRNA gene sequencing instead of shotgun sequencing, which provides little indication of pathogenicity or antibiotic resistance genes in a microbiome29. Widespread use of high-resolution metagenomic data across wild microbiome studies and the validation of microbial assays are necessary steps before microbiome-based tools can be used to make management recommendations for wild populations5,25.
This study examines the gut microbiome of the Galapagos penguin (Spheniscus mendiculus), a highly range-restricted species which occurs only in the Galapagos Islands30. The Galapagos penguin forages near the shore, consuming schooling fish (such as mullets and sardines) and crustaceans31,32,33. This penguin faces regular population bottleneck events when the nutrient-rich Cromwell Current is disrupted periodically by warm El Niño weather patterns, leading to reduced fish availability and ultimately penguin starvation33. The species is classified as endangered by the International Union for Conservation of Nature (IUCN) due to the severe declines associated with these events as well as their highly restricted range30,33. The frequent population bottlenecks are likely the reason for the genetic homogeneity found in this species—both microsatellite markers and major histocompatibility complex (MHC) sequences demonstrate a low degree of genetic variation34,35,36.
The low genetic variation may leave the penguin population vulnerable to introduced diseases37,38. Previous studies have found evidence of prior infections by Chlamydophila psittaci and Toxoplasma gondii39,40, as well as infections by microfilariae (species unknown) and a lineage of Plasmodium (Lineage A)38,41. However, microbiome characterization and a thorough assessment of enteric pathogens using high-throughput sequencing tools has not been completed and would provide valuable insight into the health of this species. Furthermore, widespread antibiotic resistance genes have been found previously in the Galapagos Islands9,42,43, but the extent to which antibiotic resistance is associated with the Galapagos penguin was unknown as we began this study.
This research thus has four goals: (1) establish a baseline for gut microbiome taxonomy and function in the Galapagos penguin, (2) identify putative enteric pathogens, (3) characterize the resistome; and (4) explore drivers of community structure. We hypothesized that hormonal variation and contrasting foraging habits may lead to distinctive microbiomes, and so microbial community structure would vary depending on both sex and age, respectively. Due to the largely homogeneous environment of the western coast of Isabela Island, we hypothesized that different locations may be a minimal factor in gut microbiome communities, particularly since the penguin population shows significant movement between sites and the penguins tend to forage near the coast32,36. Finally, we hypothesized that gut pathogens -if found- may be associated with community changes facilitated through either disruptive or opportunistic invasion.
Methods
All Galapagos penguin fecal samples used in this study were collected in July of 2018 from Isabela Island and the Marielas Islets in Galapagos, Ecuador. Sample collection methods were performed in accordance with relevant guidelines and regulations; this study was approved by the University of Missouri-St. Louis Institutional Animal Care and Use Committee, United States Department of Agriculture, Galapagos National Park Directorate, the Agency for the Regulation and Control of Biosecurity and Quarantine for Galapagos, and the Ecuadorian Ministry of the Environment and Water. Penguins were sampled at three sites on Isabela and the Marielas, with two sample days per site. To collect samples, wild penguins were safely captured on land at each site using long-handled nets and brought to a large boat for processing. Processing included morphological measurements and opportunistic fecal collection. The fecal collection procedure involved harvesting feces from clean plastic sheets placed beneath the penguins during transport and handling, immediately following capture. Fecal samples were preserved in 95% ethanol at room temperature44. Males and females were identified based on bill depth measurements and size45,46. Each penguin was tagged with a Passive Integrated Transponder tag in the web of one foot (as part of a separate study), enabling the identification of recaptured individuals. For this study, each sample corresponds to an individual penguin. Fecal DNA was extracted within four weeks of sample collection using Qiagen PowerFecal DNA extraction kits (Qiagen, LLC, Germantown, Maryland). Manufacturer instructions were followed for the extraction protocol. DNA was extracted from 0.1 g of feces from each sample (total feces for each sample ranged from 0.25 to 1000 g) based on manufacturer suggestions for avian fecal samples, and homogenization was performed using a vortex adapter at maximum speed for 10 min. DNA was quantified using a Qubit fluorometer. Many samples had low DNA yield (less than 20 ng of total DNA), despite multiple extraction attempts, and only 40 samples with total yield higher than 20 ng were used for sequencing. Extracted DNA was stored at − 20 °C.
Targeted sequencing
Sequencing of the 16S rRNA gene V4 region was performed with Illumina MiSeq on 40 of the DNA samples, excluding samples with insufficient DNA (< 20 ng), at the University of Michigan Medical School Microbiome Core. The Microbiome Core used the dual indexing sequencing strategy and primers described in Kozich et al. 201347. The V4 region was selected because its length of 250 bp allows forward and reverse reads to fully overlap when sequenced with the cost-efficient Illumina platform, reducing the error rate47. Method standardization also facilitates comparisons between microbiome studies, and the V4 region has widespread use, notably through the Earth Microbiome Project and the Mothur Standard Operating Procedure (SOP)47,48. Negative sampling and extraction controls were included to assess contamination during processing, and a water sample and a mock community (ZymoBIOMICS Microbial Community Standard) were added by the Microbiome Core prior to library preparation as negative and positive PCR amplification and sequencing controls, respectively. Seven penguin samples were later resequenced alongside a ZymoBIOMICS Gut Microbiome Standard, which had been stored in 95% ethanol prior to extraction to serve as a positive control for the preservation/extraction methods. The resequenced samples were examined to ensure bacterial composition was comparable to the originally sequenced samples; however, all data included in the downstream analysis was generated from the first sequencing run to avoid any potential batch effects. The ZymoBIOMICS Microbial Community Standard was also added to this second sequencing run by the Microbiome Core as a sequencing positive control.
Read analysis was conducted in Mothur, following the Schloss MiSeq standard operating procedure47,49. Reads were filtered by base quality and length, aligned with the SILVA 132 reference database50 and filtered for alignment quality and chimeras. Samples with less than 10,000 reads following data cleaning steps were excluded, leaving 34 samples. Operational taxonomic unit (OTU) clustering was based on a 97% similarity threshold. Unclassified reads and reads that matched mitochondria, chloroplasts, or archaea were filtered out. Filtered reads were subsampled in Mothur to match the sample with the lowest read count (11,295 reads). Community data were imported into R version 3.6.3 to analyze alpha and beta diversity51.
Whole genome sequencing
Whole genome sequencing (WGS) was performed on 20 of the extracted fecal samples by the University of Michigan Microbiome Core using the Illumina Nextera DNA Flex kit52. Four of these samples were later resequenced with the ZymoBIOMICS Gut Microbiome Standard (Zymo Research, Irvine, CA) included in the same run as a positive control to ensure comparable results—the resequenced samples were examined to make sure the bacterial compositions were similar to the original sequence results, but downstream analysis included only data generated in the initial sequencing run. Quality control was performed by trimming reads using Trimmomatic with a sliding window of 4, minimum quality score of 20, and minimum length of 7053. These cleaned reads were used directly for read mapping with KMA (k-mer alignment) against two databases, VFDB (Virulence Factor Database) and CARD (Comprehensive Antibiotic Resistance Database), on the platform PATRIC (Pathosystems Resource Integration Center)54,55,56. They were categorized taxonomically using the k-mer based program Kraken 2, also available on PATRIC57.
The trimmed reads were also assembled with SPAdes, using the recommended K-mer lengths of 21, 33, 55, 77, 99, and 127, with the BayesHammer module for error correction enabled58,59. SPAdes output was assessed with MetaQuast60. Compared to single genome assemblies, lower quality is expected for metagenomic assemblies, but two samples with the worst assemblies were ultimately excluded from much of the functional analysis due to poor downstream annotation results; alternate tools relying on reads rather than contigs were similarly problematic for those two samples. The contigs from the SPAdes assembly were classified taxonomically using Kraken 2 on PATRIC.
Finally, parallel read-based and assembly-based approaches were used to achieve a robust picture of the functional profile in these communities. Cleaned reads were categorized functionally using the program HUMAnN 3.0, while contigs were annotated using the program MetaErg52,61. Pfam (Protein Family Database) annotations were obtained through both pipelines, which notably annotated virulence-associated proteins in addition to the virulence factors obtained via PATRIC62. Pathways were only computed for a minority of gene families in both HUMAnN 3.0 and MetaErg, leaving a majority unclassified. Protein families and metabolism-related KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways detected from the metagenomic assemblies were imported to R to examine differences in metabolic profiles between groups (sex, age, and sampling location)51,63,64,65. Protein family abundances and pathway abundances were subsampled to match the lowest sample (22,259 and 19,684 reads, respectively), after excluding one sample with insufficient protein families and two samples with insufficient pathway results, to allow comparisons between samples without depth-biased results.
Control assessment
Since negative controls contained ≤ 30 reads following batch filtering steps for the 16S dataset, and < 15 classified fragments following Kraken 2 classification of trimmed shotgun sequencing reads, potential contamination (from sampling, extraction kits, sequencing crossover, etc.) was considered negligible. 16S sequences from the Zymo preservation/extraction control (ZymoBIOMICS Gut Microbiome Standard) were mapped to manufacturer-provided reference sequences using the “seq.error” function in Mothur, which calculated an error rate of 0.0079%. Sequences from the Zymo sequencing control (ZymoBIOMICS Microbial Community Standard) were mapped to 16S reference sequences provided by the University of Michigan Microbiome Core, with an error rate of 0.015% and 0.005% for the first and second sequencing runs. All expected bacterial species with theoretical abundances > 0.01% were detected in the mock communities following processing; some abundance skew was apparent, but this is unlikely to affect the reported results as all study samples were processed in the same batch and comparable to each other. Shotgun sequences of the ZymoBIOMICS Gut Microbiome Standard were directly mapped to the whole genome reference sequences provided by the manufacturer using Bowtie266. Reads successfully mapped to all expected bacteria, archaea, and yeasts.
Diversity calculations and statistical analysis
Alpha diversity was calculated for each sample from the subsampled 16S rRNA gene sequencing data using the R package phyloseq for three metrics: observed richness, Simpson’s Index, and Shannon Diversity Index67. Significant differences based on alpha diversity values were assessed between groups using Kruskal Wallis tests51. Two common measures of beta diversity are Jaccard distance, which relies on presence/absence data, and Bray–Curtis dissimilarity, which includes abundance data68. Both Jaccard distance and Bray–Curtis dissimilarity were initially used to calculate beta diversity, with similar results; Jaccard distance was ultimately used for this analysis. Beta diversity (Jaccard distance) was calculated in the phyloseq package for both 16S and WGS datasets, and differences in beta diversity were assessed using permutational multivariate analysis of variance (PERMANOVA) in the R package vegan67,69,70. Potentially confounding variables were listed first in the model, with the variable of interest listed last. The dispersion assumption for PERMANOVA, PERMDISP, was tested for significant values using the vegan package69,70,71. Welch’s t-tests were used to examine body condition variation (measured by weight:wing ratio) between groups with and without detected virulence factors. Kruskal Wallis tests were used to assess significant differences between relative abundances of metabolic pathways and groups from the WGS dataset, and false discovery correction was applied with the Benjamini–Hochberg method72.
Ethical approval
Sampling procedures and sample export were approved by the University of St. Louis´s Institutional Animal Care and Use Committee (Protocol #1211796), USDA (Permit #47418), Galapagos National Park Directorate (#PC-05-18), the Agency for the Regulation and Control of Biosecurity and Quarantine for Galapagos (#ABG-CT-2019-0019-O), and the Ecuadorian Ministry of the Environment and Water (Contract #MAE-DNB-CM-2016-0043, Export Authorization #128-2019-EXP-CM-MBI-DNB/MA).
Results
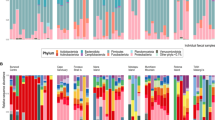
The primary bacterial phyla found in the Galapagos penguin fecal samples were Fusobacteria, Epsilonbacteraeota, Firmicutes, and Proteobacteria, and the most common families were Fusobacteriaceae, Helicobacteraceae, Clostridiaceae, Pasteurellaceae, and Peptostreptococcaceae (Fig. 1). WGS samples were profiled functionally using level II of the KEGG pathway classification hierarchy. Most identified KEGG pathways were involved in metabolic activity, primarily amino acid metabolism, carbohydrate metabolism, energy metabolism, and nucleotide metabolism (Fig. 1).

Alpha and beta diversity were calculated to assess diversity within and between communities, respectively. Overall, alpha diversity was low; no raw sample exceeded 100 identified OTUs and most contained fewer than 50 OTUs. Alpha diversity was significantly lower in juveniles compared to adults in each of the three diversity measures used (Fig. 2, observed richness P = 0.0068, Simpson’s P = 0.0046, Shannon P = 0.0071). Beta diversity significantly differed between age classes after controlling for location and sex in the model (PERMANOVA, R2 = 0.06427, P = 0.019, Supplementary Information; PERMDISP, P = 0.816). Adult penguins did not vary by sex when comparing either alpha or beta diversity. No functional differences based on age or sex were apparent in the WGS dataset with either metabolic pathways or protein families.

We also examined the importance of location for microbiome composition. Due to the uneven sample distribution across sites, single samples from El Muñeco (at the northern end of Isabela Island) and Playa Perros (near Puerto Pajas) were first excluded. The remaining three sites—Caleta Iguana, Puerto Pajas, and Marielas—represent the largest penguin colonies. Caleta Iguana is only represented by four samples following filtering steps, constraining our ability to compare between all three sites.
Slight clustering by location was apparent using Principal Coordinates Analysis (PCoA) (Fig. 3). Compared to the more exposed sample sites, Marielas was slightly different (PERMANOVA, 999 permutations, R2 = 0.05970, P = 0.038, Supplementary Information; PERMDISP, P = 0.293). However, when the sites were limited to Marielas and Puerto Pajas there was no significant difference (PERMANOVA, 999 permutations, R2 = 0.06588, P = 0.061, Supplementary Information). Shannon Diversity Index values significantly differed between the three sites (P = 0.03857), with the lowest Shannon Diversity seen at Marielas, but observed richness and Simpson’s Index did not significantly vary (Fig. 3).

Alpha and beta diversity showed minor variation between samples from the three primary sampling sites on Isabela Island: Caleta Iguana (CI), Puerto Pajas (PP), and Marielas (M). Observed richness and Simpson’s Index were similar between sites (A, B), but Shannon Diversity Index significantly differed (C). Principal Coordinates Analysis illustrated slight clustering between locations (D). Sample sites are shown along the western coast of Isabela Island, with the Galapagos Archipelago in the map inset (E). Figure created in R using tidyverse (v. 1.3.1)96, phyloseq (v. 1.40.0)67, cowplot (v. 1.1.1)97, ggpubr (v. 0.4.0)98, ggmap (v. 3.0.0)99, ggsn (v. 0.5.0)100, and ggrepel101 (v. 0.9.1).
Antibiotic resistance screening
Screening shotgun sequencing reads from each sample with the CARD database revealed a total of nine putative antibiotic resistance genes (Table 1). Two resistance genes corresponding to Helicobacter pylori reference genomes were particularly widespread, occurring in almost all samples (19/20); these genes confer resistance to Tetracyclines and Macrolides. Genes resistant to Aminoglycosides and Lincosamides were also common in this small sample set, and one Peptide-resistant gene corresponding to C. perfringens SM101 was found in six samples.
Pathogen screening
WGS samples were screened for virulence factors using three tools with two databases: PATRIC (VFDB), HUMAnN 3.0 (Pfam), and MetaErg (Pfam). Virulence factors across all three tools were associated with a single bacterium, Clostridium perfringens. Most of the reference genome matches from the VFDB were from strain 13, classified as type A (Table 2). C. perfringens virulence factors from the VFDB were detected in 12/20 penguins, though the taxonomic search using Kraken 2 showed the presence of the bacterium in an additional seven penguins (19/20). The virulence-associated BrkB protein family was also classified with C. perfringens in both HUMAnN 3.0 and MetaErg (BrkB Pfam accession: PF0361), though to varying degrees—read-based HUMAnN 3.0 detected BrkB with C. perfringens in 8/20 samples, while contig-based MetaErg detected it in 10/20 samples. All eight of the samples with virulence factors detected by HUMAnN 3.0 were also highlighted by MetaErg, and all ten of the samples with virulence factors detected by MetaErg were also found by PATRIC.
Additional bacterial taxa were associated with virulence factors in Pfam, though none appeared in the VFDB search. Cetobacterium ceti, Clostridium baratii, Clostridium thermobutyricum, Paeniclostridium sordellii, and Photobacterium damselae were detected with virulence-associated protein families by both HUMAnN 3.0 and MetaErg. The contig-based approach, MetaErg, found an additional 28 virulence-associated species; the most common were Helicobacter brantae, Gallibacterium anatis, Helicobacter sp. 002,287,135, Helicobacter sp. 001,693,335, and Fusobacterium sp. 900,015,295. Most taxa were associated with the Pfam virulence factor BrkB, but others matched with a haemolysin (SMP_2), virulence-associated protein E, or virulence protein RhuM family, among others.
Since C. perfringens was the only bacterium consistently highlighted as a pathogen by all pipelines, the presence of C. perfringens virulence factors was examined as a potential structuring force for the microbial communities. Samples grouped by C. perfringens virulence factors from the VFDB revealed signals of dysbiosis (Fig. 4). Beta diversity was calculated from protein families and from metabolic pathway abundances, and in both cases samples with virulence factors clustered away from samples without virulence factors on a PCoA. After controlling for location and including age and sex in the models, both protein families (PERMANOVA, 999 permutations, R2 = 0.14156, P = 0.020, Supplementary Information; PERMDISP, P = 0.076) and metabolic pathways (PERMANOVA, 999 permutations, R2 = 0.22465, P = 0.014, Supplementary Information; PERMDISP, P = 0.081) were significantly different based on the presence of C. perfringens virulence factors. Three metabolic groups and 17 metabolic KEGG pathways significantly differed between samples divided by virulence factors (adjusted P-values < 0.05). This pattern held true in the 16S dataset, revealing taxonomic clustering based on the presence of C. perfringens virulence factors (PERMANOVA, 999 permutations, R2 = 0.17409, P = 0.001; PERMDISP, P = 0.27, Supplementary Information). However, alpha diversity did not significantly vary between groups. No significant relationship was found between C. perfringens status with age, sex, location, or body condition.

The penguin microbiome varied with C. perfringens virulence factors (VFs). Principal Coordinates Analysis showed distinct clustering based on OTUs from 16S rRNA gene sequencing data (A), protein families from shotgun sequencing data (B), and KEGG metabolic pathways from shotgun sequencing data (C) when separated by Jaccard distance and sorted by the presence of C. perfringens virulence factors from the VFDB. Relative abundance of three metabolic groups significantly varied in the presence of C. perfringens (D). Figure created in R using tidyverse (v. 1.3.1)96, phyloseq (v. 1.40.0)67, and cowplot (v. 1.1.1)97.
Discussion
Overall, our study indicates that developmental stage, location, and pathogen presence are structuring the gut microbiome in the Galapagos penguin. The taxonomic profile of this community was similar to previously published penguin microbiomes, though the gut microbiome of the Galapagos penguin was notably dominated by Fusobacteria and lacking in Bacteroidetes22,73,74. We found adult Galapagos penguins had significantly higher alpha diversity (observed richness, Shannon Diversity, and Simpson’s Index) in their gut microbiomes compared to juvenile penguins. Differences in foraging behaviors and movement between adults and juveniles may be an explanatory factor31,32. Non-breeding adults and juveniles tend to travel longer distances as they forage for schooling fish and crustaceans in shallow water along the shore, while breeding adults travel shorter distances and stay near the nesting sites31,32. However, juveniles may return to the nest area as they gradually learn how to forage, and adults occasionally demonstrate extended parental care during this learning period by feeding fully-fledged juveniles, complicating movement-based interpretations of the reduced microbial diversity found in juveniles31,75. Hormonal differences could also play a role since some adults were in breeding condition—this is known to increase alpha diversity in males in other avian species—but surprisingly, no sex-based differences were found19. The lack of sex-based differences may again be related to foraging habits, as males and females exhibit similar foraging behaviors and are likely exposed to similar dietary and environmental microbes32. It is important to understand how factors such as developmental stage can influence the avian microbiome, as this could indicate a varied degree of vulnerability to disease depending on host age.
Though location did not appear to be a strong force structuring the microbiome, both alpha and beta diversity showed some differences at the Marielas Islets compared to the other primary sampling sites. The similar environment along the coast of Isabela and the movement of penguins between sites are likely factors behind the general homogeneity of microbiomes between locations31,32. The small differences observed may be explained by unique diets between sites or exposure to different environmental microbes. While sociality can also influence microbiomes, determining relatedness or pair bonds within the sample set was beyond the scope of this study76. Perhaps significantly, the Sierra Negra shield volcano on Isabela Island was erupting during the 2018 sampling trip. Lava flowed down the volcano’s northwestern flank and reached the sea near the Marielas sampling site77. This contributed to warmer water temperatures at that site and likely altered pH levels as well78. Changes in pH can influence aquatic microbial communities, and this proximity to volcanic activity may have led to the slight variation in microbial signatures found between sites79. Differing amounts of environmental heavy metal may also play a role, as this is known to alter microbiome compositions in some systems—a previous study found variation in heavy metal concentrations in Galapagos penguin feathers from different sites, with significantly higher levels of lead in feathers from Marielas80,81. An additional factor may be the relatively exposed position of the southern sites compared to the more sheltered location of the Marielas away from the primary current.
We determined that the putative resistome for this species contains at least nine resistance genes. Two resistance genes associated with Helicobacter pylori were almost ubiquitous, detected in all but one penguin. Antibiotic resistant genes have been previously found in the Galapagos Islands in marine water, tortoise feces, and both land iguana and marine iguana feces, but this is the first time to our knowledge that they have been detected in Galapagos penguins9,42,43. Some antibiotic resistance occurs naturally, in areas as remote as Antarctica, and resistance genes could potentially be found in Galapagos even in the absence of human activity82. However, the increasing amount of antibiotic resistance found in the wild is broadly attributed to selection from the heavy use of antibiotics in modern agricultural and clinical settings83. In one example of likely anthropogenic effects, a study in Galapagos found that proximity to humans (e.g. ports, towns) was generally associated with the antibiotic resistance found in seawater and reptile feces, with increased resistance detected in most populated areas and no resistance detected at certain isolated sites9.
The exchange of antibiotic resistance genes happens readily among bacteria through horizontal gene transfer, making it challenging to prevent resistance genes from spreading. Antibiotic resistance is found at high levels in bacteria from human waste, and even after waste treatment sewage remains a potent source of antibiotics or resistance genes84. Resistance genes can even be transferred by wildlife across large distances, such as through resident bacterial flora of migratory birds82. In Galapagos, sewage contamination from towns and boats is a likely way in which antibiotics and/or bacteria with resistance genes could be introduced into the environment (which can also be detrimental to human health), though resistance genes may also arrive from other sources42. Increased wastewater control is thus an essential factor to limit the spread of antibiotic resistance in wild communities. Finding antibiotic resistance consistently in the penguin samples indicates that it is present even in more isolated areas along Isabela Island, emphasizing a need for further investigation into the extent of resistance genes associated with anthropogenic activity (such as cruise ships) and whether wastewater management changes should be considered in the islands. Collecting water samples in parallel with penguin fecal samples in future studies would provide greater insight into where environmentally derived antibiotic resistance genes may have originated.
Finally, our pathogen screening highlighted a few potential enteric pathogens—Clostridium perfringens, Paeniclostridium sordelli, Clostridium baratii, Gallibacterium anatis, and Photobacterium damselae were among the most common bacteria linked with virulence-associated protein families in this microbiome. The class Clostridia contains some of the primary agents of enteric disease in birds, and C. perfringens, P. sordellii and C. baratii have all been associated with enteric disease85,86. Gallibacterium anatis (previously Pasteurella anatis) has also been implicated as a pathogen in several avian species and is considered an emerging poultry disease87. However, the detection of virulence-associated factors is by itself no guarantee that a microbe is actually pathogenic to the host, and microbes such as Gallibacterium anatis are also commonly found as members of the normal bacterial flora88. Further, several bacteria in the microbiome were associated with virulence proteins but are unlikely to be pathogenic to penguins—for example, Photobacterium damselae was linked to several different virulence-associated protein families in this microbiome, though this bacterium is recognized as a pathogen in taxa such as fish and marine mammals rather than birds89.
Clostridium perfringens was the only putative pathogen detected with the Virulence Factor Database in addition to the Protein Family Database (Pfam). C. perfringens is an extremely widespread bacterium and a normal member of many microbiomes, but it is also notorious for causing necrotic enteritis in poultry and other birds (as well as enteric diseases in humans, dogs, and a number of other taxa)90,91. In poultry, C. perfringens injects toxins into the intestines, resulting in intestinal lesions and a range of clinical signs including lethargy, loss of appetite, and mortality (though lethal cases may also occur without any observable symptoms)86. Outbreaks of C. perfringens leading to fatalities have also been documented in captive penguin populations92. The pathogenicity of the detected strain of C. perfringens in Galapagos penguins is unknown, and we notably did not detect the netB gene encoding a pore-forming toxin which is associated with most occurrences of necrotic enteritis in poultry93. However, the detected virulence factors correspond to type A and the genes plc and cpe, which are associated with toxin production and avian enteric disease91. Coupled with the apparent dominance of C. perfringens in the observed microbial communities and the strong structural changes observed in the presence of virulence factors, this suggests a level of pathogenicity at the time of sampling. While pathogen presence was not significantly associated with body condition in this study, a larger sample size would provide more conclusive results. Resampling the population is necessary to shed light on the role C. perfringens plays in this species’ microbiome.
The Galapagos penguin faces many threats, largely from anthropogenic sources. Climate change may lead to increased frequency of El Niño events, which would significantly increase the species’ odds of extinction31,33,94. Overfishing in the area surrounding the Galapagos National Park could influence food availability31. Introduced predators such as cats have been known to kill Galapagos penguins in the archipelago31. The low genetic variation found in these penguins may also increase their vulnerability to introduced diseases, and the small population size and limited range leave little room for species resilience in the event of an invading pathogen30,31,35,38. This study contributes to disease surveillance in this species and to understanding the degree of human influence reaching isolated penguin breeding sites. Future studies would benefit from parallel environmental microbiome samples, a larger sample size, and seasonal sampling to quantify temporal patterns of bacterial pathogens in this population.
Conclusions
This work establishes a baseline microbiome for an endangered penguin, identifies two primary drivers of microbial community structure, and emphasizes the importance of minimizing interaction between wildlife and humans. Even in a site as remote and well-protected as the Galapagos Islands, human influence is still visible through factors such as antibiotic resistance genes. The human-inhabited islands also have some domesticated animals, which increases the possibility of disease spillover occurring between domestic and wild species—the apparent pathogenicity of C. perfringens found in Galapagos penguins is concerning when considering the proximity of the penguin population to domestic chickens95. Thus, monitoring and limiting anthropogenic effects on wildlife is critical to the continued long-term preservation of Galapagos endemic species.
Data availability
The raw sequencing files corresponding to this article are available in the NCBI Sequence Read Archive (SRA) repository under BioProject ID PRJNA794207 (https://www.ncbi.nlm.nih.gov/sra/PRJNA794207). R scripts and associated files are available at https://github.com/sd784/Rohrer_etal_GAPEmicrobiome.
Abbreviations
- PCR:
-
Polymerase chain reaction
- KMA:
-
K-mer alignment
- VF:
-
Virulence factor
- VFDB:
-
Virulence factor database
- CARD:
-
Comprehensive antibiotic resistance database
- PATRIC:
-
Pathosystems resource integration center
- Pfam:
-
Protein family database
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PCoA:
-
Principal coordinates analysis
- PERMANOVA:
-
Permutational multivariate analysis of variance
References
Hills, R. D. et al. Gut microbiome: Profound implications for diet and disease. Nutrients 11, 1613 (2019).
Lazar, V. et al. Aspects of gut microbiota and immune system interactions in infectious diseases, immunopathology, and cancer. Front. Immunol. 9, 1830 (2018).
Brown, S. P., Cornforth, D. M. & Mideo, N. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends Microbiol. 20, 336–342 (2012).
Krismer, B., Weidenmaier, C., Zipperer, A. & Peschel, A. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat Rev Microbiol 15, 675–687 (2017).
Trevelline, B. K., Fontaine, S. S., Hartup, B. K. & Kohl, K. D. Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proc. R. Soc. B 286, 20182448 (2019).
Bahrndorff, S., Alemu, T., Alemneh, T. & Lund Nielsen, J. The microbiome of animals: Implications for conservation biology. Int. J. Genomics 2016, 7 (2016).
Videvall, E. et al. Early-life gut dysbiosis linked to juvenile mortality in ostriches. Microbiome 8, 147 (2020).
Gu, W. et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat. Med. 27, 115–124 (2021).
Wheeler, E., Hong, P.-Y., Bedon, L. C. & Mackie, R. I. Carriage of antibiotic-resistant enteric bacteria varies among sites in Galápagos reptiles. J. Wildl. Dis. 48, 56–67 (2012).
Roggenbuck, M. et al. The microbiome of New World vultures. Nat. Commun. 5, 5498 (2014).
Carmody, R. N. et al. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84 (2015).
Waite, D. W. & Taylor, M. W. Exploring the avian gut microbiota: Current trends and future directions. Front Microbiol. 6, 673 (2015).
Michel, A. J. et al. The gut of the finch: uniqueness of the gut microbiome of the Galápagos vampire finch. Microbiome 6, 167 (2018).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014).
Tasnim, N., Abulizi, N., Pither, J., Hart, M. M. & Gibson, D. L. Linking the gut microbial ecosystem with the environment: Does gut health depend on where we live?. Front Microbiol. 8, 1935 (2017).
Hu, X. et al. High-throughput analysis reveals seasonal variation of the gut microbiota composition within forest musk deer (Moschus berezovskii). Front. Microbiol. 9, 1674 (2018).
Song, S. J. et al. Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. MBio 11, e02901-19 (2020).
Vemuri, R. et al. The microgenderome revealed: Sex differences in bidirectional interactions between the microbiota, hormones, immunity and disease susceptibility. Semin. Immunopathol. 41, 265–275 (2019).
Escallón, C., Belden, L. K. & Moore, I. T. The cloacal microbiome changes with the breeding season in a wild bird. Integr. Org. Biol. 1, oby009 (2019).
Bolnick, D. I. et al. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat. Commun. 5, 4500 (2014).
Markle, J. G. M. et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339, 1084–1088 (2013).
Dewar, M. L. et al. Microbiota of little penguins and short-tailed shearwaters during development. PLoS ONE 12, e0183117 (2017).
Bolnick, D. I. et al. Major Histocompatibility Complex class IIb polymorphism influences gut microbiota composition and diversity. Mol. Ecol. 23, 4831–4845 (2014).
Taylor, M. J. et al. Age-related variation in the oral microbiome of urban Cooper’s hawks (Accipiter cooperii). BMC Microbiol. 19, 1–10 (2019).
Pannoni, S. B., Proffitt, K. M. & Holben, W. E. Non-invasive monitoring of multiple wildlife health factors by fecal microbiome analysis. Ecol. Evol. 12, e8564 (2022).
Amato, K. R. et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 7, 1344–1353 (2013).
Murray, M. H. et al. Gut microbiome shifts with urbanization and potentially facilitates a zoonotic pathogen in a wading bird. PLoS ONE https://doi.org/10.1101/718213 (2019).
Lee, K. et al. Mobile resistome of human gut and pathogen drives anthropogenic bloom of antibiotic resistance. Microbiome 8, 2 (2020).
Smith, O. M., Snyder, W. E. & Owen, J. P. Are we overestimating risk of enteric pathogen spillover from wild birds to humans?. Biol. Rev. Camb. Philos. Soc. 95, 652 (2020).
Vargas, H., Lougheed, C. & Snell, H. Population size and trends of the Galápagos Penguin Spheniscus mendiculus. Ibis 147, 367–374 (2005).
Borboroglu, P. G. & Boersma, P. D. Penguins: Natural History and Conservation (University of Washington Press, 2013).
Steinfurth, A., Vargas, F., Wilson, R., Spindler, M. & Macdonald, D. Space use by foraging Galápagos penguins during chick rearing. Endang. Species Res. 4, 105–112 (2008).
Vargas, F. H., Harrison, S., Rea, S. & Macdonald, D. W. Biological effects of El Niño on the Galápagos penguin. Biol. Conserv. 127, 107–114 (2006).
Akst, E. P., Boersma, P. D. & Fleischer, R. C. A comparison of genetic diversity between the Galapagos Penguin and the Magellanic Penguin. Conserv. Genet. 3, 375–383 (2002).
Bollmer, J. L., Vargas, F. H. & Parker, P. G. Low MHC variation in the endangered Galápagos penguin (Spheniscus mendiculus). Immunogenetics 59, 593–602 (2007).
Nims, B. D., Vargas, F. H., Merkel, J. & Parker, P. G. Low genetic diversity and lack of population structure in the endangered Galápagos penguin (Spheniscus mendiculus). Conserv. Genet. 9, 1413–1420 (2008).
Sommer, S. The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool. 2, 16 (2005).
Levin, I., Outlaw, D., Vargas, F. & Parker, P. Plasmodium blood parasite found in endangered Galapagos penguins (Spheniscus mendiculus). Biol. Conserv. 142, 3191–3195 (2009).
Travis, E. K. et al. Hematology, serum chemistry, and serology of Galápagos penguins (Spheniscus mendiculus) in the Galápagos Islands. Ecuador. J Wildl. Dis. 42, 625–632 (2006).
Deem, S. L. et al. Exposure to Toxoplasma gondii in Galapagos Penguins (Spheniscus mendiculus) and Flightless Cormorants (Phalacrocorax harrisi) in the Galapagos Islands. Ecuador. J. Wildl. Dis. 46, 1005–1011 (2010).
Merkel, J. et al. Microfilariae in Galàpagos penguins (Spheniscus mendiculus) and flightless cormorants (Phalacrocorax harrisi): Genetics, morphology, and prevalence. J. Parasitol. 93, 495–503 (2007).
Overbey, K. N., Hatcher, S. M. & Stewart, J. R. Water quality and antibiotic resistance at beaches of the Galápagos Islands. Front. Environ. Sci. 3, 64 (2015).
Nieto-Claudin, A., Esperón, F., Blake, S. & Deem, S. L. Antimicrobial resistance genes present in the faecal microbiota of free-living Galapagos tortoises (Chelonoidis porteri). Zoonoses Public Health 66, 900–908 (2019).
Song, S. J. et al. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSystems 1, e00021-16 (2016).
Cappello, C. & Boersma, P. Sexing Galápagos penguins Spheniscus mendiculus by morphological measurements. Endang. Species. Res. 35, 169–173 (2018).
Jiménez-Uzcátegui, G. et al. Normal physical parameters characterizing Galapagos marine birds. Mar. Ornithol. 49, 257–263 (2021).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K. & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 79, 5112–5120 (2013).
Thompson, L. R. et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 551, 457–463 (2017).
Schloss, P. D. et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Yilmaz, P. et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648 (2014).
R Core Team. R: A language and environment for statistical computing. Foundation for Statistical Computing, Vienna, Austria https://www.R-project.org/ (2020).
Franzosa, E. A. et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968 (2018).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Liu, B., Zheng, D., Jin, Q., Chen, L. & Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692 (2019).
Alcock, B. P. et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525 (2020).
Davis, J. J. et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 48, D606–D612 (2020).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
van der Walt, A. J. et al. Assembling metagenomes, one community at a time. BMC Genomics 18, 521 (2017).
Mikheenko, A., Saveliev, V. & Gurevich, A. MetaQUAST: Evaluation of metagenome assemblies. Bioinformatics 32, 1088–1090 (2016).
Dong, X. & Strous, M. An integrated pipeline for annotation and visualization of metagenomic contigs. Front. Genet. 10, 999 (2019).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 51, D587–D592 (2023).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Kers, J. G. & Saccenti, E. The power of microbiome studies: Some considerations on which alpha and beta metrics to use and how to report results. Front. Microbiol. 12, 4366 (2021).
Anderson, M. J. & Walsh, D. C. I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing?. Ecol. Monogr. 83, 557–574 (2013).
Oksanen, J. et al. vegan: Community ecology package. https://CRAN.R-project.org/package=vegan (2019).
Anderson, M. J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 26, 32–46 (2001).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 57, 289–300 (1995).
Dewar, M. L. et al. Interspecific variations in the gastrointestinal microbiota in penguins. MicrobiologyOpen 2, 195–204 (2013).
Lee, W. Y. et al. Faecal microbiota changes associated with the moult fast in chinstrap and gentoo penguins. PLoS ONE 14, e0216565 (2019).
Boersma, P. D., Cappello, C. D. & Merlen, G. First observations of post-fledging care in Galapagos Penguins (Spheniscus mendiculus). Wilson J. Ornithol. 129, 186–191 (2017).
Archie, E. A. & Tung, J. Social behavior and the microbiome. Curr. Opin. Behav. Sci. 6, 28–34 (2015).
Vasconez, F. J. et al. The different characteristics of the recent eruptions of Fernandina and Sierra Negra volcanoes (Galápagos, Ecuador). Volcanica 1, 127–133 (2018).
González-Santana, D., Santana-Casiano, J. M., González, A. G. & González-Dávila, M. Coastal carbonate system variability along an active lava–seawater interface. Front. Mar. Sci. 9, 1588 (2022).
Wang, J. et al. Response of bacterial communities to variation in water quality and physicochemical conditions in a river-reservoir system. Global Ecol. Conserv. 27, e01541 (2021).
Jiménez-Uzcátegui, G. et al. Lead and cadmium levels in Galapagos penguin Spheniscus mendiculus, flightless cormorant Phalacrocorax harrisi, and waved albatross Phoebastria irrorata. Mar. Ornithol. 45, 159–163 (2017).
Xia, J. et al. Chronic exposure to low concentrations of lead induces metabolic disorder and dysbiosis of the gut microbiota in mice. Sci. Total Environ. 631–632, 439–448 (2018).
Marcelino, V. R. et al. Meta-transcriptomics reveals a diverse antibiotic resistance gene pool in avian microbiomes. BMC Biol. 17, 31 (2019).
Ventola, C. L. The antibiotic resistance crisis. P & T 40, 277–283 (2015).
Grehs, B. W. N., Linton, M. A. O., Clasen, B., de Oliveira Silveira, A. & Carissimi, E. Antibiotic resistance in wastewater treatment plants: Understanding the problem and future perspectives. Arch Microbiol. 203, 1009–1020 (2021).
Crespo, R., Franca, M. & Shivaprasad, H. L. Ulcerative enteritis-like disease associated with Clostridium sordellii in quail. Avian Dis. 57, 698–702 (2013).
Cooper, K. K., Songer, J. G. & Uzal, F. A. Diagnosing clostridial enteric disease in poultry. J. Vet. Diagn. 25, 314–327 (2013).
Varan Singh, S. & Singh, B. R. Gallibacterium anatis: An emerging pathogen of poultry birds and domiciled birds. J. Vet. Sci. Technol. 07, 324 (2015).
Persson, G. & Bojesen, A. M. Bacterial determinants of importance in the virulence of Gallibacterium anatis in poultry. Vet. Res. 46, 1–11 (2015).
Rivas, A. J., Lemos, M. L. & Osorio, C. R. Photobacterium damselae subsp. damselae, a bacterium pathogenic for marine animals and humans. Front. Microbiol. 4, 283 (2013).
Kiu, R. & Hall, L. J. An update on the human and animal enteric pathogen Clostridium perfringens. Emerg. Microbes Infect. 7, 1–15 (2018).
Petit, L., Gibert, M. & Popoff, M. R. Clostridium perfringens: Toxinotype and genotype. Trends Microbiol. 7, 104–110 (1999).
Penrith, M., Huchzermeyer, F. W., De Wet, S. C. & Penrith, M. J. Concurrent infection with Clostridium and Plasmodium in a captive king penguin Aptenodytes patagonicus. Avian Pathol. 23, 373–380 (1994).
Lepp, D. et al. Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS ONE 5, e10795 (2010).
Vargas, F. et al. Modelling the effect of El Niño on the persistence of small populations: The Galápagos penguin as a case study. Biol. Cons. 137, 138–148 (2007).
Gottdenker, N. L. et al. Assessing the risks of introduced chickens and their pathogens to native birds in the Galápagos Archipelago. Biol. Conserv. 126, 429–439 (2005).
Wickham, H. et al. Welcome to the tidyverse. JOSS 4, 1686 (2019).
Wilke, C. O. cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’. https://CRAN.R-project.org/package=cowplot (2020).
Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. https://CRAN.R-project.org/package=ggpubr (2023).
Kahle, D. & Wickham, H. ggmap: Spatial Visualization with ggplot2. The R Journal 5, 144 (2013).
Baquero, O. S. ggsn: North Symbols and Scale Bars for Maps Created with ‘ggplot2’ or ‘ggmap’. https://CRAN.R-project.org/package=ggsn (2019).
Slowikowski, K. et al. ggrepel: Automatically Position Non-Overlapping Text Labels with ‘ggplot2’. https://CRAN.R-project.org/package=ggrepel (2023).
Acknowledgements
We are very grateful to our collaborators and assistants who helped in this research, particularly to Jane Merkel and Dr. Maricruz Jaramillo for their valuable contributions through sample collection and laboratory work. We thank the Ecuadorian Ministry of the Environment and Water and the Galapagos National Park Directorate for granting research permissions. We are grateful to the Charles Darwin Foundation and to the Agency for the Regulation and Control of Biosecurity and Quarantine for Galapagos for their support. This research was supported in part by the University of Michigan Medical School Microbiome Core. The computation for this work was performed on the high performance computing infrastructure provided by Research Computing Support Services and in part by the National Science Foundation under grant number CNS-1429294 at the University of Missouri, Columbia MO. This publication is contribution number 2405 of the Charles Darwin Foundation for the Galapagos Islands.
Funding
Funding support for this project was generously provided by the NSF Graduate Research Fellowship #1945995, Saint Louis Zoo Field Research for Conservation Grant #2018.02, Saint Louis Zoo WildCare Institute, Des Lee Collaborative Vision, NIH Grant #AI137984, Harris World Ecology Center, and by the donors of the Galapagos Penguin, Flightless Cormorant and Waved Albatross Project at the Charles Darwin Foundation: The Leona M. and Harry B. Helmsley Charitable Trust, Penguin Fund of Japan, and Mr. Seishi Sakamoto.
Author information
Authors and Affiliations
Contributions
S.D.R., P.G.P., and L.M.C conceptualized the study. G.J.-U. organized sampling logistics, and S.D.R. and G.J.-U. collected data. S.D.R. analyzed the data and wrote the manuscript in consultation with LMC and PGP. All authors revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rohrer, S.D., Jiménez-Uzcátegui, G., Parker, P.G. et al. Composition and function of the Galapagos penguin gut microbiome vary with age, location, and a putative bacterial pathogen. Sci Rep 13, 5358 (2023). https://doi.org/10.1038/s41598-023-31826-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-31826-y
- Springer Nature Limited